DOI:
10.1039/C9RA10596D
(Paper)
RSC Adv., 2020,
10, 5636-5647
A combined spectroscopic and ab initio study of the transmetalation of a polyphenol as a potential purification strategy for food additives†
Received
16th December 2019
, Accepted 20th January 2020
First published on 4th February 2020
Abstract
Recently, metal exchange (transmetalation) techniques have become popular for the post-synthesis modification of metal organic complexes (MOCs). Here, we have explored the possibility of toxic metal ion (mercury (Hg)) exchange from a model polyphenol, curcumin, which is a very important food additive, using a much less toxic counterpart (copper). While the attachment of different metals on the polyphenol was confirmed using a picosecond resolved fluorescence technique, the surface plasmon resonance (SPR) band of the Ag nanoparticle (NP) was employed as a tool to detect uncoupled Hg ions in aqueous media. Furthermore, a microscopic understanding of the experimental observations was achieved through density functional theory (DFT) based theoretical studies. The presence of Cu ions in the vicinity of Hg–curcumin, upon ground state optimization, was observed to extrude most of the Hg from the curcumin complex and replace its position in the complex. The study may find relevance in the development of a purification strategy for food additives heavily contaminated with toxic metals.
1. Introduction
Metal–organic complexes (MOCs) are a class of organic–inorganic hybrid substances composed of metals linked with an organic moiety.1 In recent times, MOCs have rapidly grown as one of the most dynamic areas of coordination chemistry research fields owing to their enormous potential application in various fields including storage, catalysis, sensing, optoelectronics, adsorption, biomedical imaging, enhanced medicinal activity and so forth.2–6 Although MOCs have many diverse applications, some toxic metal contaminated organometallic compounds can be very much harmful to the human body. Upon contact with biological fluids, various transformations can take place on the metal organic compound (MOC) containing toxic metal, which can be harmful to the human body.7,8 The metal exchange technique (MET) is a beneficial strategy used to modify MOCs by exchanging the metal node, which can change the properties of the organometallic compound.9,10 The toxic metal contaminated MOC could be transmetalated with a significantly reduced amount of toxic divalent transition metal cations to remove the toxic hazard.
Heavy metals are elements which have a very high density11 (greater than 5 g cm−3) and atomic weight. Heavy metal related pollution is extremely dangerous, as it directly affects human physiology. It also enters living organisms including microbes and plants through the food-chain. The intake of elements such as mercury (Hg), arsenic (As), lead (Pb) and cadmium (Cd), even in minute concentrations over a longer period of time, can be extremely hazardous to human health.13 These heavy metals are not biodegradable and also have a tendency to accumulate in human anatomical structures such as the bones, liver, brain and kidneys.12 Mercury is purported to be the most toxic heavy metal of them all.13 It is designated as being one of the most hazardous materials listed by the United States Environmental Protection Agency (USEPA), as it can move through the blood–brain barrier readily and damage the human brain.14,15 The main target organ for the metal is the living tissue, primarily in the brain.16 Methylmercury (MeHg) is known to be the most lethal of the mercury compounds.17,18 The main source of human exposure to MeHg comes from the consumption of sea fish and marine mammals.18,19 Almost 80–90% of MeHg contamination is due to the intake of contaminated seafood and sea fish.18 Although organic mercury sources (MeHg) have an intense toxic effect in the human body, this pollution is limited to sea food and sea fish. However, there are several other inorganic mercury sources present which is also very harmful to the human body.20,21 One of the most recurrent sources of mercury pollution is the aqueous stable oxidized divalent mercuric ion (Hg2+), possessing a high stability in normal environmental conditions.22 This water-stable form of Hg can create significant contamination in water and also becomes deposited in the soil through water. Contaminated and untreated industrial waste is the main source of stable Hg pollutants entering into the human food chain, mostly via usable water.23 Those toxic metals have a high tendency to chelate with essential food elements, and through the food chain, it directly enters the human body causing hazardous health effects.
Turmeric, one of the most regularly used spices and coloring agents, contains curcumin as its coloring pigment, which is isolated from the rhizomes of Curcuma longa.24,25 In addition to its primary use as a spice, it also has significant application as a phytomedicine. Curcumin is a very well known chelating agent as the turmeric powder is rich in curcuminoids, proteins and carbohydrates.26 There are a lot of available metal binding sites within curcumin.27 Moreover, curcumin has a substantial number of pores, in which metal ions can be trapped and adsorbed. Hence, curcumin can act as a natural chelating agent to many metals and forms metal–curcumin (M–Cur) complexes. A recent study has shown that the M–Cur complex is very useful for curing many diseases.28 In our earlier study, we found that the Cu–curcumin complex shows a higher anti-oxygen activity, whereas Zn–curcumin shows a better aqueous stability.3 However, this metal chelating tendency of curcumin sometimes exhibits adverse effects when it undergoes complexation with toxic heavy metal ions such as lead, mercury, cadmium and arsenic. In a sub-continental country like Bangladesh, turmeric is considered to be one of the most probable sources of lead toxicity.29 Lately, it has been observed that curcumin can become contaminated with other toxic metals such as arsenic and mercury. The water soluble form of mercury (Hg2+) can easily contaminate curcumin at the time of cultivation or transport. Moreover, contaminated curcumin used in beauty products may cause toxic metal contamination to the skin.30 The presence of Hg in curcumin can cause detrimental effects, as it affects the product quality and safety issues significantly. Therefore, invention of a simple strategy to remove harmful Hg from essential food elements is required.
The change in the surface plasmon resonance (SPR) band of Ag nanoparticles (NPs) can be used to detect Hg2+ ion efficiently in aqueous media.22 The SPR band of the NPs depend on various properties such as the size, shape, adsorbing material, distance between particles and so forth.31 It was proposed that Hg2+, after forming complexes with Ag-NPs, undergoes aggregation and amalgamation, which results in a change in the SPR band. Owing to strong metallophilic interactions, free Hg ions can easily interact with Ag-NPs and coagulate. Inorganic mercury usually forms a stable bond with an organic moiety. However, owing to the stable binding of inorganic Hg with curcumin, the detection of Hg using the SPR band of Ag-NP is not possible, as it does not interact with the SPR band of the Ag-NPs. To summarize, the detection of free Hg ions can be performed by employing the SPR band of Ag-NPs, when it is detached from curcumin.
In the present work, we have shown a possible way to exchange the toxic Hg from the Hg–curcumin complex with another chelating metal, copper, via a metal exchange technique. It was found that the LD50 value of Hg is significantly higher than Cu.7 The daily consumption limit of Hg is 20 μg per day, whereas for Cu the consumption limit is approximately 4 mg per day.32 Although it has been reported that Cu has some toxicity, it is only toxic to the body only at higher concentrations, whereas Hg has a well-known toxicity at very low concentrations.32–34 Moreover, being a vital part of several enzymes, Cu is an essential micronutrient up to a certain concentration.34 With proper Cu concentration and incubation time, it is possible to replace most of the Hg from the complex. Using a first principles theoretical calculation, we have found that the Hg–curcumin complex is energetically unstable, and that Cu–curcumin is energetically favorable. We have used a relatively cost effective and field deployable technique to detect the free Hg from the curcumin complex. The SPR band of an Ag-nanoparticle (NP) was employed to detect the dissociated Hg ion. The rate of suppression of the SPR band increased after the transmetalation process, which indicates the decoupling of Hg from curcumin. To date, some reports have described the detection of Hg contamination in curcumin, however, there are no reports concerning the decontamination of the toxic metal from curcumin. This study proposes a simple but effective method to remove toxic Hg metal from a well-known food additive polyphenol by using Cu metal and can thus be expected to be a promising technique to remove harmful contamination from essential food elements.
2. Experimental section
2.1 Materials
In this study, analytical grade chemicals were used as received, without further purification, for synthesis and sample preparation. Silver nitrate (AgNO3, 99.99%), sodium citrate, sodium borohydride, curcumin, mercury(II) nitrate monohydrate (Hg(NO3)2·H2O), cupric chloride dehydrate (CuCl2·2H2O) were purchased from Sigma Aldrich. Methanol (Merck) was used as a suitable solvent to synthesize the M–Cur complexes. For all other studies, Cur and M–Cur complexes were dissolved in a methanol water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture. Millipore water was used as an aqueous solvent.
:1) mixture. Millipore water was used as an aqueous solvent.
2.2 Synthesis procedure
2.2.1. Synthesis of the Hg–curcumin complex. The Hg–curcumin complex was prepared by following our earlier reports.35 Curcumin (50 mL, 2 mM) was dissolved in methanol by stirring and heating at 60 °C. 2 mM Hg(NO3)·2H2O was dissolved in 100 mL methanol. This solution was added into the curcumin. Immediately a reddish yellow color precipitation was visible upon the addition, which confirmed the complexation reaction. The reaction mixture was refluxed for 2 h. The product was filtered and washed several times to remove the residual reactants.
2.2.2. Synthesis of the Cu(II)–curcumin complex from the Hg(II)–curcumin complex. The washed Hg–curcumin complex was placed in methanol and a methanolic 2 mM solution of CuCl2·2H2O was added to the collected supernatant solution and cyclomixed for 2 h. A pale-brown solution was generated. Half of this solution was taken for further experiments and the remaining part of the solution was washed several times with water and methanol and dried for energy-dispersive X-ray spectroscopy (EDS) measurements.
2.2.3. Synthesis of silver nanoparticles. Citrate functionalized Ag-NP was synthesized at ∼0 °C in an aqueous solution (pH = 6.5) by following the synthesis technique described below .36 In a typical synthesis process, 1 mL of a 5 mM aqueous solution of AgNO3 was added to 16 mL of a 1 mM aqueous solution of sodium citrate. The solution was then placed under continuous stirring condition for 2 h within an ice-bath, maintaining the reaction temperature at 0 °C. 150 μL of a 5 mM aqueous NaBH4 solution was added to the solution dropwise. The color of the solution turned yellow. The as-synthesized NPs were kept at 0 °C until further use. The NPs were characterized using various spectroscopic techniques.
