
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Novel fatty chain-modified GLP-1R G-protein biased agonist exerts prolonged anti-diabetic effects through targeting receptor binding sites
Maorong Wangab,
Ping Yaob,
Minpeng Gaoc,
Jian Jind and
Yerong Yu*ad
aDepartment of Endocrinology, West China Hospital, Sichuan University, Chengdu, Sichuan, China 610041. E-mail: 1100312222@vip.jiangnan.edu.cn; yerongyu@scu.cn
bDepartment of Endocrinology, Affiliated Hospital of Hubei University for Nationalities, Enshi 445000, Hubei, China
cChina Pharmaceutical University, Nanjing, Jiangsu 210009, P. R. China
dJiangnan University, Wuxi, Jiangsu 214062, P. R. China
First published on 24th February 2020
Abstract
Here, we design and evaluate novel long-lasting GLP-1R G-protein-biased agonists with promising pharmacological virtues. Firstly, six GLP-1R G-protein-biased peptides (named PX01–PX06), screened by using a previous reported high-throughput autocrine-based method, were fused to the N-terminus of GLP-1(9-37) to generate six fusion peptides (PX07–PX012). In vitro surface plasmon resonance (SPR) measurements showed that PX09 exerts the highest binding affinity for both human and mouse GLP-1R extracellular domains (ECD). We further used the PX09 as a starting point to conduct site-specific modifications yielding twelve lysine-modified conjugates, termed PX13–PX24. Of these conjugates, PX17 retained relatively better in vitro GLP-1R activation potency and plasma stability compared with other ones. Preclinical studies in db/db mice demonstrated that acute treatment of PX17 exerts enhanced hypoglycemic and insulinotropic activities in a dosage dependent model within the range of 0.1–0.9 mg kg−1. Similarly, prolonged glucose-lowering abilities were exhibited in modified multiple oral glucose tolerance tests (OGTTs) and a hypoglycemic duration test. Apparently prolonged in vivo half-lives of ∼96 and ∼141 h were observed after a single subcutaneous administration of PX17 at 0.1 and 0.3 mg kg−1, respectively, in healthy cynomolgus monkeys. In addition, twice-weekly treatment of PX17 in db/db mice for 8 weeks obviously improved the hemoglobin A1C (HbA1C), and was more effective at improving the insulin resistance, glucose tolerance as well as function of pancreatic beta cells compared with Semaglutide. Furthermore, subcutaneously dosed PX17 in diet induced obese (DIO) mice achieved long-term beneficial effects on food intake and body weight control, HbA1C and inflammation-related factor level lowering. The above results indicate that PX17, as a novel GLP-1R G-protein-biased agonist, may be a promising candidate for antidiabetic therapies.
Introduction
Type 2 diabetes mellitus (T2DM) is one of the chronic, complex and progressive metabolic disorders, which is characterized by deteriorative hyperglycemia due to the insufficient or relatively insufficient insulin secretion.1–3 Although some first-line hypoglycemic agents, such as metformin and sulfonylureas, have already been widely utilized in the glycemic control, there is still an urgent and persistent demand for more effective treatments of T2DM.4–7Incretin-based therapies for T2DM treatment are rapidly gaining favor as the most effective therapeutic approach.8–10 Among these incretins, GLP-1 and its analogs, as the external GLP-1R agonists have been intensively developed.11,12 GLP-1, as an endogenous peptide hormone comprising 30-amino-acids, was secreted in response to food ingestion in gut L-cells. The secreted GLP-1 plays important roles in glucose homeostasis in a glucose-dependent model without hypoglycemic risk. In addition, GLP-1 promotes the gastric emptying, body weight loss, improves the insulin resistance and function of pancreatic β-cells. However, the GLP-1, under physiological conditions, is rapidly inactivated by dipeptidyl peptidase IV (DPP-IV) and some other endopeptidase yielding an extremely short in vivo half-life.13,14 Hence, numerous research efforts have focused on the development of more long-lasting GLP-1R agonists. For example, Semaglutide, a fatty chain-modified dipeptidyl peptidase-IV (DPP IV) resistant GLP-1R agonist and shares more than 95% homology with native GLP-1 and was developed as a clinically-approved antidiabetic agent with a stronger affinity for GLP-1R and significant longer in vivo half-life.15,16
Better understanding of GLP-1R pleiotropic signaling and the underlying physiological consequences might provide new avenues for the development of anti-diabetic agents. All of the GLP-1 and its analogs exert their actions through the GLP-1R, which was mainly expressed in central nervous system and pancreas. Historically, β-arrestins, recruited by the elevation of cAMP due to the activation of GLP-1R, were reported to play an exclusive role in desensitization of G-protein coupled receptor (Scheme 1).17–19
 | ||
| Scheme 1 Schematic depicting the identification and characterization of a novel GLP-1R-biased agonist. | ||
We previously developed an autocrine-based system and obtained thirteen peptides, comprising of random 7–10 amino acids, as G-protein-biased ligands for the development of drugs with novel modes of action.20 In this study, we selected six GLP-1R-biased peptides to the GLP-1(9-37) to generate the hybrid candidates with improved bias for GLP-1R and free of endopeptidase cutting sites. We further identified the designated fusion peptides before and after the specific fatty chain modification via in vitro receptor binding and activation assays. Both the preclinical pharmacokinetics and pharmacodynamics characteristics of selected fusion peptide were also carefully studied in cynomolgus monkeys and diabetes models, respectively, to evaluate the in vivo therapeutic potential for the T2DM treatment.
Results
Autocrine-based selection of novel GLP-1R agonists
Based on the strategy as previously reported, commercial peptide libraries were designed on the strength of the competitive GLP-1R antagonist GLP-1(9-39) sequence with 7–10 random amino acids added to the N-terminus. As a result showed in Table 1, six GLP-1R G-protein-biased peptides (named PX01–PX06), screening by these libraries in high-throughput, autocrine manner, were fused to the N-terminus of GLP-1(9-37) to generate six fusion peptides (PX07–PX12). All of these hybrid peptides were identified to exhibit higher affinity for GLP-1R ECD after fusion (Table 2). Nevertheless, these fusion peptides were predictably considered the short half-life in vivo. To enhance the bioavailability of these candidate molecules, PX09 was selected as the lead peptides for further acylation because of the significant higher binding affinity (KD = 0.22 × 10−6 M) for human GLP-1R in SPR measurements (Table 2).| Peptide | Sequence | Frequency (%) |
|---|---|---|
| PX01 | ACACIEVDCDI | 1.4 |
| PX02 | ACDNAV | 1.3 |
| PX03 | ELVDCAV | 1.3 |
| PX04 | ACSYVKFPCVL | 1.2 |
| PX05 | ACASYEVDCVL | 0.7 |
| PX06 | ACCIDSVQVI | 0.6 |
| Peptide | Human GLP-1R ECD | Mouse GLP-1R ECD | ||||
|---|---|---|---|---|---|---|
| ka (M−1 s−1) | kd (s−1) | KD (M) | ka (M−1 s−1) | kd (s−1) | KD (M) | |
| PX07 | 1.38 × 104 | 1.83 × 10−2 | 1.33 × 10−6 | 2.18 × 104 | 5.34 × 10−2 | 2.44 × 10−6 |
| PX08 | 1.14 × 104 | 3.60 × 10−2 | 3.16 × 10−6 | 0.85 × 104 | 6.12 × 10−2 | 7.21 × 10−6 |
| PX09 | 1.28 × 104 | 0.28 × 10−2 | 0.22 × 10−6 | 1.94 × 104 | 2.26 × 10−2 | 1.16 × 10−6 |
| PX10 | 1.24 × 104 | 5.33 × 10−2 | 4.31 × 10−6 | 1.18 × 104 | 2.81 × 10−2 | 2.37 × 10−6 |
| PX11 | 1.13 × 104 | 2.49 × 10−2 | 2.21 × 10−6 | 1.10 × 104 | 1.92 × 10−2 | 1.74 × 10−6 |
| PX12 | 2.01 × 104 | 6.27 × 10−2 | 3.12 × 10−6 | 1.20 × 104 | 6.27 × 10−2 | 5.21 × 10−6 |
Synthesis and characterization of the fatty chain-modified GLP-1R agonists
Lysine scanning mutagenesis of PX09 and further pharmacological characterization were conducted to determine the final modification site and suitable length of fatty acid chain, respectively, for better biological activities in vitro and in vivo.As shown in Fig. 1, two selected sites (Gly11 and Gly38) were replaced by lysine accompanied with Lys27, Lys35 were modified with different lengths of fatty chain maleimide to generate twelve new conjugates (termed PX13–PX24). These conjugates were prepared by using solid phase synthesis method, and then identified by high performance liquid chromatography and mass spectrometry.