2.3 Characterization
The absorbance density of all of the samples was measured by using a Shimadzu UV-2600 spectrophotometer with a quartz cuvette of 1 cm path length. High resolution transmission electron microscopy (HRTEM) was employed to characterize the particle size and determine exhaustive structural and morphological information of the NPs. Transmission electron microscopy (TEM) samples were prepared by the dropwise addition of the diluted samples on to carbon-coated copper grids. The particle size was determined by the dynamic light scattering (DLS) method by using a Zetasizer Nano S DLS instrument. Scanning electron microscopy (SEM) images were recorded using a field emission scanning electron microscopy (FESEM, QUANTA FEG 250) electron microscope operating at 20 kV and equipped with an EDS. A picosecond resolved fluorescence study was carried out using a time correlated single photon counting (TCSPC) setup purchased from Edinburgh Instruments with an instrument response function (IRF = 80 ps) upon excitation from a 409 nm laser using the method reported previously by our group.37,38
2.4 General procedure used to determine Hg2+
To detect the Hg2+ ions in the system, we used UV-Vis spectroscopy as a tool. Approximately, 0.15 nM of Ag-NPs were placed in 2 mL of a water:methanol mixture (1:1) so that the SPR O.D. (optical density) of the Ag-NPs achieved 0.5 a.u. 20 μL of 2 mM Hg–Cur, Cu–Hg–Cur and the Hg2+ ion only were added to three different cells containing Ag-NPs. The changes in the SPR absorption peak of the Ag-NPs in the presence of Hg2+ were monitored using the UV-Vis spectrometer at ambient temperature.
3. Computational details
For the density functional calculations, we used projector augmented wave (PAW) potentials under generalized gradient approximation (GGA) with a Perdew–Burke–Ernzerhof exchange correlation functional as implemented in the Vienna ab initio simulation package (VASP).39 The detailed calculation methodology is depicted in our previous work.40,41 All control and assembled systems were placed in a rectangular box with the dimensions 30 × 16 × 14 Å3, with a sufficient vacuum so as to avoid influence from periodic replication. Owing to a large volume of the simulation cell for these cluster systems, single k-point (Γ point) calculations were performed for these systems with ionic optimization under a conjugate gradient algorithm until the Hellmann–Feynman force on each ion was less than 0.01 eV Å−1 van der Waals corrections were included in this simulation process using the Grimme DFT-D2 formalism.42
4. Results and discussion
4.1. Characterization of curcumin and the metal curcumin complex
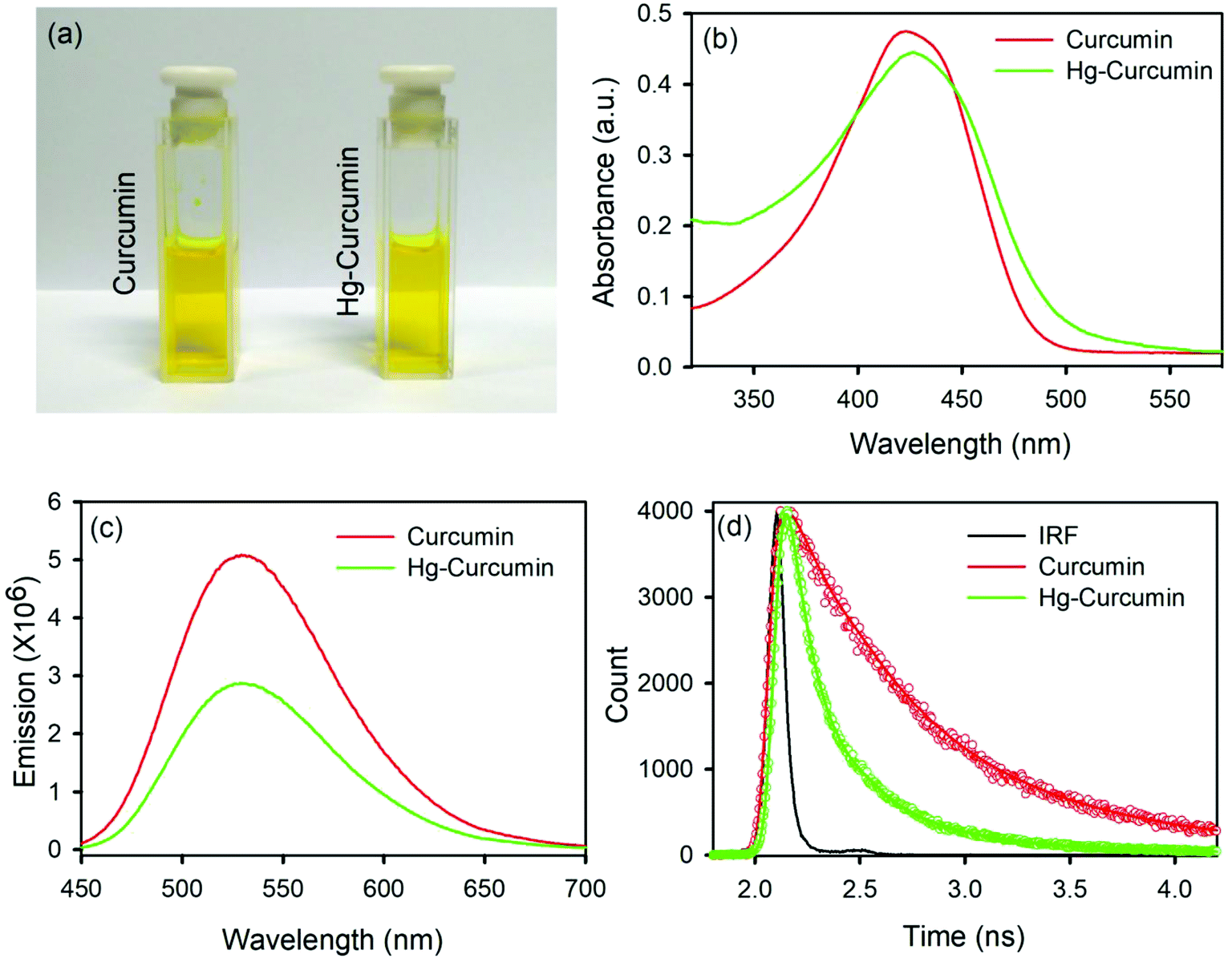
Metal complexation of curcumin causes significant transformation in the electronic structure. As a result of this, colorimetric changes may be observed in the metal curcumin with respect to pure curcumin. However, the Hg–curcumin complex cannot be distinguished from curcumin by simple visualization, as it is the same color. Fig. 1a shows the colorimetric image of Hg–curcumin and curcumin of the same concentration. The absorption peak of curcumin and Hg–curcumin using the UV-Vis spectrometer is depicted in Fig. 1b. The absorption peak of curcumin is at 424 nm owing to the π–π* transition, whereas Hg–curcumin shows an absorption maxima at 427 nm. Hg–curcumin does not show a significant change in absorption after complexation. Hence, it is very challenging to detect, as well as remove Hg, from the curcumin complex to avoid hazardous contamination. In the present work, we have described a new strategy to remove toxic Hg from the Hg–curcumin complex. The O.D. normalized steady state emission spectra of curcumin and Hg–curcumin are shown in Fig. 1c. A significant decrement for the emission intensity of curcumin was found after metal attachment, indicating the generation of new non-radiative processes. The fluorescence quenching of curcumin in the presence of Hg metal is a clear indication of the attachment of curcumin to the metal. To further confirm the attachment of metal in the curcumin complex, the picosecond resolved fluorescence decay of curcumin and Hg–curcumin was monitored in a glycerol medium at 530 nm using a 409 nm laser. Fig. 1d depicts the decay profile of curcumin and Hg–curcumin. The decay profile of curcumin shows a bi-exponential component for which the shorter component (446 ps) corresponds to the solvation dynamics and the longer component (1.08 ns) is due to the excited-state intramolecular hydrogen atom transfer.28 An additional shorter component of ∼100 ps was generated upon the metalation of curcumin. The positively charged metal ion in the metal curcumin complex behaves as an electron acceptor and the ligand curcumin in the metallo–curcumin complex behaves as an electron donor to the metal in the complex system.43 The shorter lifetime could be attributed to the photoinduced electron transfer process from curcumin to metal ions.44 Details of the time scale are tabulated in Table 1.