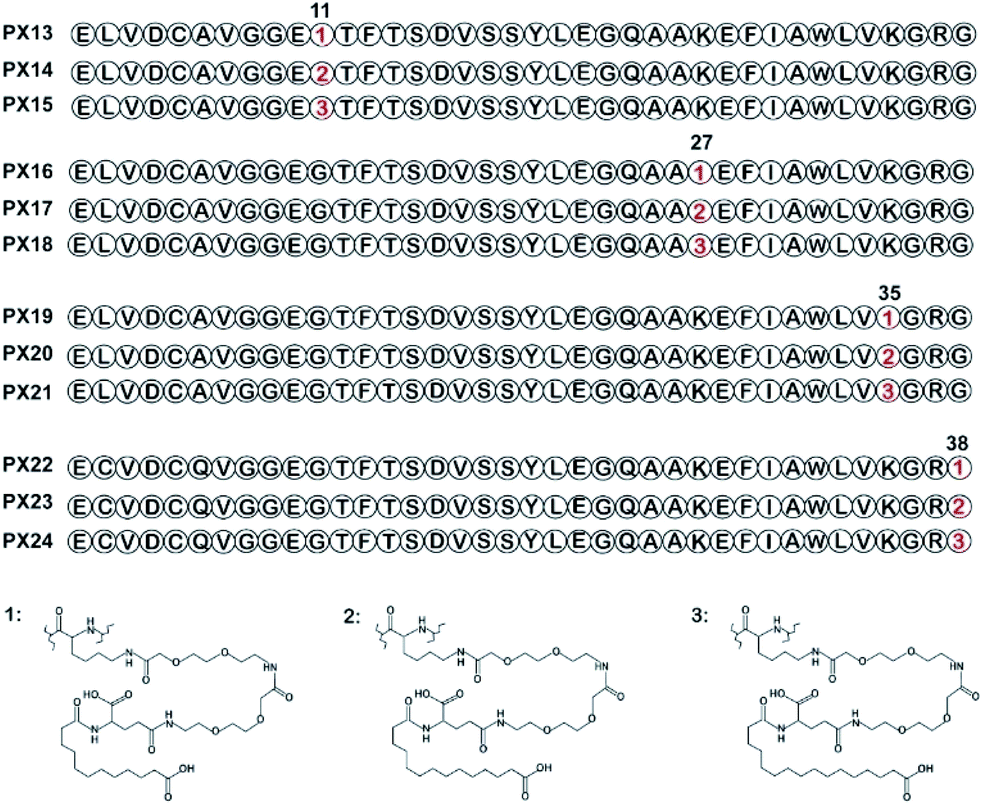 | ||
| Fig. 1 Structure of fatty chain-modified GLP-1 analog conjugates. | ||
In vitro activity of GLP-1R agonists
The receptor activation assay was designed to evaluate the potency of PX13–PX24 in HEK239 cells stably expressing human GLP-1R. As summarized in Table 3, several modified conjugates (e.g., PX13–15, PX22–24) were observed with a relatively obvious reduction in the activation potency for the GLP-1R compared with PX09 which may partially due to modification sites of fatty chain. Remarkably, PX16–18 exert better EC50 values than other modified conjugates, suggesting that conjugation on position 27 with different lengths of fatty chainmaleimide could retained most of the receptor activation potency of PX09 which were relatively far away from the probable active domain (N- and C-terminal regions). Furthermore, PX16–18 with three lengths of fatty chains showed similar activation potency of GLP-1R indicating that the activation potency of GLP-1R was not closely related to the length of the fatty chain in this study.| Peptides | Human GLP-1R | |
|---|---|---|
| EC50 (nM) | Emax (%) | |
| Semaglutide | 0.392 ± 0.095 | 100 ± 8.5 |
| GLP-1 | 0.742 ± 0.082 | 74.1 ± 6.1 |
| PX13 | 0.385 ± 0.129 | 103.3 ± 8.9 |
| PX14 | 0.595 ± 0.091 | 90.0 ± 6.8 |
| PX15 | 0.611 ± 0.094 | 88.1 ± 8.2 |
| PX16 | 0.355 ± 0.045 | 105.7 ± 7.7 |
| PX17 | 0.228 ± 0.019 | 115.8 ± 7.9 |
| PX18 | 0.409 ± 0.091 | 96.9 ± 10.2 |
| PX19 | 0.399 ± 0.035 | 99.1 ± 7.2 |
| PX20 | 0.611 ± 0.099 | 85.9 ± 10.9 |
| PX21 | 0.602 ± 0.113 | 86.1 ± 12.1 |
| PX22 | 0.753 ± 0.073 | 73.1 ± 9.3 |
| PX23 | 0.652 ± 0.102 | 84.3 ± 9.2 |
| PX24 | 0.535 ± 0.092 | 92.2 ± 8.2 |
Then the preliminary in vitro stabilities of PX16–PX18, compared with GLP-1, were assessed in plasma of wistar rats at 37 °C. As shown in Fig. 2, PX17 exert an approximate half-life of 78.6 h which was longer than PX16 (t1/2 ∼ 36.4 h) and PX18 (t1/2 ∼ 64.6 h). As a result, PX17 was selected to conduct the further in vivo efficacy evaluation.
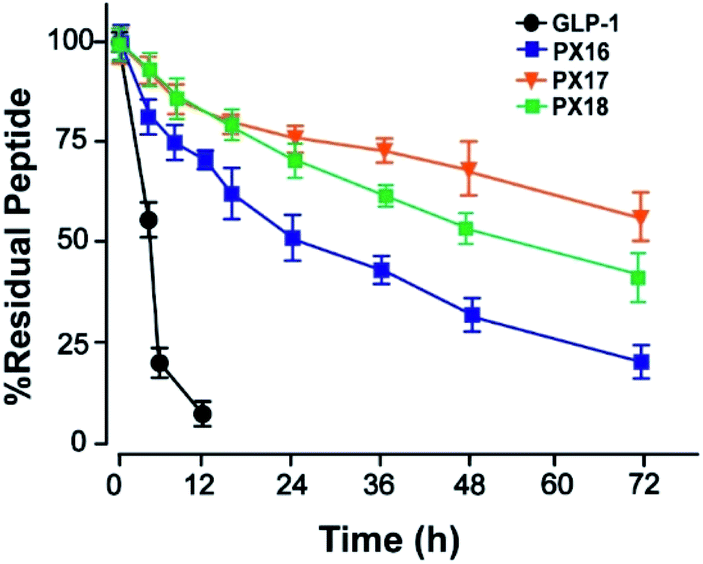 | ||
| Fig. 2 The in vitro stability test of PX16–18 in rat plasma. | ||
Glucose tolerance and insulin secretion test
To further evaluate the in vivo glucoregulatory activities of PX17, OGTT was performed at 1 h after the single subcutaneous administration of PX17 (0.1, 0.3 and 0.9 mg kg−1) and Semaglutide (0.3 mg kg−1) in diabetic db/db mice. As shown in Fig. 3A, PX17 treatment at three doses all notably decreased the blood glucose levels (BGLs) to normal level (<16.6 mmol L−1) at 30 min after a 2 g kg−1 oral glucose challenge owing to the insulinotropic activities, while BGLs of saline-treated mices rapidly increased to 28.6 ± 3.1 mmol L−1 at 30 min and then slowly decreased to normal level within 120 min. Moreover, the glucose AUC0–120 min of PX17 at all three doses (0.1, 0.3 and 0.9 mg kg−1) were significantly lower than that of the saline treated group (P < 0.01), and slightly better than Semaglutide group at same dose (Fig. 3B). Meanwhile, at each time point after the oral glucose challenge, PX17 treatment remarkably increased the plasma insulin levels to the similar extent as Semaglutide and significantly higher than saline (Fig. 3C and D). Remarkably, both glucoregulatory and insulinotropic effects of PX17 in db/db mice exhibited a dose dependent manner indicating a promising dose–effect relationship which is beneficial to the follow-up preclinical researches or even clinical dosage design. | ||
| Fig. 3 Glucose tolerance and insulin secretion test in diabetic db/db mice. Blood glucose (A) and insulin (B) level–time curve. (C) AUCglucose and (D) AUCinsulin after oral glucose administration. P < 0.01, 0.001 using one-way ANOVA (*, **, ***) vs. placebo. All data are expressed as mean ± SD (n = 8). | ||
Multiple oral glucose tolerance test
Sustained glucose-stabilizing ability of PX17 was further assessed using a modified multiple OGTTs in db/db mice. PX17 at both two doses (0.3 mg kg−1 and 0.9 mg kg−1) were subcutaneously administrated 1 hour before the each glucose challenge, and then the BGLs were detected before and 15, 30, 45, 60 and 120 min after glucose administration. As shown in Fig. 4, the BGLs of saline treated mice rapidly reached peak at 30 min, then slowly reach baseline (∼10 mmol L−1) more than 120 min. While the glucose lowering ability of PX17 at 0.3 mg kg−1 maintained during the first OGTT (0–5 h), decreased slightly after 96 h and was still effective in third period (192–194 h). In contrast, Semaglutide at the same dose was almost ineffective at 192 h. In particular, AUC192–194 h of PX17 (0.3 mg kg−1) was still significant better than that of the Semaglutide at same dose (P < 0.05). In addition, the glucose-stabilizing duration of PX17 at 0.9 mg kg−1 was sustained to 192 h, and its AUC192–194 h was obviously lower than the saline or Semaglutide treated group (P < 0.01). These results indicated that PX17 at both two doses exert obvious better glucose lowering ability and duration (more than 192 h) compared with saline or Semaglutide in db/db mice.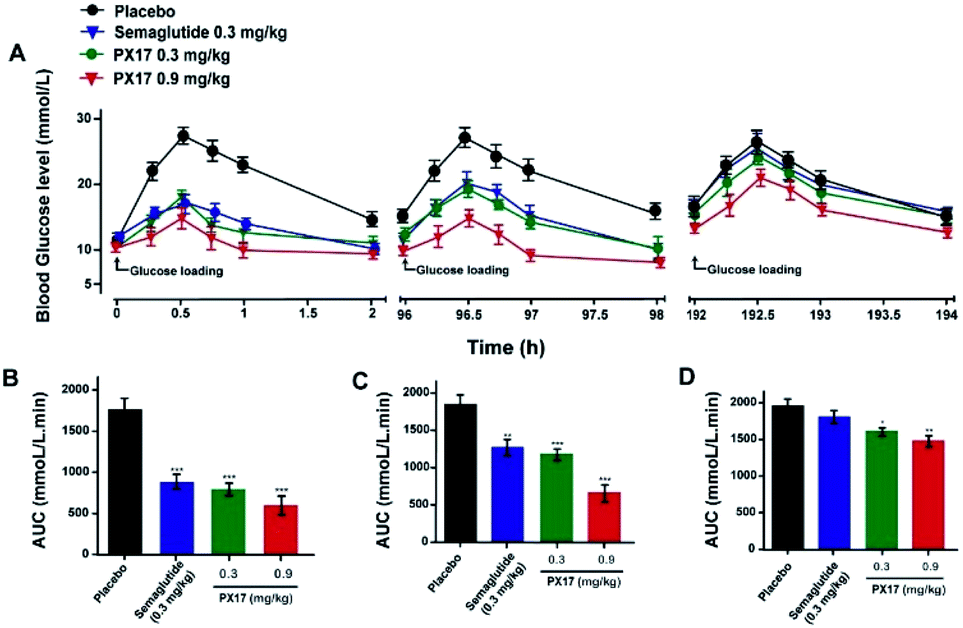 | ||
| Fig. 4 Glucose-stabilizing capabilities of PX17 were evaluated using multiple OGTTs in male db/db mice. (A) Time-course average BGLs and (B–D) AUC values of db/db mice with saline, Semaglutide (0.3 mg kg−1) and PX17 (0.3, 0.9 mg kg−1) for 194 h. P < 0.01, 0.001 using one-way ANOVA (*, **, ***) vs. placebo. All data are expressed as mean ± SD (n = 8). | ||
Hypoglycemic duration test