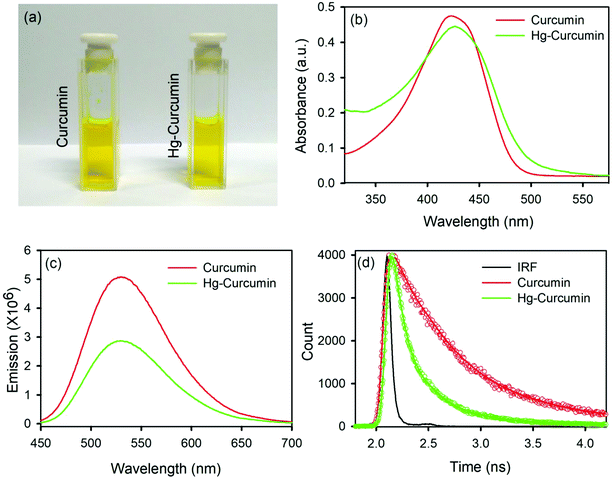 |
| | Fig. 1 (a) A pictorial image of Hg–curcumin and curcumin; (b) absorption spectra of curcumin and Hg–curcumin; (c) emission spectra of curcumin and Hg–curcumin; and (d) time resolved fluorescence decay of curcumin and Hg–curcumin. | |
Table 1 Picosecond resolved fluorescence transient data for curcumin and metallo–curcumin samples (the numbers in parentheses indicate the relative contributions)
| |
τ1 (ps) |
τ2 (ps) |
τ3 (ps) |
τavg (ps) |
| Curcumin |
— |
446 (65.51) |
1081 (34.49) |
664.96 |
| Hg–curcumin |
85 (58.82) |
274 (35.29) |
855 (5.88) |
197.0 |
| Cu–curcumin |
126 (66.22) |
521 (31.08) |
2457 (2.7) |
311.7 |
| Cu–Hg–curcumin |
87 (59.25) |
342 (33.33) |
1309 (7.41) |
262.51 |
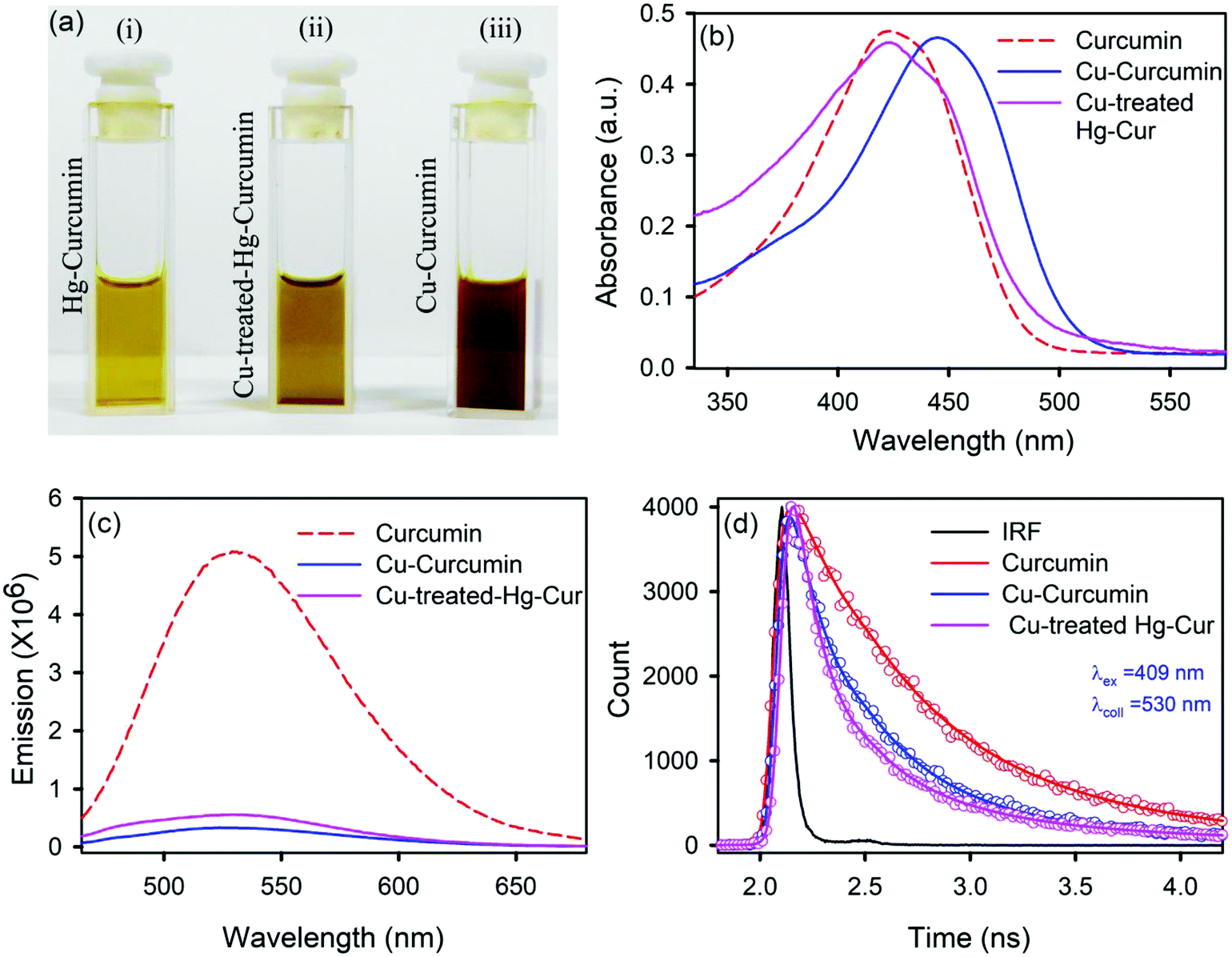
We employed a wet chemical transmetalation technique to replace the Hg metal attached to curcumin with Cu metal. The transformation process was evaluated by allowing the Hg–curcumin to react in the aqueous solution of copper(II) acetate. The detailed mechanism of MET is described in the next section. The pictorial image of Hg–curcumin, Cu–curcumin and Cu-treated Hg–curcumin at the same concentration is displayed in Fig. 2a. The color of Hg–curcumin is light yellow and Cu–curcumin is deep brown, whereas Cu-treated Hg–curcumin has a color that is in between the two. Fig. 2b shows the absorbance spectra of the pure Hg–curcumin, Cu–curcumin and Cu–Hg–curcumin complex. There is a huge change in the absorption spectra of the Hg–curcumin and Cu–curcumin. Cu–curcumin shows a red shifted absorption peak at ∼447 nm. This redshift can be attributed to the generation of new electronic states, which are lower in energy. Another reason for the shift may be the participation of the carbonyl group in the metal curcumin complex formation.45 The Cu-treated Hg–curcumin shows two peaks at ∼424 and ∼445 nm which correspond to the contribution of both Hg–curcumin and Cu–curcumin respectively. This observation suggests that a part of Hg–curcumin converts to Cu–curcumin after the transmetalation reaction. The O.D. normalized steady state emission spectra of Hg–curcumin, Cu-treated Hg–curcumin and Cu–curcumin are shown in Fig. 2c. The excited state decay of Cu–curcumin and Hg–curcumin after Cu treatment are shown in Fig. 2d. It should be noted that the decay pattern of Hg–curcumin after Cu-treatment resembles the decay of Cu–curcumin. This result is a clear indication that Hg–curcumin changes to Cu–curcumin after modification by copper.
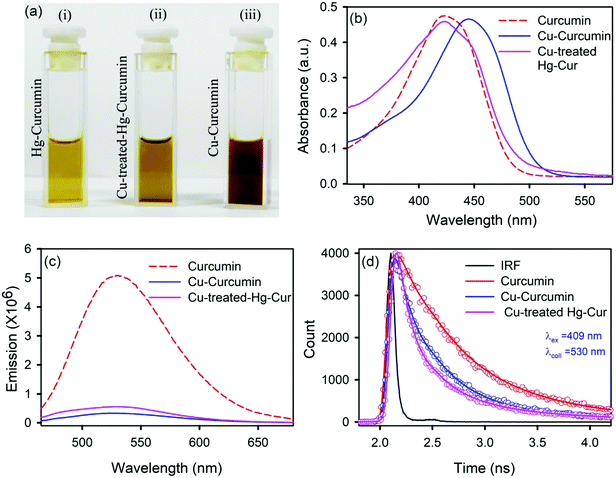 |
| | Fig. 2 (a) A pictorial image of Hg–curcumin, Cu-treated Hg–curcumin and Cu–curcumin; (b) absorption spectra of curcumin, Cu–curcumin and Cu-treated Hg–curcumin; (c) emission spectra of curcumin, Hg–curcumin and Cu–curcumin; and (d) the excited state fluorescence decay of curcumin, Cu-treated curcumin and Cu–curcumin. | |
4.2. Possible transmetalation mechanism to remove toxic Hg metal from the curcumin complex
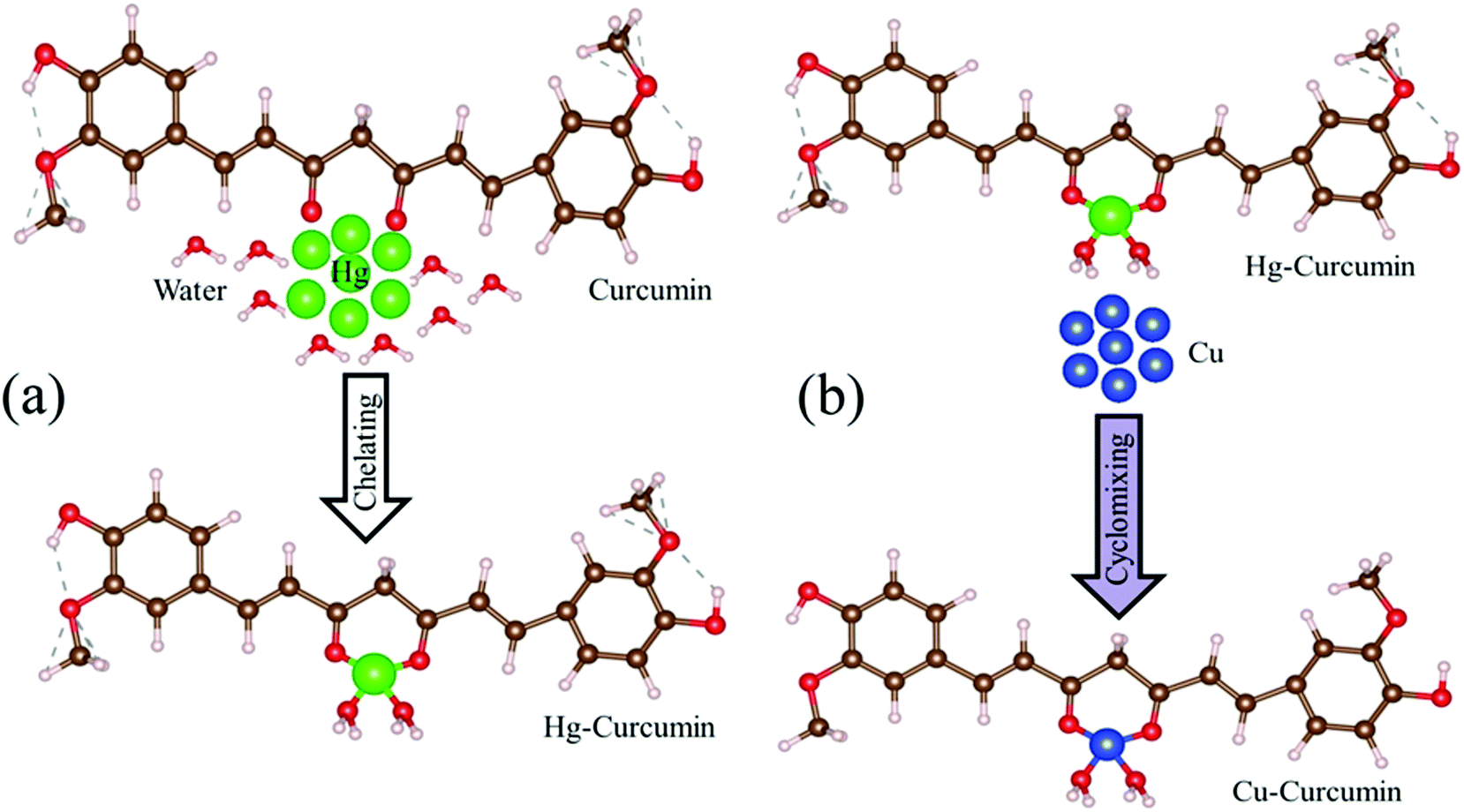
Mercury is reported to be one of the most toxic heavy metals, and can easily enter our body through the food chain. Scheme 1a represents the complexation of the Hg ion with curcumin to form Hg–curcumin. We have utilized MET to remove the toxic Hg ion in the curcumin complex by replacing it with a Cu ion which is toxic only at a much higher concentration. According to the activity series of metals in chemistry, copper is more reactive than mercury. Hence, for a single-replacement reaction (in which one element replaces another element in a compound), Cu can replace Hg in a compound system.46 Crystal field theory (CFT) describes the electrostatic interaction between a transition metal and ligand, which originates owing to the attraction between the positively charged metal cation center and the negatively charge ligand site.47 CFT can approximately illustrate the strength of the metal–ligand bonds in the system. According to CFT, the stability of the metal–ligand complexes will increase with the increasing ionic potential. Cu2+ is a d9 system which exhibits a much higher stability, as the six coordinate Cu2+ complexes undergo a Jahn–Teller distortion to form a system with a lower symmetry and lower energy.48 The transmetalation process can also be explained using Lewis acid–base theory.49 According to HSAB (hard and soft (Lewis) acids and bases) theory, the hard acid reacts faster with a hard base and forms much stronger bonds among themselves. In the present case, Hg2+ is a soft acid, whereas Cu2+ is harder acid and the beta-diketone of curcumin is a hard base. Therefore, attachment of Cu2+ with curcumin is much more favorable compared to Hg2+.39 The above discussion demonstrates that for the Cu metal, the formation of the metal–organic complex is energetically more stable than that of the Hg metal. The metal exchange reaction is shown in eqn (1).| | |
Hg–curcumin + Cu2+ = Cu–curcumin + Hg2+
| (1) |
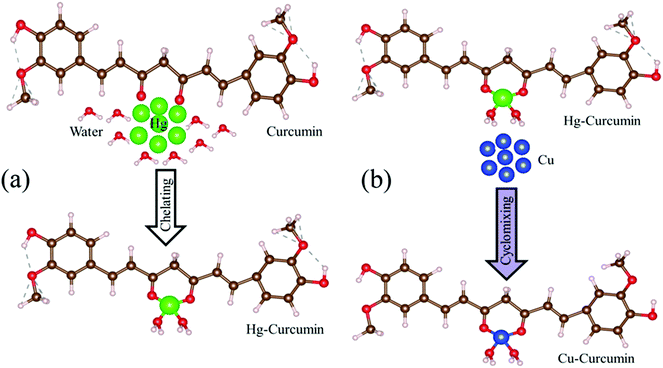 |
| | Scheme 1 (a) The natural route to Hg chelation with curcumin to form the Hg–curcumin complex. (b) A pictorial representation of Cu replacement in Hg–curcumin, which results in the formation of Cu–curcumin. | |
The graphical representation of the process is described in Scheme 1b. This Cu–curcumin removes the hazardous Hg metal, and at the same time, it has a much higher antioxidant activity than pure curcumin.3 Therefore, it is possible to convert a heavily toxic contaminated material to a much less toxic one, and a more health friendly material, using a simple transmetalation process.