The hypoglycemic effects of PX17 (0.3 and 0.9 mg kg−1, s.c.) were further investigated in db/db mice using Semaglutide (0.3 mg kg−1, s.c.) as the positive control. As illustrated in Fig. 5A, administration of PX17 at the doses of 0.3 and 0.9 mg kg−1 both rapidly maintained the normal BGLs within 6 h and the time required to rebound to 8.35 mmol L−1, which was considered as the hypoglycemic duration, were nearly 40 h and 60 h, respectively. By contrast, the saline treated db/db mice exhibited a hyperglycemic state (>20 mmol L−1) during the entire experiment (0–192 h). In addition, the hypoglycemic effects of PX17 at 0.3 mg kg−1 and 0.9 mg kg−1 were maintained up to at least 144 h and 192 h, which were comparable and significant better than Semaglutide (0.3 mg kg−1), respectively. Furthermore, single acute treatment of PX17 significantly lowered the 60.8% and 70.2% of AUC0–192 h at 0.3 mg kg−1 and 0.9 mg kg−1, respectively, compared with the saline group, while the Semaglutide treatment was only 52.3%. Above results indicated that PX17 exert relatively greater antidiabetic duration than Semaglutide, one of the currently best GLP-1R agonists.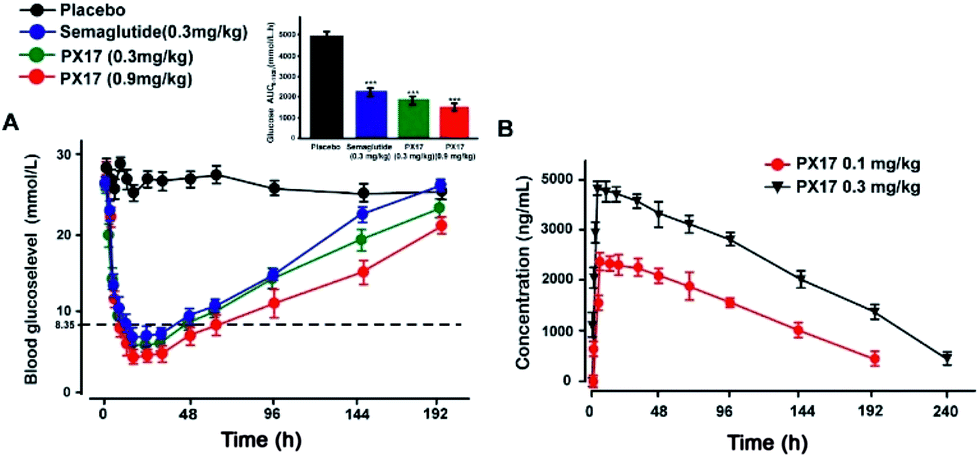 | ||
| Fig. 5 Hypoglycemic duration test of PX17 (A) in non-fasted db/db mice (n = 8) and pharmacokinetic concentration–time curve (B) in cynomolgus monkeys (n = 4). Results are presented as means ± SD. P < 0.001, using one-way ANOVA (***) vs. placebo. All data are expressed as mean ± SD. | ||
In vivo pharmacokinetics
The drug concentration–time curve of PX17 at two doses (0.3 mg kg−1 and 0.9 mg kg−1) in cynomolgus monkeys after a single s.c. injection were showed in Fig. 5B. As a result, the t1/2 of PX17 at dose of 0.1 mg kg−1 and 0.3 mg kg−1 were approximately 96.3 h and 141.8 h respectively. Combined with the results of hypoglycemic duration test, PX17 has the potential to be developed into a weekly antidiabetic agent.Chronic studies in db/db mice
PX17 was subcutaneously injected twice-weekly at the dose of 0.3 mg kg−1 using Semaglutide as positive control in db/db mice. It was observed that body weight gains and food intake of db/db mice were effectively suppressed in PX17 treatment group, while the saline treated ones maintained a continuous upward trend (Fig. 6A and B). Particularly, PX17 treatment exert a more significant suppression effects than that of Semaglutide (P < 0.05) at the end of treatment.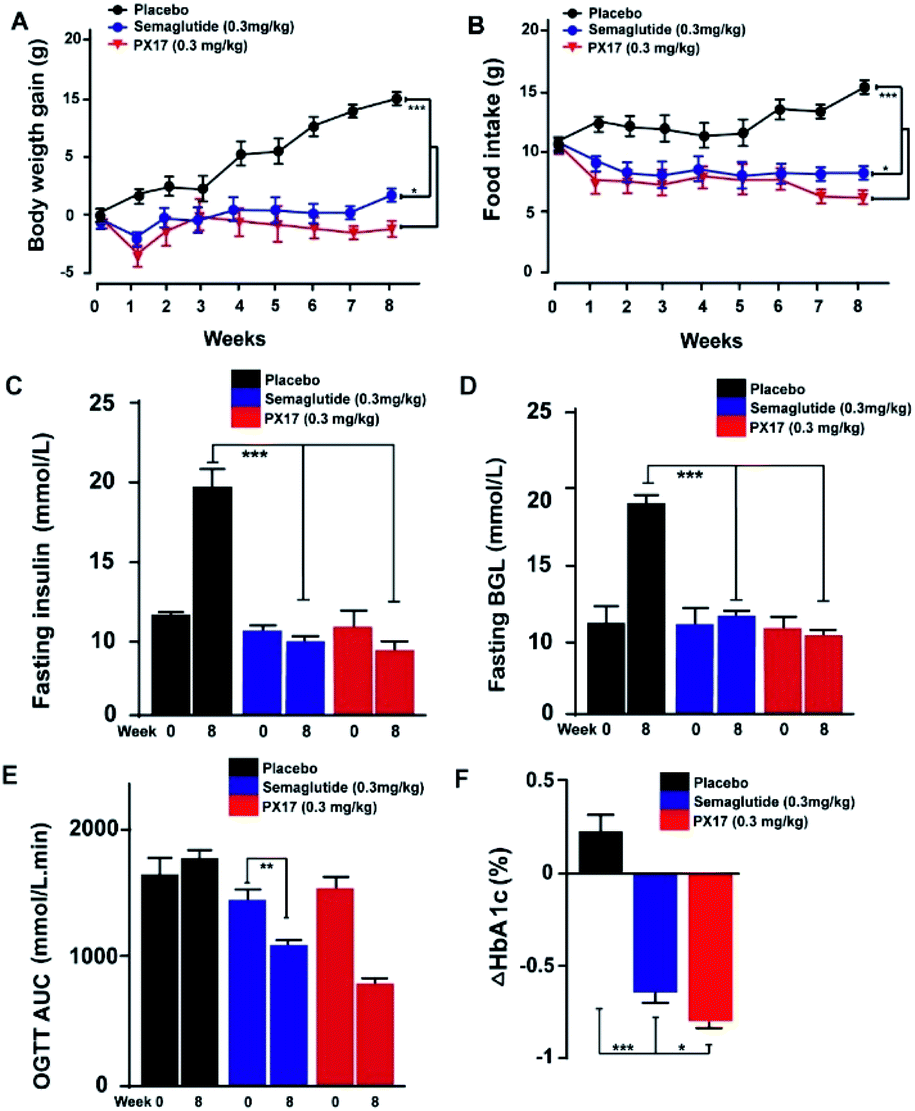 | ||
| Fig. 6 Chronic in vivo studies on male db/db mice. Effects of longterm administration of PX17 on (A) body weight gain, (B) food intake, (C) fasting insulin, (D) BGL, OGTT AUC0–120 min and (F) % HbA1C change in male DIO mice. *P < 0.05, **P < 0.01, ***P < 0.001. All results are present as mean ± SD (n = 8). | ||
Similarly, chronic treatment of PX17 also exhibited a stably reduction in fasting BGLs and insulin of db/db mice (Fig. 6C and D). To further evaluate whether a long-term treatment of PX17 effectively improved the glucose metabolism in db/db mice, OGTT were performed before and after the chronic study (week 0 and 8, respectively). Interestingly, consecutive 8 weeks treatment of PX17 obviously lowered the OGTT AUC by more than 50% compared with its value at week 0, while it was 23.4% after the Semaglutide treatment, suggesting that PX17 may had greater chronic improvement on the glucose tolerance than Semaglutide at same dose (Fig. 6E). According to the numerous reports, % HbA1C was a more sensitive index of glycemic control than real-time glucose concentration.21 As shown in Fig. 6F, chronic treatment of PX17 for 8 weeks resulted an obvious reduction (>0.8%) of % HbA1C value in db/db mice compared with saline group (P < 0.001) and also better than Semaglutide treated ones (P < 0.01).
In addition, histological analyses of the pancreas revealed that the db/db mice chronically treated with PX17 preserved proper insulin immunoreactivity (Fig. 7A), and the islet number and area were also significantly increased compared with the saline treated group (Fig. 7B and C). Together, these results suggested that chronic treatment of PX17 for consecutive 8 weeks significantly exerted improvement on the weight control, intake reduction, glucose metabolism and pancreas islet function in db/db mice.
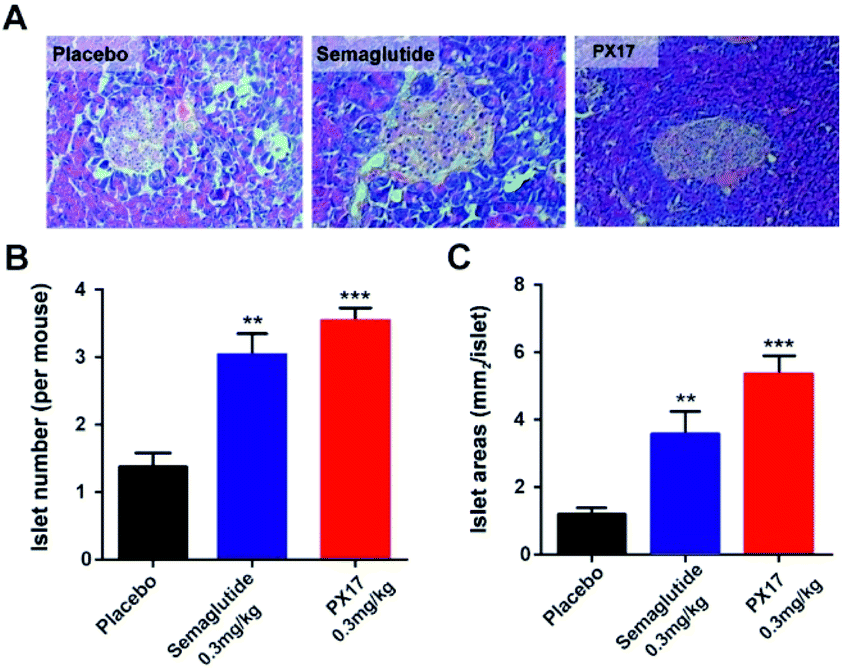 | ||
| Fig. 7 Effects of chronic PX17 treatment on pancreatic islets in db/db mice. (A) Representative images of db/db mice histologic samples. The (B) number and (C) area pancreatic islet in placebo, Semaglutide and PX17 treated groups. P < 0.01, 0.001 using one-way ANOVA (**, ***) vs. placebo. All results are present as mean ± SD (n = 8). | ||
Chronic studies in DIO mice
To further confirm the appetite suppression and body weight control effects of PX17, a consecutive 12 weeks treatment was conducted in DIO mice. During the 12 weeks experiment course, PX17 decreased food intake and body weight gain by up to 43% and 40%, respectively (Fig. 8A and B). Nevertheless, by consecutive 12 weeks treatment, PX17 at 0.3 mg kg−1 significantly affect % fat/body when compared with the saline treated mice (Fig. 8C). Results of % HAb1c showed that single subcutaneously injected of PX17 at 0.3 mg kg−1 exert a inhibition by up to 1.3% at the end of treatment, compared with the saline group and also obvious higher than Semaglutide (0.9%) at same dose (Fig. 8D).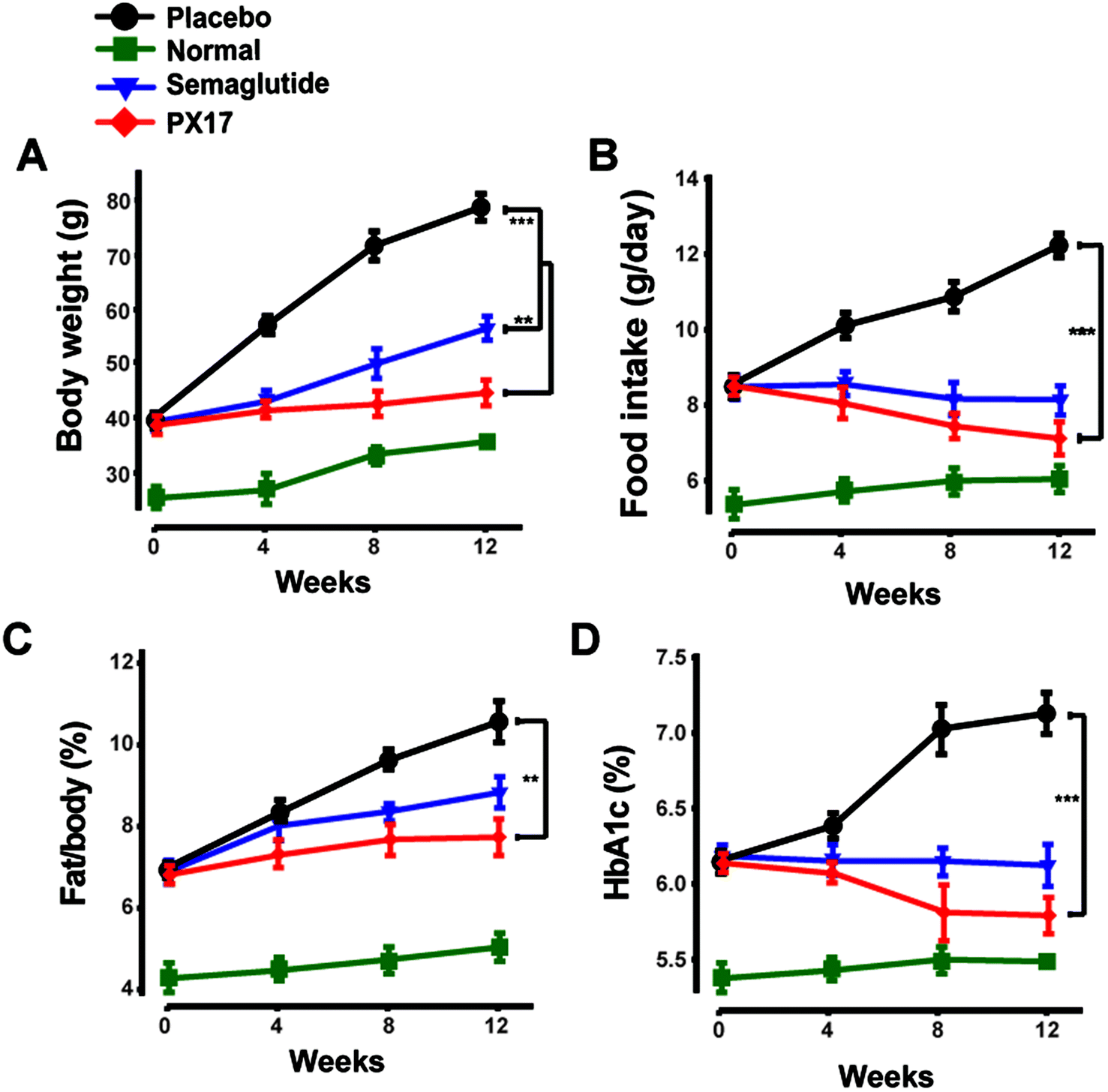 | ||
| Fig. 8 Effects of chronic PX17 treatment on pancreatic islets in DIO mice. The (A) fat weight, (B) food intake, (C) % fat/body and (D) % HbA1c in placebo, Semaglutide and PX17 treated groups in male DIO mice. **P < 0.01, ***P < 0.001. All results are present as mean ± SD (n = 8). | ||
We further investigated the effects of 12 weeks treatment of PX17 on the circulating levels of GIP. As a result, treatment with PX17 at 0.3 mg kg−1 resulted in a significant increase in GIP level compared with saline (P < 0.01) and Semaglutide (P < 0.05). Nevertheless, PX17 at 0.3 mg kg−1 significantly decreased the levels of leptin and resistin (Fig. 9B and C). By contrast, PX17 exert no obvious effects on both circulatory levels of leptin and resistin in DIO mice (Fig. 9C). We further examined the expression of peroxisome proliferator-activated receptor gamma (PPARγ), which was closely related to the insulin resistance.22 In this study, protein expression level of PPARγ was significantly upregulated nearly 1.5-fold in mice treated with PX17 when compared with saline-treated db/db mice (Fig. 9D). In addition, a significant reduction in expression levels of TNF-α and IL-6 were observed in the PX17 group (Fig. 9E and F).
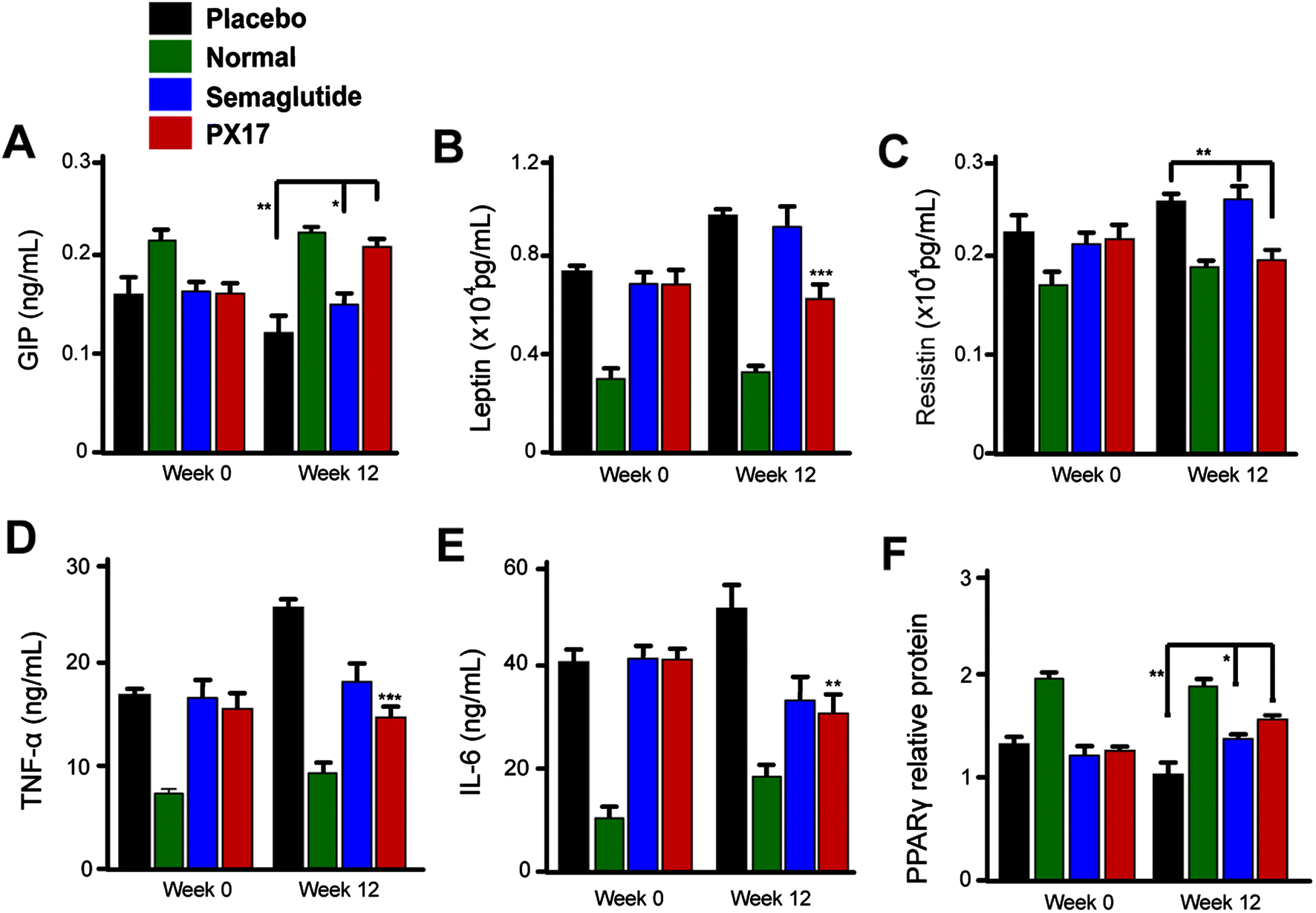 | ||
| Fig. 9 Chronic in vivo studies on male DIO mice. Effects of long-term administration of PX17 on the expression of (A) GIP, (B) leptin, (C) resistin, (D) TNF-α, (E) IL-6, (F) PPARγ in male DIO mice. *P < 0.05, **P < 0.01, ***P < 0.001. All results are present as mean ± SD (n = 8). P < 0.5, 0.02, 0.001 using one-way ANOVA (*, **, ***) vs. placebo. All results are present as mean ± SD (n = 8). | ||
After 12 weeks treatment, blood biochemical indexes of DIO mice were measured and results were listed in Table 4. As a result, PX17 treatment exert an obvious decline in serum levels of TC, TG and LDL-C (P < 0.01) compared with saline. Furthermore, similar reduction in the ALT and AST level were also observed in PX17 treated mice indicating effective improvement on hepatocyte damage compared with saline treated ones. In addition, the LDL/HDL ratio, which was considered as calculated index to quantize the increase magnitude of the two lipoproteins among different groups, and results showed that 12 weeks exposure of PX17 to DIO mice decreased this ratio up to 45% while Semaglutide treatment only resulted in a 29% reduction.