4.3. Theoretical prediction of the removal technique of toxic Hg-metal from the Hg–curcumin complex
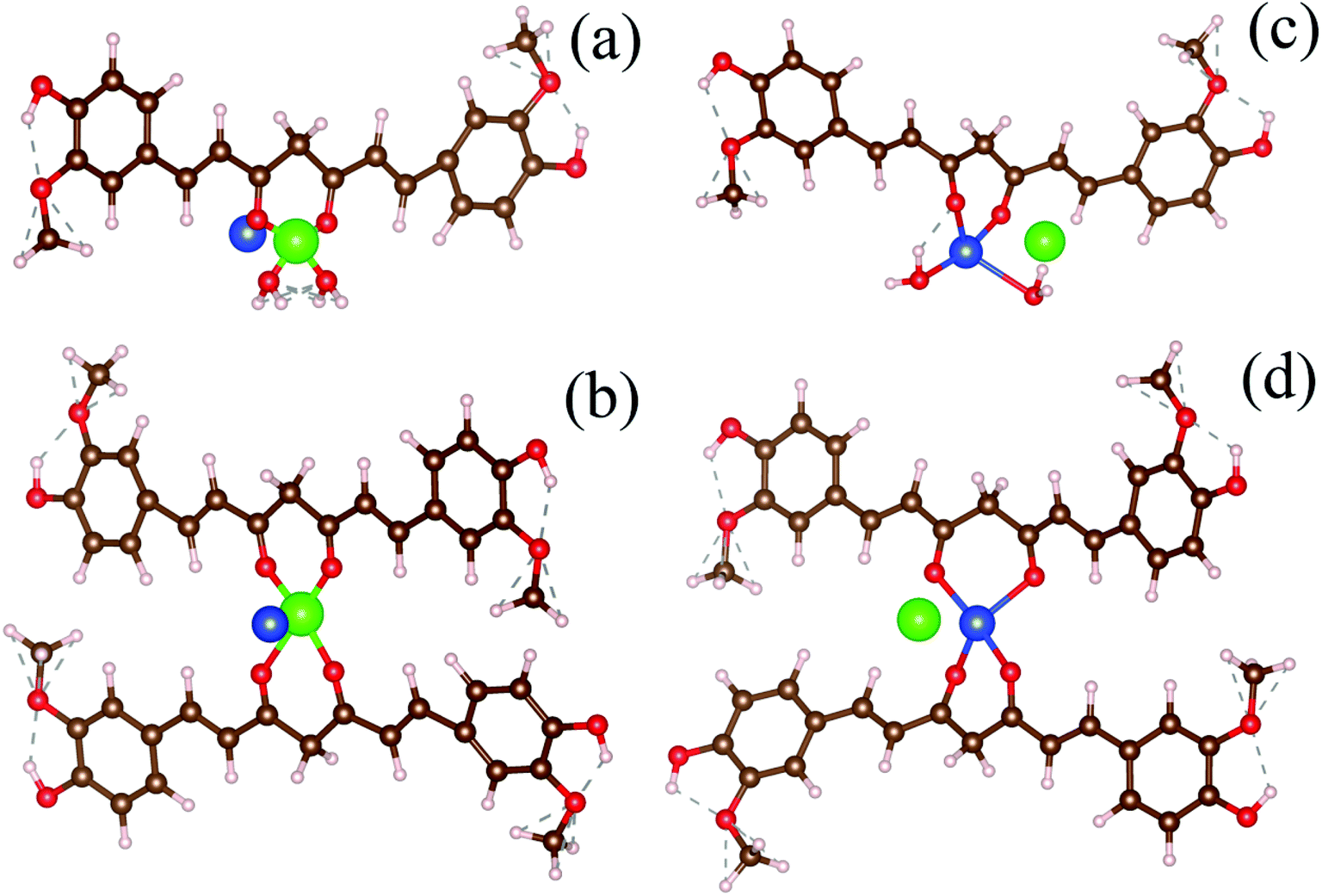
To simulate the present condition, we placed the Hg–curcumin in an aqueous environment inside a box with the dimensions mentioned earlier. Deprotonation of the hydroxyl group in the curcumin system leads to the formation of a bidentate β-diketonate moiety, which has a strong tendency to chelate with transition metal ions in two probable geometries forming monodentate (1:1) and bidentate (1:2) complexes.50 We have considered the possibility of both types of metal chelation. In the monodentate condition, one metal ion is attached to one curcumin, and in the bidentate condition, one metal ion is attached to two curcumin. Free Cu-ion was placed in the vicinity of the Hg–curcumin. Fig. 3a and b shows the initial structure of the system, from two different orientations. It is clear from the figure that Hg is attached to the curcumin, whereas the Cu is free within the system. Next, this complex system was allowed to undergo complete structural relaxation via minimization of the force and energy. The energy optimized relaxed systems are reproduced in Fig. 3c and d. It was found that upon optimization, the Cu ion replaces the Hg from the Hg–curcumin complex and forms the Cu–curcumin complex and sets the Hg free in the environment. The bond length shown in Table 2 clearly indicates that the distance of Hg from the curcumin chain increased, whereas the distance of the Cu bond decreased. The ESI Video file SV1† shows the trajectory of the system during the relaxation process. It is very evident from the video file, that the mercury atom has been run off gradually from the Hg–curcumin complex in the presence of copper, and the copper is replaced at the center of the curcumin chain after attaining structural relaxation. We have calculated the formation energy of the Hg–curcumin and Cu–curcumin. The formation energy of Cu–curcumin is −1.4 eV, which is energetically more favorable than that of Hg–curcumin (0.4 eV). This explains why the formation of Cu–curcumin is more energetically favorable than Hg–curcumin. Mercury is a filled d10 system, whereas Cu is just one electron away from a completely filled d-orbital. Therefore, the unfilled d-orbital of Cu has a higher affinity to fully fill itself and thus promotes easier chelation than Hg.36 Thus, Cu–curcumin complexation is much energetically feasible than Hg–curcumin. The final and initial bond lengths of the Hg and Cu atoms are tabulated in Table 1. This free Hg ion mixes with water and increases the amount of Hg ions in the water. It is possible to detect the increment of Hg ions in the aqueous solution using the experimental technique to verify the detoxification of curcumin.
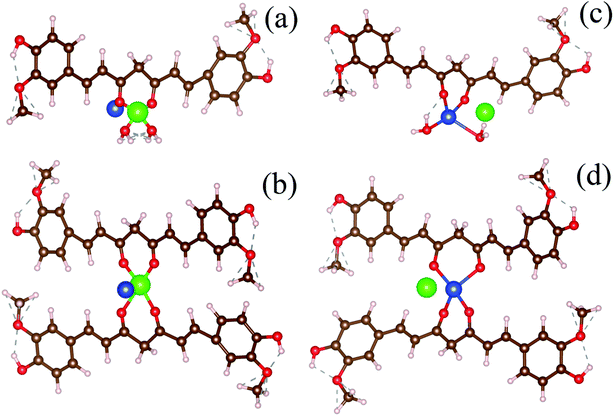 |
| | Fig. 3 (a and b) The initial structures of monodentate and bidentate Hg–curcumin in a Cu environment. (c and d) The energy optimized final structures of the systems showing the Cu-ion replacing Hg from curcumin in the monodentate and bidentate metal-curcumin systems. | |
Table 2 Distance of the Cu and Hg atoms from the curcumin-chain
| System |
Distance from the curcumin chain |
| Initial (Å) |
Final optimized (Å) |
| Hg |
1.32 |
3.73 |
| Cu |
2.39 |
1.92 |
4.4. Sensing detection of Hg2+
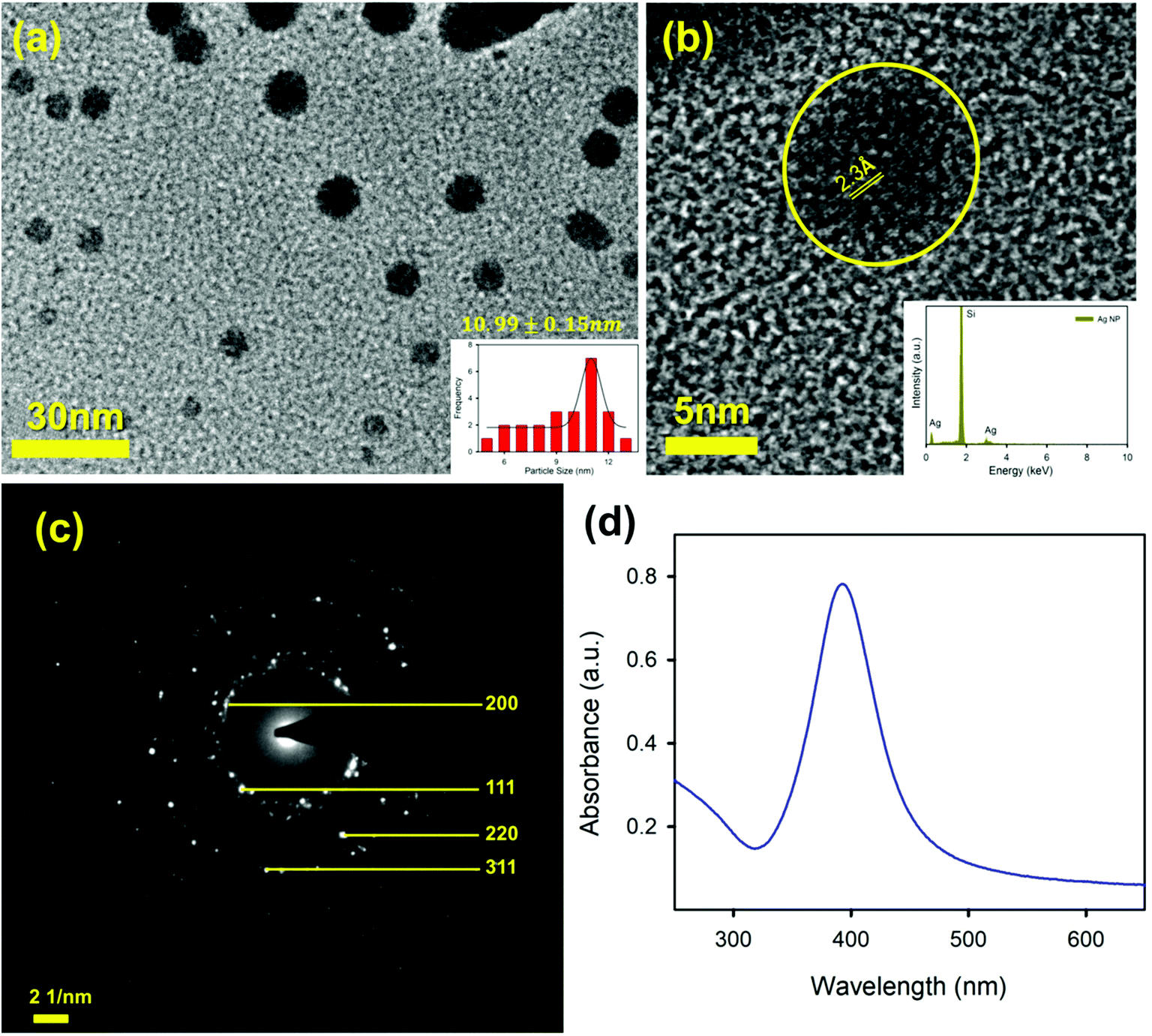
To sense the free Hg2+ ion in the aqueous medium we employed the SPR band of the Ag-NP as a probe for the detection. Citrate capped Ag-NPs were prepared by using a simple chemical synthesis process. The size and shape of the NPs were characterized by using TEM analysis. Fig. 4a shows the TEM image of the citrate capped Ag-NPs. It is clearly evident from the figure that the NPs are spherical in shape. The particle size distribution is shown in the inset of Fig. 4a. The average particle size of the NPs was found to be ∼9.12 ± 0.09 nm. The HRTEM image of the Ag-NPs shows detailed crystallographic information about the structure. The inter-planar distance calculated from the fringes-width is around 0.23 nm, which corresponds to the [111] lattice plane of the Ag-NPs.51 The inset of the figure shows the EDS spectra of the Ag-NPs, which confirms the presence of Ag-NPs in the system. Fig. 4c shows the selected area electron diffraction (SAED) patterns of the Ag-NPs. The diffraction rings on the SAED patterns can be well indexed with previous literature, which corresponds to the FCC structure of the Ag-NPs. The spacings of the diffraction rings are 0.20, 0.23, 0.14 and 0.12 nm, which correspond to the (200), (111), (220) and (311) lattice planes of the FCC Ag particle respectively.52 The citrate capped Ag-NPs show a yellowish-brown color in aqueous solution. The as-prepared citrate capped Ag-NPs have a yellowish color and show a characteristic SPR band ∼395 nm (shown in Fig. 4d).53 This absorption peak is generated by the excitation of the SPR band vibrations of the Ag-NPs. The average particle size calculated from the absorbance band of the synthesized NPs was roughly ∼11 nm,36 which is consistent with the particle size obtained from the TEM images.
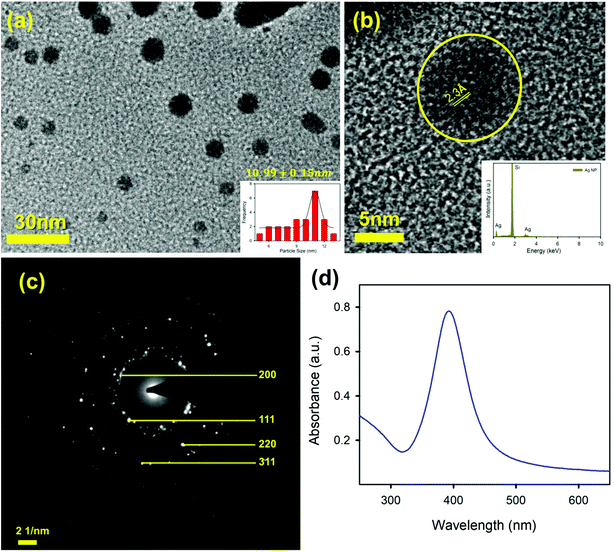 |
| | Fig. 4 (a) A TEM image of the Ag-NPs; (b) a HRTEM image of an Ag-NP showing the fringe distance of 2.3 Å; the inset shows the X-ray EDS image of the Ag-NP; (c) an SAED image of the Ag-NP; and (d) the absorption spectrum of the Ag-NPs. | |
As the mercury(II) ion has a d10 configuration, it has no optical spectroscopic signature. In order to understand how the Hg2+ ion interacts with the SPR band of the Ag-NPs, a control study was carried out without adding anything to the system. To perform the sensing experiments, the initial concentration of the Ag-NPs was taken as ∼0.15 nM for all cases. The absorbance spectrum of the aqueous system remained constant (data not shown) for more than 1 h demonstrating that without the presence of any ion, the Ag-NPs remain invariant with respect to time. To detect the aqueous mercury solutions, a time dependent spectral study of the SPR spectra of the Ag-NPs was executed before and after introducing the Hg2+ aqueous solutions with known concentrations. The spectrum (depicted in Fig. 5a) instantly shows a significant change in the presence of the Hg2+ ion. In addition to the suppression of the intensity of the SPR band, there is a distinct blue shift that occurs instantly after the addition of the Hg2+ ion. This phenomena is known as the Mie blue shift, in which the distance between the Ag-NPs become closer owing to the interaction with Hg(II), thus they aggregate with each other and form an Ag–Hg–Ag complex.54–56 As a result of this, Ag/Hg amalgam formation can occur. The aggregated Ag-NPs lose the SPR band (decrease in absorbance) and increase the overall particle size.55 In the presence of the cationic Hg2+ ion, the citrate capped Ag-NPs start aggregating owing to the hydrophobic effect,42 as a result of that the average particle size increases.