| Parameters | Normal | Saline | Semaglutide | PX17 |
|---|---|---|---|---|
| a P < 0.001 using one-way ANOVA versus saline treated group (*, **, ***). All data are expressed as mean ± S.E.M. (n = 8). | ||||
| TG (mmol L−1) | 0.6 ± 0.1 | 1.3 ± 0.1 | 0.9 ± 0.2 | 0.7 ± 0.1* |
| TC (mmol L−1) | 3.4 ± 0.2 | 4.8 ± 0.5 | 4.2 ± 0.6 | 3.5 ± 0.5* |
| HDL (mmol L−1) | 3.2 ± 0.5 | 2.5 ± 0.2 | 2.4 ± 0.2 | 2.6 ± 0.3 |
| LDL (mmol L−1) | 0.5 ± 0.1 | 1.6 ± 0.2 | 1.2 ± 0.3 | 1.0 ± 0.1** |
| LDL/HDL | 0.26 ± 0.05 | 0.63 ± 0.08 | 0.45 ± 0.06 | 0.35 ± 0.07*** |
| ALT (IU L−1) | 24.5 ± 2.5 | 62.7 ± 3.5 | 45.1 ± 5.1 | 50.5 ± 3.4** |
| AST (IU L−1) | 121 ± 12.1 | 161 ± 13.4 | 132 ± 9.9 | 128 ± 10.5** |
Discussion
T2DM, as a chronic and deteriorative metabolic disease, was characterized by progressive hyperglycemia caused by dysfunctional insulin secretion and/or its impaired utilization.11,23 GLP-1 is an endogenous peptide hormone secreted from intestinal endocrine cells in response to nutrient ingestion, and the GLP-1-based therapies are considered as the most effective approaches for the treatment of T2DM.9,10 However, there are many challenges in developing the GLP-1 into therapeutic agents, in particular the short half-life due to the rapid degradation and renal filtration.24,25 Thus, considerable efforts have focused on improving the pharmacokinetic properties of GLP-1, such as modification of a fatty acid chain or polyethylene glycol (PEG), as well as linking to human albumin or Fc fragment.26–29 Although the currently approved treatments with GLP-1R agonists exert remarkable antidiabetic effects, such as Exenatide or Semaglutide, further improvements on the GLP-1R agonists are still expected such as longer glucose control, better weight loss or more cardiovascular benefits.15,30 In addition, currently all the GLP-1R agonist drugs were developed according to the only two native GLP-1R agonists (GLP-1 and Exendin-4) which may induce severely homogenization and limit the development of more potential agents.A better understanding of the signaling and the underlying physiological consequences of GLP-1R, as a classical G-protein-coupled receptor, might provide more new avenues for developing candidate drugs with pleiotropic therapeutic values. Recently, affinity-based screening technologies like phage and yeast display are commonly used for the screening of high-throughput large combinatorial peptide libraries, but they rely on the use of recombinant protein or membrane preparations and do not distinguish between agonists, inverse agonists, and antagonists.20 According to our previous research,20 a potent and selective GLP-1R G-protein-biased agonist, P5, was screened by a functional cell-based autocrine system, and its efficacy was also proved in mouse models. However, our previously designed P5, as an Exendin-4 analog, exert short-time antidiabetic effects duo to the rapid degradation and renal filtration rendering higher doses and frequent injections, which will also negatively affects patient compliance. In addition, P5 only shares a less than 50% homology with GLP-1 which will inevitably result in a high immunogenicity. Thus, we hope that a novel GLP-1R agonist with higher homology and prolonged in vivo half-life could be discovered in this study via the previously reported autocrine selection system.
In this research, six peptides were selected from commercial peptide libraries using the autocrine-based selection system, and defined as PX01–PX06. Then these six GLP-1R G-protein-biased peptides were fused to the N-terminus of GLP-1(9-37) to generate six fusion GLP-1 analog peptides (named PX07–PX12), and PX07–12 were confirmed with high affinity for GLP-1R ECD (Table 2). In order to enhance the in vivo stability of these candidate molecules, PX09, with the highest affinity for GLP-1R, was selected as the lead peptide for further side chain acylation. A series of fatty chain conjugates (termed PX13–24) with different modification sites and side chain length, were then synthesized and evaluated by in vitro GLP-1R-based cell assays and plasma stability assays. As a result, PX17, with the same fatty chain modification site in the GLP-1 sequence with Semaglutide, exhibited the best GLP-1R activation potency and in vitro plasma stability. It's worth mentioning that, Semaglutide, as a recently approved once-weekly type 2 diabetes drug, exerts the best anti-diabetic and anti-obesity efficacies in clinical practice.31 Therefore, PX17 was selected to conduct the further in vivo efficacy evaluation using Semaglutide as a positive control.
As is showed in Fig. 3A, glucose tolerance tests revealed that acute treatment of PX17 at three doses (0.1, 0.3 and 0.9 mg mL−1) all remarkably decreased the blood glucose levels and stimulated the secretion of insulin compared with saline in a dose dependent model. This will be beneficial to increase the drug safety for future preclinical efficacy study or even clinical application without the risk of hypoglycemia. Moreover, the glucose AUC0–120 min of PX17 (0.1, 0.3 and 0.9 mg kg−1) were also significantly lower than that of the saline treated group (P < 0.01) and relatively better than Semaglutide group at same dose (Fig. 3B).
The long-acting glucose-lowering effects of PX17 were then confirmed by multiple OGTTs as well as the hypoglycemic duration test. As shown in Fig. 4 and 5, PX17 exerts a better glucose lowering ability and sustained hypoglycemic effect than Semaglutide at same dose in both two assays, probably due to its improved in vivo stability and enhanced hypoglycemic effect resulting from the fatty chain modification and re-engineering of the GLP-1 N-terminal which is critical for the GLP-1R activation, respectively.32,33 The in vivo pharmacokinetic profiles of PX17 were further evaluated in cynomolgus monkey and results showed a t1/2 of PX17 at dose of 0.1 mg kg−1 and 0.3 mg kg−1 were approximately 96.3 h and 141.8 h, respectively, indicating that PX17 has the potential to be developed into a weekly antidiabetic agent (Fig. 5B).