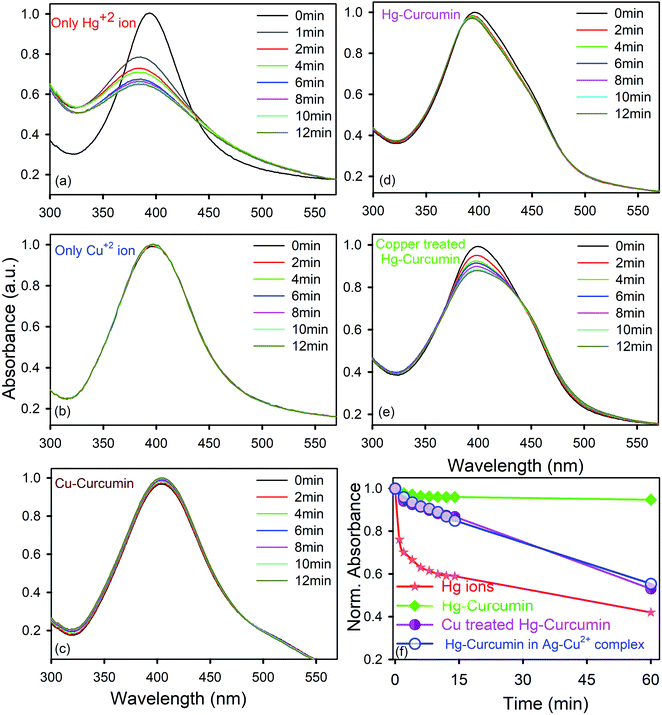 |
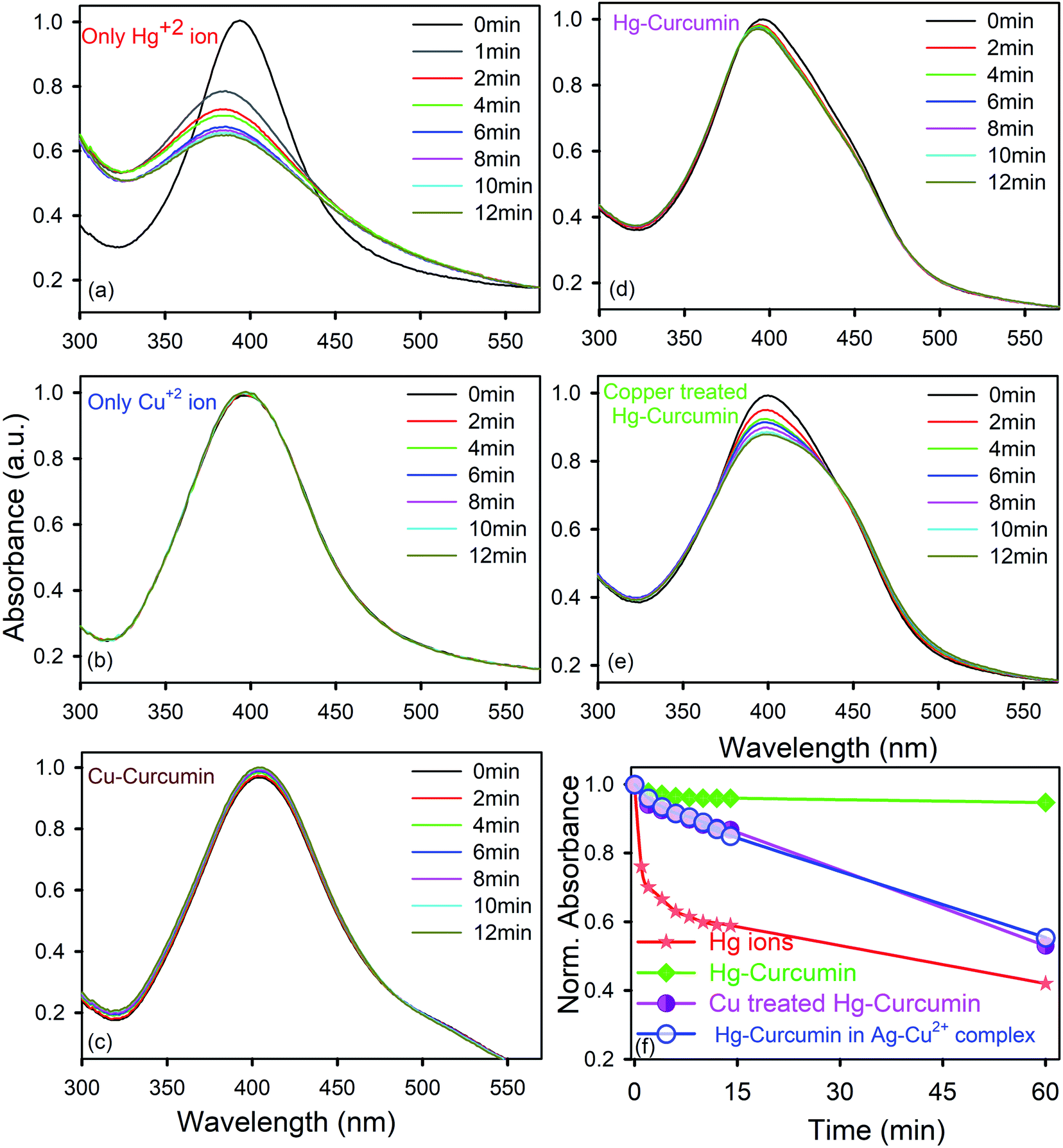
| | Fig. 5 Temporal variations of the SPR spectra of Ag-NPs immersed in aqua-methanolic solution in the presence of (a) the Hg2+ ion only; (b) the Cu2+ ion only; (c) Cu–curcumin; (d) Hg–curcumin; and (e) Cu-treated Hg–curcumin. The absorbance at zero time was normalized to 1.0 in each case. (f) Relative variations of the SPR spectra of the Ag-NPs with respect to time, immersed in aqua-methanolic solution in the presence of Hg2+ ions, Hg–curcumin and Cu-treated Hg–curcumin. | |
In order to determine how the SPR band of the nanoparticles are disturbed by the free Hg2+ ion being removed from the Hg–curcumin complex, first we studied the nature of the SPR band quenching upon interaction with the free Hg2+ ion. It is clear from Fig. 5a that amalgamation or partial oxidation of the Ag-NPs in the presence of the Hg2+ ion is completed within 14 min. The aggregated or oxidized Ag-NPs lose the characteristic SPR band and the particle sizes increase. Therefore, the fast response time and simple technique make this method an efficient probe for the detection of the Hg2+ ion in a real-life system. On the other hand, in the presence of the Cu2+ ion and Cu–curcumin, the SPR band of the Ag-NPs does not show any visible change.
In the present case, we utilized this efficient technique to detect the amount of decontamination of Hg–curcumin upon treatment with Cu-ions. Mercury interacts differently with the SPR band when it is in a complex form with other particles, as compared to free-standing Hg2+ ions. In the Hg–curcumin system, Hg is chelated with curcumin, as a result of this almost no, or a negligible amount, of free Hg ions are available in the system. However, upon Cu-treatment, the Cu-ion squeezes out the Hg-ion from the Hg–curcumin and makes it available in the aqueous solution. Keeping the other parameters constant, we have brought in Hg–curcumin in the Ag-NPs containing aqueous solution. We found there is an almost negligible subduing of the SPR band, in the presence of Hg–curcumin. However, in the case of the Cu-treated Hg–curcumin, the SPR band of Ag-NPs again started decreasing (Fig. 5e). The decrement is similar in nature to the decrement of the free Hg2+ ion. The copper-treated Hg–curcumin has a greater impact on the SPR peak than the Hg–curcumin because of the released Hg ions from curcumin after being replaced by Cu. Furthermore, we tried to investigate the time required for the Cu2+ ion to replace the Hg2+ ion from the Hg–curcumin complex. At first, the free Cu2+ ion is intermixed with the Ag-NPs, then the Hg-curcumin is added into the Cu2+–Ag-NPs system. The temporal variation in the present system is almost similar to that of the previous case, implying the free Cu2+ ion reacts with the Hg–curcumin very fast and replaces it instantly in the curcumin complex, which supports the computational result. The relative change of the absorption spectra with respect to time is shown in Fig. 5f. It is clearly visualized that, for the free Hg2+ ion, the suppression rate of the SPR band is the highest (41% in 12 min). The suppression rate is almost negligible in the case of Hg–curcumin. Interestingly, Cu-treated Hg–curcumin shows a higher suppression rate compared to Hg–curcumin. Almost 15% of the SPR band has been decreased in the treated curcumin within 14 min. The amount of suppression and the rate constants are tabulated in Table 3. The systems were then stirred, and we monitored the SPR of the Ag-NPs in the system after 1 h of incubation time. We observed the SPR of the Cu treated Hg–curcumin was suppressed up to 47% in comparison to 58% for the free Hg2+ ion after 1 h. The presence of the Cu2+ ion removes more Hg atoms from the curcumin with the increasing reaction time. To obtain further quantitative details about how many Cu ions are replacing the Hg from the Hg–curcumin, we have performed EDS measurements of these samples (shown in Fig. S1a†). The Cu-treated Hg–curcumin was washed several times prior to the EDS measurement to ensure that there was no free Hg or Cu ions present in the system. Fig. S1b and c† shows the mapping of the Cu and Hg atoms in the system, confirming the increment of Cu in the complex system, which indicates that Cu replaces the Hg in the system. Our modified system shows the presence of both Hg and Cu. It was found that upon Cu-treatment, 75% of the Hg–curcumin converted to the more efficient Cu–curcumin. The elemental amounts are tabulated in detail in Table 4. This result is consistent with the result obtained using the Ag SPR quenching detection method.
Table 3 Relative degradation of the SPR of the Ag-NPs with respect to the system after 14 min and 1 h
| System |
Relative O.D. degradation of Ag SPR |
| After 14 min |
After 1 h |
| Free Hg2+ ions |
41% |
58% |
| Hg–curcumin |
4% |
5.5% |
| Cu–Hg–curcumin |
14% |
47% |
Table 4 Relative atomic percentages of the Cu and Hg atoms for different systems measured using EDS
| System |
Relative atomic% |
| Cu |
Hg |
| Hg–curcumin |
0.1 |
99.9 |
| Cu–curcumin |
0.0 |
100 |
| Cu–Hg–curcumin |
75.1 |
24.9 |
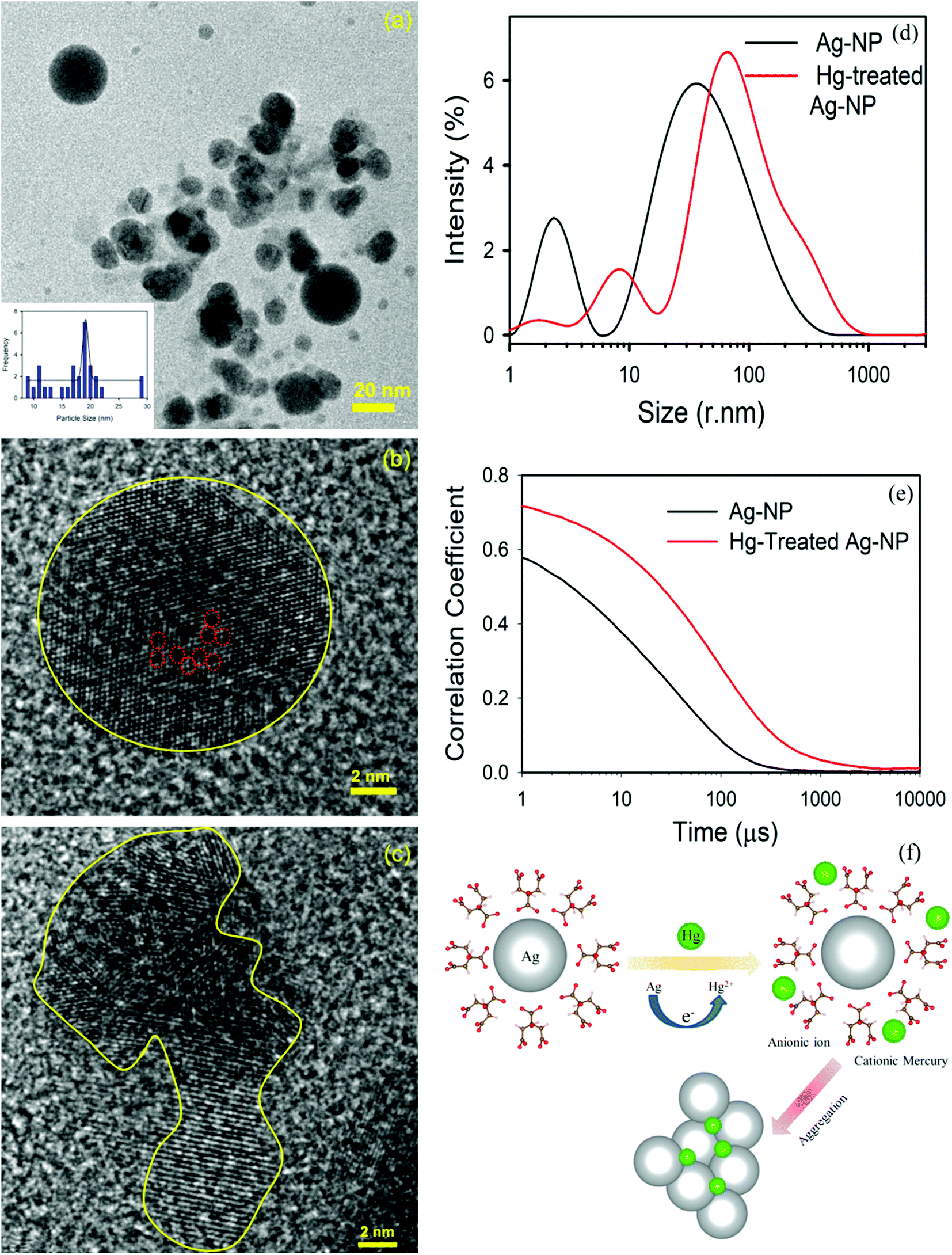
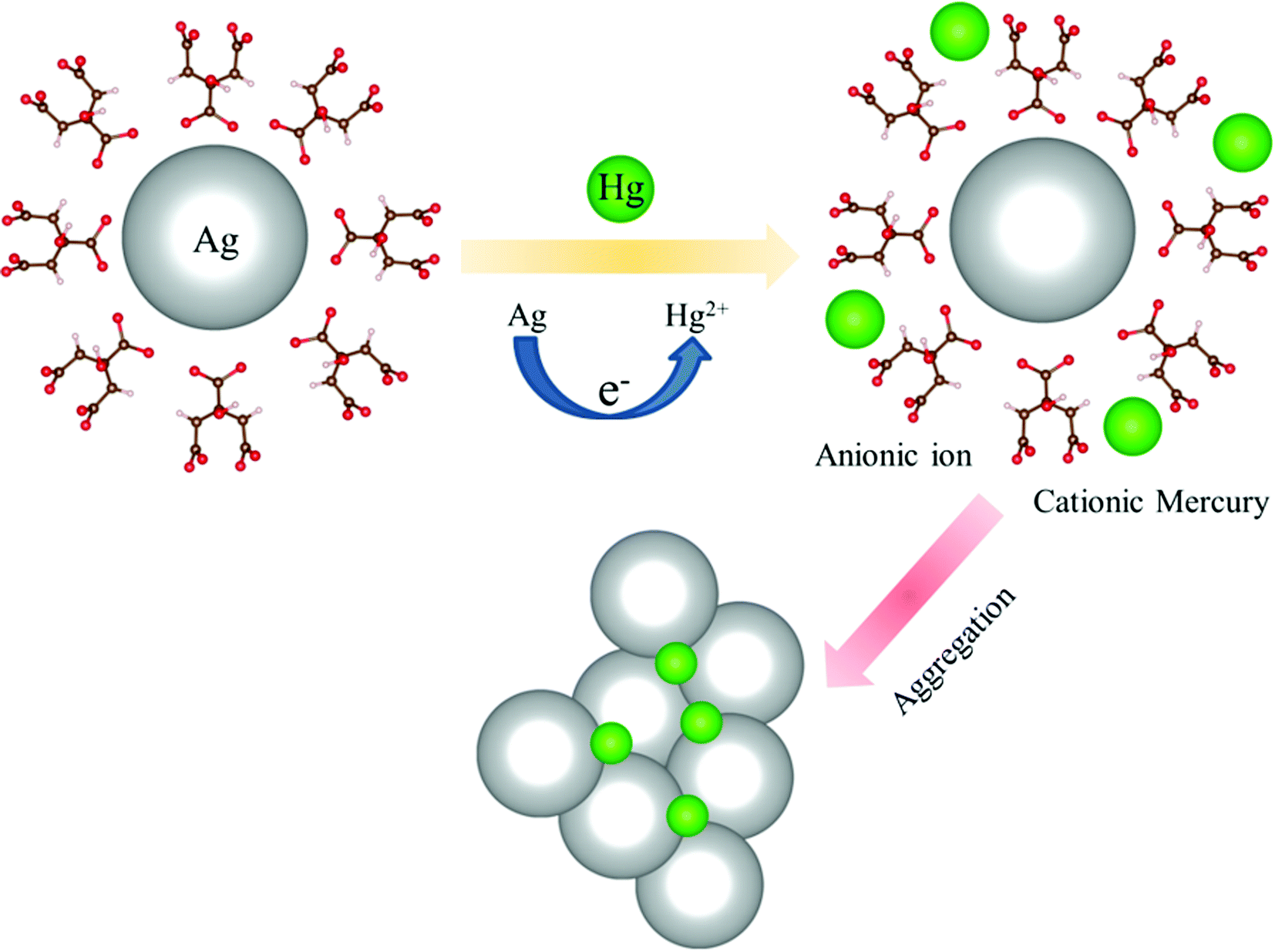
The TEM image of an Ag-NP after interaction with Hg is depicted in Fig. 6a. The measured average size of the Ag-NP was found to be ∼19.62 nm, which is larger than the pure Ag-NPs. Fig. 6b shows the HRTEM image of an Ag-NP after agglomeration with a free Hg2+ ion. Interestingly, a distinct contrast difference (red marked area) was observed within the Ag-NP, which was absent in the pure Ag-NPs. We propose that there is a possibility of decorating Hg0/Hg2+ particles on the surface of Ag-NP during the amalgamation process. The aggregation of the Ag-NPs in the presence of cationic Hg2+ ion is further confirmed in Fig. 6c. The hydrodynamic diameter (dH) of the particles can be estimated using DLS. The average dH measured for the Ag-NPs was ∼16.8 nm (Fig. 6d) which is slightly larger than the particle size obtained using TEM, because of the presence of the ligand citrate. The hydrodynamic diameter of the Ag-NPs increases to ∼23.2 nm upon amalgamation with the Hg2+ ion. Organic compounds, such as citrate are electron transparent, and therefore, they cannot be detected in the TEM micrograph. The autocorrelation coefficient is also plotted in Fig. 6e. The decay of the Ag-NPs is much more rapid than the Hg-treated Ag-NPs, which is consistent with the size increment of dH. The schematic representation of the aggregation mechanism of the Ag-NPs in the presence of the Hg ion is illustrated in Fig. 6f (Scheme 2).
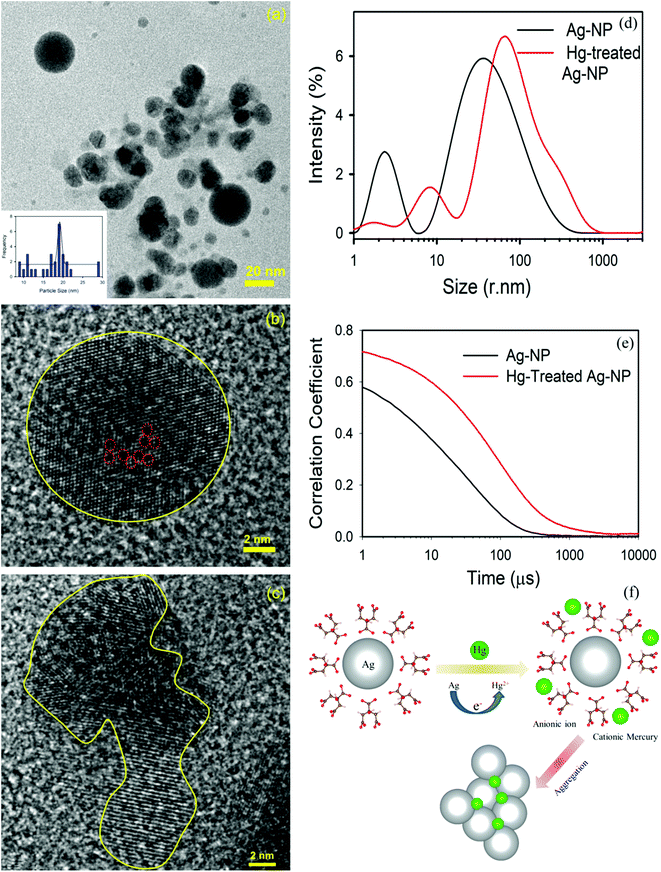 |
| | Fig. 6 (a) A TEM image of the Ag-NPs after amalgamation with Hg2+ ions. (b) A HRTEM image of the Ag-NP after treatment. Hg ions are decorated (marked by the red dots) inside the Ag-NP. (c) A HRTEM image of the agglomeration of the Ag-NPs. (d) The size of the Ag-NPs measured before and after detection. (e) The correlation coefficient of the Ag-NPs before and after treatment. (f) A schematic diagram showing the mechanism of the aggregation of the Ag-NPs in the presence of a cationic mercury ion. | |
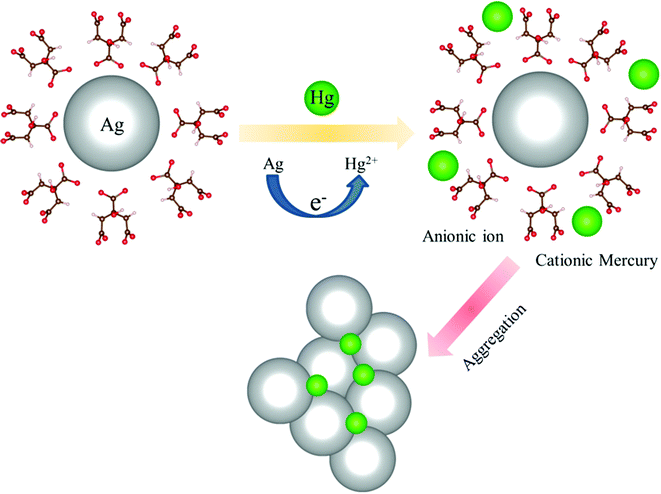 |
| | Scheme 2 A possible aggregation mechanism of the Ag-NPs in the presence of a cationic mercury ion. | |
In summary, a simple wet chemical transmetalation technique was invented to exchange the toxic Hg metal from the contaminated curcumin complex by employing a Cu ion. We propose that the Cu ion turns the Hg–curcumin complex into a Cu–curcumin complex and releases free Hg into the medium. The free Hg ion is monitored by utilizing the SPR band of the Ag-NPs. Using a first principles DFT calculation, we have corroborated our experimental findings. This result suggests that the proposed mechanism may have great potential to decontaminate heavily toxic Hg from curcumin complexes.
5. Conclusions
Briefly, a simple, efficient and field deployable method has been proposed to remove toxic mercury from a Hg–curcumin complex. The present study depicts a strategy for heavy metal replacement from a chelated complex using a less toxic metal ion, which can exchange up to 70% of the toxic mercury ions from curcumin using copper. The shift in the absorption spectrum undoubtedly indicates the exchange of the metal ion at the attachment site. First principles DFT calculations reveal that Hg–curcumin, in the presence of a Cu environment, undergoes a process in which mercury metal is replaced by copper. The SPR band of Ag-NPs is employed to detect the uncoupled Hg2+ ions. In the case of Hg–curcumin, there was no alteration in the SPR band of the Ag-NPs. However, the Cu-treated Hg–curcumin decouples the Hg ions from curcumin, which can be detected by the suppression of the SPR band of the Ag-NPs. This simple transmetalation technique holds great potential as a novel approach to decouple toxic metals from essential food elements. The proposed method could be a very promising technique for real implementation to remove mercury contamination from food additives.
Conflicts of interest
We declare that we have no conflict of interest.
Acknowledgements
T. K. M. and D. B. want to thank DST for fellowship. D. K. wants to thank Anupam supercomputer facility BARC. We are thankful to DBT (India), DST (India) for financial grants BT/PR11534/NNT/28/766/2014, DSTTM-SERIFR-117 and EMR/2016/004698. We want to thank Anindita Bhattacharya, IIT Kharagpur India for careful reading of the manuscript and relevant scientific suggestions.
References
- A. Mohammadi, F. S. Zabihi and N. Chaibakhsh, J. Photochem. Photobiol., A, 2018, 367, 384–389 CrossRef CAS.
- P. Zhang and P. J. Sadler, J. Organomet. Chem., 2017, 839, 5–14 CrossRef CAS.
- D. Bagchi, S. Chaudhuri, S. Sardar, S. Choudhury, N. Polley, P. Lemmens and S. K. Pal, RSC Adv., 2015, 5, 102516–102524 RSC.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC.
- M. W. Johnson, S. W. Bagley, N. P. Mankad, R. G. Bergman, V. Mascitti and F. D. Toste, Angew. Chem., 2014, 53, 4404–4407 CrossRef CAS PubMed.
- C. Sanchez, B. Julián, P. Belleville and M. Popall, J. Mater. Chem., 2005, 15, 3559–3592 RSC.
- K. S. Egorova and V. P. Ananikov, Organometallics, 2017, 36, 4071–4090 CrossRef CAS.
- E. Merian, M. Anke, M. Ihnat and M. Stoeppler, Elements and their compounds in the environment: occurrence, analysis and biological relevance, Wiley-VCH Verlag GmbH & Co. KGaA, 2004 Search PubMed.
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC.
- M. Lalonde, W. Bury, O. Karagiaridi, Z. Brown, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2013, 1, 5453–5468 RSC.
- M. Oves, M. S. Khan, A. Zaidi and E. Ahmad, in Toxicity of heavy metals to legumes and bioremediation, Springer, 2012, pp. 1–27 Search PubMed.
- R. Singh, N. Gautam, A. Mishra and R. Gupta, Indian J. Pharmacol., 2011, 43, 246–253 CrossRef CAS PubMed.
- T. W. Clarkson, Environ. Health Perspect., 1992, 100, 31–38 CrossRef PubMed.
- C. Krishnamoorthi and P. Vishwanathan, Toxic Metals in the Indian Environment, Tata McGraw Hill Publishing Co. Ltd., New Delhi, 1991 Search PubMed.
- F. Berglund and M. Bertin, Chemical fallout, Thomas Publishers Spring field, IL, 1969 Search PubMed.
- M. Berlin, R. K. Zalups and B. A. Fowler, in Handbook on the Toxicology of Metals, ed. G. F. Nordberg, B. A. Fowler, M. Nordberg and L. T. Friberg, Academic Press, Burlington, 3rd edn, 2007, pp. 675–729 Search PubMed.
- P. Li, X. Feng and G. Qiu, Int. J. Environ. Res. Public Health, 2010, 7, 2666–2691 CrossRef CAS PubMed.
- Y.-S. Hong, Y.-M. Kim and K.-E. Lee, J. Prev. Med. Public Health, 2012, 45, 353 CrossRef PubMed.
- K. R. Mahaffey, R. P. Clickner and C. C. Bodurow, Environ. Health Perspect., 2004, 112, 562–570 CrossRef CAS PubMed.
- J.-D. Park and W. Zheng, J. Prev. Med. Public Health, 2012, 45, 344–352 CrossRef PubMed.
- K. M. Rice, E. M. Walker Jr, M. Wu, C. Gillette and E. R. Blough, J. Prev. Med. Public Health, 2014, 47, 74–83 CrossRef PubMed.
- K. Farhadi, M. Forough, R. Molaei, S. Hajizadeh and A. Rafipour, Sens. Actuators, B, 2012, 161, 880–885 CrossRef CAS.
- B. Wei and L. Yang, Microchem. J., 2010, 94, 99–107 CrossRef CAS.
- B. B. Aggarwal, A. Kumar and A. C. Bharti, Anticancer Res., 2003, 23, 363–398 CAS.
- N. Luo, K. Varaprasad, G. V. S. Reddy, A. V. Rajulu and J. Zhang, RSC Adv., 2012, 2, 8483–8488 RSC.
- K. Bairwa, J. Grover, M. Kania and S. M. Jachak, RSC Adv., 2014, 4, 13946–13978 RSC.
- K. I. Priyadarsini, Molecules, 2014, 19, 20091–20112 CrossRef PubMed.
- S. Wanninger, V. Lorenz, A. Subhan and F. T. Edelmann, Chem. Soc. Rev., 2015, 44, 4986–5002 RSC.
- K. Gleason, J. P. Shine, N. Shobnam, L. B. Rokoff, H. S. Suchanda, M. O. S. Ibne Hasan, G. Mostofa, C. Amarasiriwardena, Q. Quamruzzaman, M. Rahman, M. L. Kile, D. C. Bellinger, D. C. Christiani, R. O. Wright and M. Mazumdar, J. Environ. Public Health, 2014, 2014, 1–5 CrossRef PubMed.
- S. Borowska and M. M. Brzóska, J. Appl. Toxicol., 2015, 35, 551–572 CrossRef CAS PubMed.
- K. Aslan, J. R. Lakowicz and C. D. Geddes, Anal. Biochem., 2004, 330, 145–155 CrossRef CAS PubMed.
- Z. Hubicki and D. Kołodyńska, Ion Exchange Technologies, 2012, pp. 193–240 Search PubMed.
- R. A. Bernhoft, J. Environ. Public Health, 2012, 2012, 1–5 CrossRef PubMed.
- L. M. Gaetke, H. S. Chow-Johnson and C. K. Chow, Arch. Toxicol., 2014, 88, 1929–1938 CrossRef CAS PubMed.
- D. Bagchi, T. K. Maji, S. Sardar, P. Lemmens, C. Bhattacharya, D. Karmakar and S. K. Pal, Phys. Chem. Chem. Phys., 2017, 19, 2503–2513 RSC.
- D. Paramelle, A. Sadovoy, S. Gorelik, P. Free, J. Hobley and D. G. Fernig, Analyst, 2014, 139, 4855–4861 RSC.
- T. K. Maji, D. Bagchi, P. Kar, D. Karmakar and S. K. Pal, J. Photochem. Photobiol., A, 2017, 332, 391–398 CrossRef CAS.
- T. K. Maji, P. K. Sarkar, P. Kar, B. Liu, P. Lemmens, D. Karmakar and S. K. Pal, Appl. Catal., A, 2019, 583, 117124 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- T. K. Maji, K. Vaibhav, S. K. Pal, K. Majumdar, K. Adarsh and D. Karmakar, Phys. Rev. B, 2019, 99, 115309 CrossRef CAS.
- T. K. Maji, P. Kar, H. Mandal, C. Bhattacharya, D. Karmakar and S. K. Pal, ChemistrySelect, 2018, 3, 6382–6393 CrossRef CAS.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- T. J. Meyer, Pure Appl. Chem., 1986, 58, 1193–1206 CAS.
- M. H. Leung, D.-T. Pham, S. F. Lincoln and T. W. Kee, Phys. Chem. Chem. Phys., 2012, 14, 13580–13587 RSC.
- M. Subhan, K. Alam, M. Rahaman, M. Rahman and R. Awal, J. Sci. Res., 2014, 6, 97–109 CrossRef.
- W. F. Kieffer, J. Chem. Educ., 1950, 27, 659 CrossRef CAS.
- J. H. Van Vleck, Phys. Rev., 1932, 41, 208–215 CrossRef CAS.
- H. Maier and U. Scherz, Phys. Status Solidi B, 1974, 62, 153–164 CrossRef CAS.
- W. B. Jensen, Chem. Rev., 1978, 78, 1–22 CrossRef CAS.
- M. A. Addicoat, G. F. Metha and T. W. Kee, J. Comput. Chem., 2011, 32, 429–438 CrossRef CAS PubMed.
- E. Rodríguez-León, R. Iñiguez-Palomares, R. E. Navarro, R. Herrera-Urbina, J. Tánori, C. Iñiguez-Palomares and A. Maldonado, Nanoscale Res. Lett., 2013, 8, 318 CrossRef PubMed.
- S. Saravanan, R. Kato, M. Balamurugan, S. Kaushik and T. Soga, J. Sci.: Adv. Mater. Dev., 2017, 2, 418–424 Search PubMed.
- M. Forough and K. FAHADI, Turk. J. Eng. Environ. Sci., 2011, 34, 281–287 Search PubMed.
- A. Nain, S. R. Barman, S. Jain, A. Mukherjee and J. Satija, Appl. Nanosci., 2017, 7, 299–307 CrossRef CAS.
- P. K. Sarkar, A. Halder, N. Polley and S. K. Pal, Water, Air, Soil Pollut., 2017, 228, 314 CrossRef.
- L. Rastogi, R. B. Sashidhar, D. Karunasagar and J. Arunachalam, Talanta, 2014, 118, 111–117 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10596d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence bc,
Debjani Karmakar
bc,
Debjani Karmakar