Chronic in vivo studies of PX17 were further performed to comprehensively evaluate its potential therapeutic utilities in db/db and DIO mice when compared with the reference agonist Semaglutide. A daily injection of PX17 at 0.3 mg kg−1 achieved long-term beneficial effects on appetite suppression and body weight control in both db/db and DIO mice at the end of 12 weeks treatment. Nevertheless, chronic treatment of PX17 also exhibited a stably reduction in fasting BGLs, insulin and glucose tolerance of db/db mice (Fig. 6C and D). Similarly, chronic treatment of PX17 resulted an obvious reduction of % HbA1C values in both db/db and DIO mice compared with saline group (P < 0.001) and also better than Semaglutide treated ones (P < 0.05) indicating a better improvement on the long-term glucose metabolism. Results of pancreas histological analyses also revealed that the db/db mice chronically treated with PX17 preserved proper insulin immunoreactivity.
The enhanced glucoregulatory effects of PX17 could not be only attributed to the insulinotropic effects and additional mechanisms might also contribute to improve the overall glycaemia. We further investigated the effects of 12 weeks treatment of PX17 on the circulating levels of GIP, a hormone which was involved in the regulation of glucose homeostasis. As a result, a significant increase in GIP level was showed in Fig. 9A. Resistin, as an adipokine shown to negatively impact glucose metabolism and insulin sensitivity. Nevertheless, PX17 also significantly decreased the levels of leptin and resistin (Fig. 9B and C) compared with Semaglutide. As the activity of PPARγ by increasing insulin sensitivity in peripheral tissues could improve insulin resistance, regulating glucose metabolism, and lowering blood glucose. The expression of PPARγ was obviously upregulated by PX17 when compared with saline group (Fig. 9D). Furthermore, previous researches34 proved that a GIP and GLP-1 synergism may result in enhanced glycaemic control and % HbA1C compared with GLP-1R agonist alone. Thus, the enhanced glycaemic control observed in this study may result from PX17-induced GIP secretion and concomitant receptor activation. In addition, a significant reduction in expression levels of TNF-α and IL-6 were observed in the PX17 group, indicating that chronic administration of PX17 reduced the inflammatory factors levels in DIO mice (Fig. 9E and F). Similar results were also showed in the results of blood biochemical indexes, chronic treatment of PX17 significantly decreased the levels of TC, TG and LDL suggesting an enhanced improvement on the liver (Table 4).
Experimental
Materials and animals
The competitive GLP-1R antagonist GLP-1(9-39) as anchor site to GLP-1R extracellular domain was used to construct the combinatorial peptide libraries, which was subjected to biopanning for GLP-1R agonist peptides. HEK293 cells stably expressing GLP-1R were obtained from Sino Biological Biological Technology Co. (Beijing, China). Diet induced obese (DIO) mice (male C57BL/6 mice, 5–6 months old) and db/db mice (male, 6–8 weeks old) were purchased from Guangdong experimental animal center (Guangzhou, China). Male cynomolgus monkeys were bought from Chuangyao Co., Ltd. (Zhaoqing, China) and all the animals were grouped and housed in six/cage and temperature and humidity were controlled at 25 ± 2 °C and 50%, respectively, with a cycle of 12 h dark![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 h light. Glucose level was detected using a handheld one touch UltraEasy glucometer (Johnson & Johnson, USA). All animal studies were approved by the Sichuan University Animal Ethics Committee. The current study was carried out in line with the recommendations of the Guide for the Care and Use of Laboratory Animals. All experimental operations were in accordance with the International Laboratory Animal Ethics Convention and complied with relevant national regulations. The study was conducted in accordance with the Institutional Review Board of West China Hospital of Sichuan University.
:12 h light. Glucose level was detected using a handheld one touch UltraEasy glucometer (Johnson & Johnson, USA). All animal studies were approved by the Sichuan University Animal Ethics Committee. The current study was carried out in line with the recommendations of the Guide for the Care and Use of Laboratory Animals. All experimental operations were in accordance with the International Laboratory Animal Ethics Convention and complied with relevant national regulations. The study was conducted in accordance with the Institutional Review Board of West China Hospital of Sichuan University.
Autocrine selection of GLP-1R agonists
The combinatorial peptide libraries containing linear 7–10 random peptide sequences, was used to screen for specific peptide binding to GLP-1R in high-throughput, autocrine manner. A 96-well microtitre plate was coated with GLP-1R extracellular domain (ECD) in 0.01 M PBS (pH 7.0) for four rounds of screening peptides with high affinity for GLP-1 ECD conducted by the instruction of phage library kit. The selected phage was then amplified in vivo on E. coli ER 2738 strain. The accurate sequences were obtained for the subsequent DNA sequencing using the universal primer 96 gIII (5-CCCTCATAGTTAGCGTAACG 3).Synthesis and characterization of GLP-1R agonists
All the other peptides, with or without side chain modification, were obtained from Sino Biological Biological Technology Co. (Beijing, China) used a solid phase method. Molecular weight and purity of peptides were detected using an MALDITOF/MS and RP-HPLC on a SHIMADZU Inertsil ODS-SP (Shimadzu, Japan).In vitro activity of GLP-1R agonists
To evaluate the specificity of GLP-1R activation, GLP-1R agonists were incubated in the Chinese hamster ovary (CHO) cells stably expressing hGLP-1R cells for 30 min at 4 °C in PBS containing 2% FBS and 0.02% sodium azide. The binding and activation of GLP-1R were assessed for cAMP using homogeneous time-resolved fluorescence technology (Cisbio Bioassays, France) in accordance with established protocols.Oral glucose tolerance test
Oral glucose tolerance test (OGTT) was conducted in male db/db mice. The male db/db mice were grouped with average BGLs and body weights, and then received a subcutaneous administration of PX17 at 0.1, 0.3 and 0.9 mg kg−1. The db/db mice were challenged with a glucose dose of 2.0 g kg−1 at 1 h after injection, and then the BGLs were collected and detected before and 15, 30, 60 and 120 min after each administration. Multiple OGTTs were performed at an interval of 96 h after s.c. injection of PX17 (0.3 and 0.9 mg kg−1) or Semaglutide (0.3 mg kg−1). Antihyperglycemic effects of PX17 were evaluated by calculating the area under the curve from 0 to 2 h, 96 to 98 h and 192 to 194 h separately.Hypoglycemic efficacy test
Hypoglycemic effects of PX17 were tested in male diabetic db/db mice using a classical hypoglycemic duration test after single s.c. injection. The db/db mice were free access to food and water during the entire experimental period. The blood samples were collected and detected before and 1, 2, 3, 6, 12, 24, 36, 48, 72, 96, 144 and 192 h by using a handheld glucometer. Moreover, the hypoglycemic durations (BGL < 8.35 nmol L−1) were also carefully calculated to evaluate the antidiabetic effects.Pharmacokinetics in cynomolgus monkey
The pharmacokinetic profiles of PX17 were studied in male cynomolgus monkeys (0.1 and 0.3 mg kg−1, n = 4) after a subcutaneous injection. Blood samples were collected at pre-dose, 1, 6, 12, 24, 36, 48, 72, 96, 144, 192 and 240 h. Plasma concentrations of PX17 were determined by a liquid chromatography-mass spectrometry (LC-MS) method. Assay sensitivity was estimated to be 10 ng mL−1 and detection range was between 20–3000 ng mL−1.Chronic in vivo studies
Data analysis
Data were analyzed using GraphPad Prism 5 software (San Diego, USA). All measured variables were presented by mean ± SD and parameter differences were tested using one-way ANOVA. P < 0.05 considered significant.Conclusions
In summary, our previously designed autocrine-based functional screening strategy enabled the rapid discovery of high-potency GLP-1R agonists. A series of in vitro bioactivity studies demonstrated that the fusion of selected GLP-1R agonist and GLP-1(9-37) showed a significant enhancement in the affinity for GLP-1R. Subsequently, the further side fatty chain modified hybrid peptide PX17 showed well preserved in vitro receptors activation activity and long-acting in vivo incretin-based antidiabetics. Moreover, chronic in vivo studies in db/db and DIO mice also showed that PX17 is a promising agent deserving further investigation to treat obesity patients with diabetes. Furthermore, this autocrine-based functional screening strategy may also be applicable to other therapeutic ligands of G-protein coupled receptors (GPCRs) rather than only applied to GLP-1 for the treatment of T2DM.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by research grant from The Ministry of Science and Technology of China (No. 2012ZX10002006-003).Notes and references
- N. Leon, R. LaCoursiere, D. Yarosh and R. S. Patel, P&T, 2017, 42, 676–711 Search PubMed.
- M. Markowicz-Piasecka, J. Sikora, A. Szydlowska, A. Skupien, E. Mikiciuk-Olasik and K. M. Huttunen, Pharm. Res., 2017, 34, 2614–2627 CrossRef CAS PubMed.
- X. Xu, G. H. Wang, T. T. Zhou, L. L. Chen, J. Chen and X. Shen, Expert Opin. Drug Discovery, 2014, 9, 1047–1058 CrossRef CAS PubMed.
- M. R. Raee, A. A. Nargesi, B. Heidari, M. A. Mansournia, M. Larry, S. Rabizadeh, M. Zarifkar, A. Esteghamati and M. Nakhjavani, Arch. Iran. Med., 2017, 20, 141–146 Search PubMed.
- C. L. Roumie, A. M. Hung, R. A. Greevy, C. G. Grijalva, X. Liu, H. J. Murff, T. A. Elasy and M. R. Griffin, Ann. Intern. Med., 2012, 157, 601–610 CrossRef PubMed.
- P. Weissman, B. J. Goldstein, J. Rosenstock, B. Waterhouse, A. R. Cobitz, M. J. Wooddell and L. J. Strow, Curr. Med. Res. Opin., 2005, 21, 2029–2035 CrossRef CAS PubMed.
- M. Bodmer, C. Meier, S. Krahenbuhl, S. S. Jick and C. R. Meier, Diabetes Care, 2008, 31, 2086–2091 CrossRef CAS PubMed.
- S. Farr and K. Adeli, Curr. Opin. Lipidol., 2012, 23, 56–61 CrossRef CAS PubMed.
- A. Wick and K. Newlin, J. Am. Acad. Nurse Pract., 2009, 21(suppl 1), 623–630 CrossRef PubMed.
- A. Penfornis, S. Borot and D. Raccah, Diabetes Metab., 2008, 34(suppl 2), S78–S90 CrossRef CAS.
- D. K. Arulmozhi and B. Portha, Eur. J. Pharm. Sci., 2006, 28, 96–108 CrossRef CAS PubMed.
- T. Redij, R. Chaudhari, Z. Li, X. Hua and Z. Li, ACS Omega, 2019, 4, 961–970 CrossRef CAS PubMed.
- M. Malm-Erjefalt, I. Bjornsdottir, J. Vanggaard, H. Helleberg, U. Larsen, B. Oosterhuis, J. J. van Lier, M. Zdravkovic and A. K. Olsen, Drug Metab. Dispos., 2010, 38, 1944–1953 CrossRef CAS PubMed.
- A. Plamboeck, J. J. Holst, R. D. Carr and C. F. Deacon, Diabetologia, 2005, 48, 1882–1890 CrossRef CAS PubMed.
- J. Lau, P. Bloch, L. Schaffer, I. Pettersson, J. Spetzler, J. Kofoed, K. Madsen, L. B. Knudsen, J. McGuire, D. B. Steensgaard, H. M. Strauss, D. X. Gram, S. M. Knudsen, F. S. Nielsen, P. Thygesen, S. Reedtz-Runge and T. Kruse, J. Med. Chem., 2015, 58, 7370–7380 CrossRef CAS PubMed.
- J. Blundell, G. Finlayson, M. B. Axelsen, A. Flint, C. Gibbons, T. Kvist, E. Q. Bergan and J. Hjerpsted, Diabetes Res. Clin. Pract., 2016, 120, S119 Search PubMed.
- M. V. Hager, L. Clydesdale, S. H. Gellman, P. M. Sexton and D. Wootten, Biochem. Pharmacol., 2017, 136, 99–108 CrossRef CAS PubMed.
- Y. Zhang, Y. Ding, X. Zhong, Q. Guo, H. Wang, J. Gao, T. Bai, L. Ren, Y. Guo, X. Jiao and Y. Liu, Mol. Cell. Endocrinol., 2016, 430, 89–96 CrossRef CAS PubMed.
- M. V. Hager, L. M. Johnson, D. Wootten, P. M. Sexton and S. H. Gellman, J. Am. Chem. Soc., 2016, 138, 14970–14979 CrossRef CAS PubMed.
- H. Zhang, E. Sturchler, J. Zhu, A. Nieto, P. A. Cistrone, J. Xie, L. He, K. Yea, T. Jones, R. Turn, P. S. Di Stefano, P. R. Griffin, P. E. Dawson, P. H. McDonald and R. A. Lerner, Nat. Commun., 2015, 6, 8918 CrossRef CAS PubMed.
- A. K. Saleh and M. A. A. Moussa, Clin. Chem., 1985, 31, 1872–1876 CrossRef CAS.
- D. A. Sarruf, F. Yu, H. T. Nguyen, D. L. Williams, R. L. Printz, K. D. Niswender and M. W. Schwartz, Endocrinology, 2009, 150, 707–712 CrossRef CAS PubMed.
- K. M. Heppner and D. Perez-Tilve, Front. Neurosci., 2015, 9, 92 Search PubMed.
- B. D. Green, V. A. Gault, M. H. Mooney, N. Irwin, P. Harriott, B. Greer, C. J. Bailey, F. P. O'Harte and P. R. Flatt, Biol. Chem., 2004, 385, 169–177 CAS.
- R. Mentlein, Best Pract. Res., Clin. Endocrinol. Metab., 2009, 23, 443–452 CrossRef CAS PubMed.
- S. Choi, M. Baudys and S. W. Kim, Pharm. Res., 2004, 21, 827–831 CrossRef CAS PubMed.
- E. Jimenez-Solem, M. H. Rasmussen, M. Christensen and F. K. Knop, Curr. Opin. Mol. Ther., 2010, 12, 790–797 CAS.
- C. N. Li, M. M. Yang, G. J. Hou, S. N. Liu, Y. Huan, D. G. Yu, S. J. Sun, Q. Liu, S. S. Yan and Z. F. Shen, Biol. Pharm. Bull., 2017, 40, 1399–1408 CrossRef CAS PubMed.
- X. Cai, L. Sun, Y. Dai, Y. Avraham, C. Liu, J. Han, Y. Liu, D. Feng, W. Huang and H. Qian, Bioorg. Med. Chem., 2018, 26, 2599–2609 CrossRef CAS PubMed.
- J. B. Buse, R. M. Bergenstal, L. C. Glass, C. R. Heilmann, M. S. Lewis, A. Y. M. Kwan, B. J. Hoogwerf and J. Rosenstock, Ann. Intern. Med., 2011, 154, 103 CrossRef PubMed.
- S. Dhillon, Drugs, 2018, 78, 275–284 CrossRef CAS PubMed.
- X. H. Bai, Y. H. Niu, J. J. Zhu, A. Q. Yang, Y. F. Wu and X. S. Ye, Bioorg. Med. Chem., 2016, 24, 1163–1170 CrossRef CAS PubMed.
- C. Ørskov, H. Kofod, L. Rabenhøj, A. Wettergren and J. J. Holst, Regul. Pept., 1992, 40, 223 CrossRef.
- B. Finan, T. Ma, N. Ottaway, T. D. Muller, K. M. Habegger, K. M. Heppner, H. Kirchner, J. Holland, J. Hembree, C. Raver, S. H. Lockie, D. L. Smiley, V. Gelfanov, B. Yang, S. Hofmann, D. Bruemmer, D. J. Drucker, P. T. Pfluger, D. Perez-Tilve, J. Gidda, L. Vignati, L. S. Zhang, J. B. Hauptman, M. Lau, M. Brecheisen, S. Uhles, W. Riboulet, E. Hainaut, E. Sebokova, K. Conde-Knape, A. Konkar, R. D. DiMarchi and M. H. Tschop, Sci. Transl. Med., 2013, 5, 209ra151 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |