
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-yield synthesis of Ce modified Fe–Mn composite oxides benefitting from catalytic destruction of chlorobenzene†
Anqi Lib,
Hongming Long*ab,
Hongliang Zhanga and
Haijin Li *ac
*ac
aKey Laboratory of Metallurgical Emission Reduction & Resource Recycling (Anhui University of Technology), Ministry of Education, Ma’anshan, Anhui 243002, PR China. E-mail: yaflhm@126.com; lihaijin@ahut.edu.cn
bSchool of Metallurgical Engineering, Anhui University of Technology, Ma’anshan, Anhui 243002, China
cSchool of Energy and Environment, Anhui University of Technology, Ma’anshan, Anhui 243002, China
First published on 10th March 2020
Abstract
Ce–Fe–Mn catalysts were prepared by an oxalic acid assisted co-precipitation method. The influence of Ce doping and calcination temperature on the catalytic oxidation of chlorobenzene (as a model VOC molecule) was investigated in a fixed bed reactor. The Mn3O4 phase was formed in Ce–Fe–Mn catalysts at low calcination temperatures (<400 °C), which introduced more chemisorbed oxygen, and enhanced the mobility of O atoms, resulting in an improvement of the reduction active of Mn3O4 and Fe2O3. Additionally, CeO2 has strong redox properties, and Ce4+ would oxidize Mnx+ and Fex+. Therefore, the interaction of Ce, Fe and Mn can improve the content of surface chemisorbed oxygen. As compared with Fe–Mn catalysts, the catalytic conversion of chlorobenzene over Ce(5%)–Fe–Mn-400 was about 99% at 250 °C, owing to high specific surface area, Mn3O4 phase, and Ce doping. However, with the increase in roasting temperature, the performance of the catalysts for the catalytic combustion of chlorobenzene was decreased, which probably accounts for the formation of the Mn2O3 phase in Ce–Fe–Mn-500 catalysts, leading to a decrease in the specific surface area and concentration of chemically adsorbed oxygen. As a result, it can be expected that the Ce–Fe–Mn catalysts are effective and promising catalysts for chlorobenzene destruction.
1. Introduction
Volatile organic compounds (VOCs) mainly originate from municipal burning and industrial manufacturing and are an important class of pollutants in the atmosphere. Among them, chlorinated aromatic compounds have been always considered as the most harmful contaminants.1–4 The widespread emissions of chlorinated aromatic compounds not only cause the pollution of the ecological environment but also damage human health due to their acute toxicity and strong bioaccumulation potential.5,6 Thus, the elimination of chlorinated aromatic compounds has received more and more attention. Currently, chlorinated aromatic compounds are usually removed by absorption, thermal oxidation, and catalytic oxidation.7–10 The subsequent treatment of carbon materials adsorbed with a large amount of chlorinated aromatic compounds is very complex and can easily form secondary pollutions.11 Direct thermal oxidation usually requires high temperatures exceeding 850 °C and leads to small amounts of more toxic polychlorinated aromatic compounds.12 By comparison, catalytic oxidation is regarded as one of the most promising techniques for elimination of chlorinated aromatic compounds at much lower temperatures (<500 °C), due to its high efficiency, less harmful by-products (e.g., CO2, HCl, Cl2), and low energy consumption.11,13–16Catalysts based on noble metals, perovskites, and transition metal oxides have been developed for the catalytic oxidation of chlorinated aromatic compounds.11,16,17 Noble metals are expensive and always deactivate owing to chlorine (Cl) poisoning during catalytic oxidation processes, which limits their practical large-scale application.11 The temperature of catalytic oxidation for perovskites is usually above 500 °C, thus they were not the ideal catalysts for low-temperature catalytic degradation technology.18 Previous reports showed that transition metal oxides as proper catalysts have the advantages of low-temperature catalytic activity, thermal stability, and low cost.19 Mn-based oxides, such as Mn3O4, Mn2O3, and MnO2, are capable of providing the mobile-electron environment required by redox catalysts, owing to polymorphism and polyvalence, and have exhibited remarkable activities in the removal of gaseous pollutants (CO, NOx and VOCs).20–24 The catalytic activity is in the order Mn3O4 > Mn2O3 > MnO2, which are benefits from oxygen mobility in the catalyst.24 Nevertheless, the individual MnOx catalysts are still some way from reaching a satisfactory catalytic performance in terms of stability and activity to some kinds of VOCs, for example, the poor activity to catalytically oxidize chlorobenzene (CBz). Moreover, with the addition of metal ions (such as Ce, Fe, Co, and Cu), the catalytic activity of Mn-based oxides at low temperature could be obviously enhanced, due to possible complementary advantages of different metals in the catalytic activity.25–27 Among the alternative transition metal oxides, Fe–Mn binary oxides are common choices for catalytic combustion.20,28 The Mn–Fe–O catalysts exhibit high catalytic activity for toluene oxidation, arising from high amounts of Mn4+ ions, and high concentrations of lattice defects and oxygen vacancies.11 However, Fe–Mn catalysts were not capable of removing chlorinated pollutants.29 The active sites of Fe–Mn oxides were easily poisoned due to the strong adsorption of Cl, which could be overcome by the addition of other metals.12 Recently, CeO2 was used in the catalytic combustion of low concentration chlorobenzene, owing to its high oxygen storage capacity, abundant oxygen vacancies and strong redox property.30
To meet these challenges and further improve low-temperature performance for the catalytic destruction of chlorobenzene, herein, we prepare a series of Ce modified Fe–Mn oxides by a co-precipitation process. The influence of calcination temperature and the amount of the doped Ce on the catalytic destruction of chlorobenzene were investigated. It was found that Ce–Fe–Mn oxides consisting of Mn3O4, Fe2O3, and CeO2, exhibited high activity for the catalytic oxidation of chlorobenzene. The inner relationship between catalytic behavior and structural properties was investigated by a combination of performance and characterization studies (XRD, BET, TEM, H2-TPR, XPS, etc.).
2. Experimental
2.1 Catalysts preparation
The Ce–Fe–Mn catalysts were prepared by an oxalic acid assisted co-precipitation method. For a typical synthesis, firstly, a certain amount of FeSO4·4H2O, MnCl2·4H2O (the molar ratio of Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn of 1:4) and Ce(NO3)3·6H2O were dissolved in deionized water, and stirred for 30 min in an ice water bath. Then, the oxalic acid solution was added into the metal ion solution to form a precipitate. Then the mixture was stirred in an ice water bath for 1 h. The precipitate was collected by suction filtration, and washed with distilled water and alcohol three times. Finally, the obtained products were dried in a vacuum freeze drier and calcined at 300 °C (Ce–Fe–Mn-300), 400 °C (Ce–Fe–Mn-400) and 500 °C (Ce–Fe–Mn-500), respectively. After grinding to 60 mesh, the catalysts were named as Ce–Fe–Mn catalysts. As a contrast, CeO2 and Fe–Mn oxides were prepared by the same process.
:Mn of 1:4) and Ce(NO3)3·6H2O were dissolved in deionized water, and stirred for 30 min in an ice water bath. Then, the oxalic acid solution was added into the metal ion solution to form a precipitate. Then the mixture was stirred in an ice water bath for 1 h. The precipitate was collected by suction filtration, and washed with distilled water and alcohol three times. Finally, the obtained products were dried in a vacuum freeze drier and calcined at 300 °C (Ce–Fe–Mn-300), 400 °C (Ce–Fe–Mn-400) and 500 °C (Ce–Fe–Mn-500), respectively. After grinding to 60 mesh, the catalysts were named as Ce–Fe–Mn catalysts. As a contrast, CeO2 and Fe–Mn oxides were prepared by the same process.
2.2 Catalytic performance test
The chlorobenzene oxidation experiments were performed in a fixed bed reactor as shown in Fig. S1.† The reaction consists of a gas distribution system, a flue gas preheating system, a selective catalytic reduction reactor and a flue gas analysis system. The flow rate of each gas in the mixing part was controlled by the mass flow controller. The content of the chlorobenzene in the flue gas was controlled by the adjustment of the inlet concentration. The simulated gas was heated to a required temperature by preheating for the reaction, and the catalysts were in the reactor. The temperature of catalyst bed was controlled by an E-type thermocouple in the reactor. In this experiment, N2 was used as the equilibrium gas. The specific operating conditions were shown in Table S1.†Unless otherwise specified, in this experiment the weight of catalysts was 0.2 g, which was prepared by oxalic acid co-precipitation. The ratio of chlorobenzene conversion was calculated as follows:
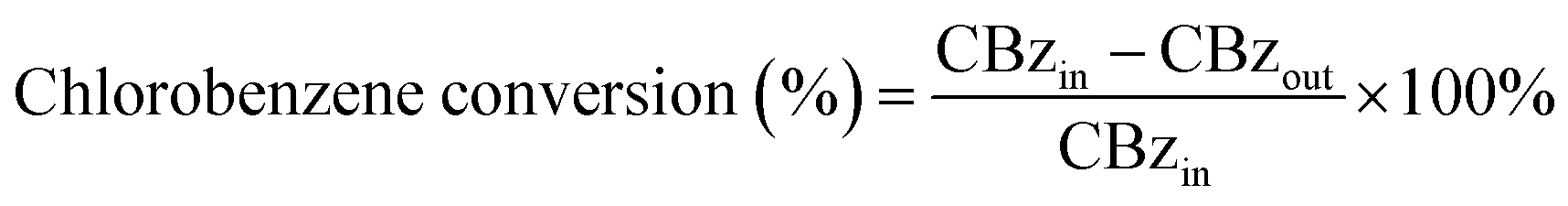
3. Results and discussion
The XRD patterns of Ce–Fe–Mn catalysts are shown in Fig. 1. Pure CeO2 in the form of cerianite with a cubic fluorite structure exhibited diffraction peaks at 28.5°, 33.1°, 47.5°, and 56.3°, which correspond with reflections of the (111), (200), (220), and (311) crystallographic planes, respectively (JCPDS no. 34-0394). The XRD pattern of the Fe–Mn catalyst calcinated at 400 °C is attributed to the combination of Mn3O4 (JCPDS no. 24-0734, 2θ = 18.0°, 28.9°, 32.3°, and 36.1°) and Fe2O3 (JCPDS no. 39-1346, 2θ = 30.2°, 35.6°, 43.2°, and 57.3°). The diffraction peaks of 2% Ce modified Fe–Mn catalysts were not observed, owing to low concentration of CeO2 in a highly dispersed phase. The low intensity diffraction peak at 47.5° 2θ appeared in the high Ce content (5, 7.5, and 10%) catalysts, coinciding with the face centred cubic (fcc) fluorite structure CeO2. The XRD patterns of Ce(5%)–Fe–Mn-300 and Ce(5%)–Fe–Mn-400 were similar, which exhibited the diffraction peaks of Mn3O4. When the calcination temperature of Ce–Fe–Mn was increased up to 500 °C, the diffraction peaks of MnOx were those of Mn2O3 (JCPDS no. 41-1442, 2θ = 18.8°, 33.0°, 38.2°, and 55.2°). The crystalline phase transition of MnOx was consistant with the TEM and H2-TPR results. In the meantime the particle sizes of Ce(5%)–Fe–Mn-300, Ce(5%)–Fe–Mn-400 and Ce(5%)–Fe–Mn-500 were estimated by the Scherrer equation to be 1.1, 8.1 and 21.4 nm, respectively.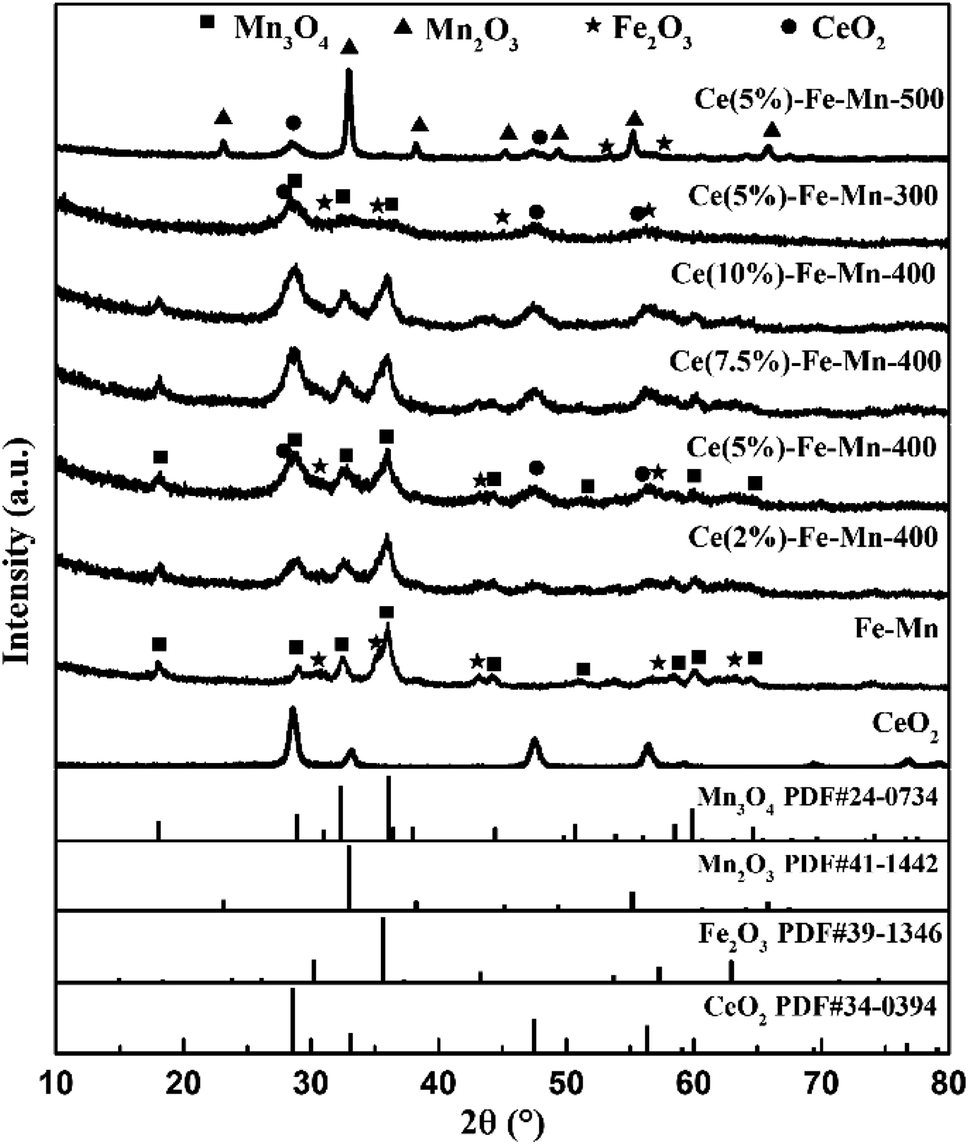 | ||
| Fig. 1 XRD patterns of as-synthesied CeO2, Fe–Mn, Ce–Fe–Mn catalysts. | ||
It is well known that a high surface area is beneficial to catalytic performance. The BET surface area, total pore volume and pore size of the Ce–Fe–Mn catalysts are summarized in Table 1. All Ce–Fe–Mn catalysts exhibited higher surface areas than the Fe–Mn catalyst. With increasing amounts of Ce doping, the specific surface area firstly increased and then decreased at the same roasting temperature. 5% Ce modified Fe–Mn catalysts had the largest surface area (190.8 m2 g−1), which might be caused by the pore structure and good dispersion of Ce. In addition, the sintering temperature had an obvious influence on the BET surface area of the Ce–Fe–Mn catalysts. When the roasting temperature was 300 °C, Ce(5%)–Fe–Mn-300 catalysts had a wonderful surface area (290.6 m2 g−1). With the increasing the calcination temperature to 500 °C, the BET surface area decreased significantly, which is attributed to the increase in the size of the particles, which agrees well with the sample morphology (Fig. 2 and S2†).
| Catalysts | Surface area (S/m2 g−1) | Pore volume (V/cm3 g−1) | Pore diameter (D/nm) |
|---|---|---|---|
| Fe–Mn | 33.4616 | 0.226514 | 266.9446 |
| Ce(2%)–Fe–Mn-400 | 188.1677 | 0.321711 | 68.3881 |
| Ce(5%)–Fe–Mn-400 | 190.8000 | 0.301823 | 61.1414 |
| Ce(7.5%)–Fe–Mn-400 | 85.7452 | 0.208402 | 94.6542 |
| Ce(10%)–Fe–Mn-400 | 73.7974 | 0.238484 | 126.6648 |
| Ce(5%)–Fe–Mn-300 | 290.6160 | 0.319763 | 42.3521 |
| Ce(5%)–Fe–Mn-500 | 47.0542 | 0.206230 | 178.4679 |
 | ||
| Fig. 2 TEM images of Ce–Fe–Mn-400 (a)–(c), and HRTEM image of Ce–Fe–Mn-400 (d). | ||
The morphology of samples was characterized by TEM. As shown in Fig. 2 and S2,† the Ce–Fe–Mn catalysts consisted of crumb-like nanoparticles with shaggy surface. In addition, it is found that the particle size of the samples obviously increased with increasing the calcination temperature, which was consistent with the average crystallite size estimated from the Scherrer equation. Additionally, the spatial chemical compositions of Ce–Fe–Mn catalysts were further identified by EDS spectroscopy (Fig. S3†), confirming the uniform distribution of Ce, Fe, Mn and O elements on the catalysts. The high dispersion of Mn, Fe, and Ce on the surface of the Ce–Fe–Mn-400 catalysts provided a large number of oxidative active sites, which improved the oxidation efficiency of chlorobenzene.31
In order to check the redox properties of Ce–Fe–Mn catalysts, H2-TPR of the catalysts was carried out, and the results are shown in Fig. 3. The H2-TPR patterns strongly depend on the oxidation state of Ce–Fe–Mn catalysts. As presented in Fig. 3, for Fe–Mn and Ce–Fe–Mn catalysts, the reduction temperature process of Fe2O3 to Fe3O4 occurred in the range 350–450 °C.32 Meanwhile, for Fe–Mn, Ce–Fe–Mn-300, and Ce–Fe–Mn-400 catalysts, two main reduction peaks (TTP: 290 °C and 430 °C) were observed, which were ascribed to the reduction of manganese from MnO2 to Mn2O3 and Mn2O3 to Mn3O4, respectively.33 Compared with Ce–Fe–Mn-400 catalysts, the reduction temperature 230 °C appeared in Fe–Mn and Ce–Fe–Mn-300 catalysts, which is consistent with the H2-TPR results previously reported.24 Therefore, Fe–Mn and Ce–Fe–Mn-300 catalysts were reduced at a lower temperature and they consumed more H2 than Ce–Fe–Mn-400 catalysts, demonstrating the weaker Mn–O bonds of Fe–Mn and Ce–Fe–Mn-300 catalysts,34 whereas, for the Ce–Fe–Mn-500 catalyst, the first reduction peak changed from 290 °C to 325 °C. The reduction temperature shifting to a higher temperature probably means a decrease in the lattice oxygen mobility on the Ce–Fe–Mn catalyst.35 Therefore, the catalytic activity of chlorobenzene oxidation was correlated with the oxygen mobility.36 In other words, the higher the oxygen mobility, the higher the catalytic activity. According to previous reports, CeO2 samples showed two reduction peaks at 530 °C and 800 °C, which could be assigned to the reduction of surface and bulk Ce4+, respectively.37 However, no obvious reduction peaks attributable to CeO2 were observed for the Ce–Fe–Mn catalysts, implying that the interaction between the Ce, Fe and Mn in the catalysts was large, which coincided with He’s observations.38
 | ||
| Fig. 3 TPR profiles of Ce–Fe–Mn catalysts. | ||
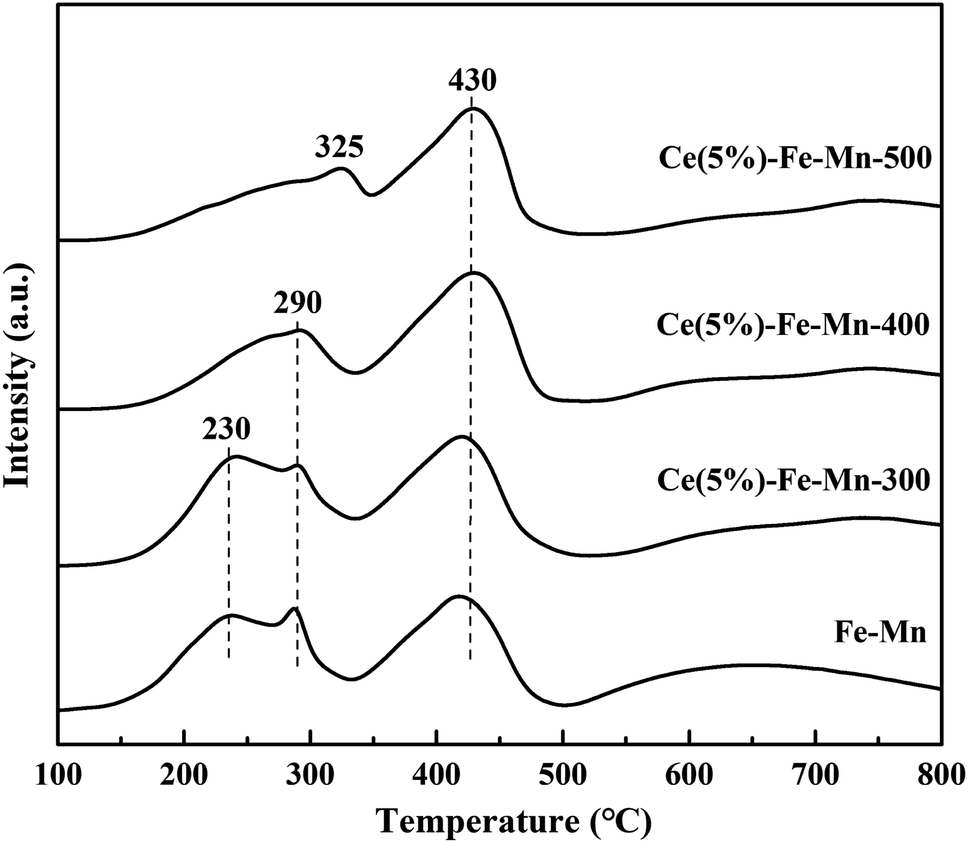
On the basis of the XPS analysis, the surface chemical states of Ce, Fe, Mn, and O species for the Ce–Fe–Mn catalysts were evaluated and are shown in Fig. 4, and the atomic concentrations on the surface of the catalysts are shown in Table 2. As shown in Fig. 4A, two main peaks of the Mn 2p spectra can be identified as Mn 2p3/2 and Mn 2p1/2. The deconvolution procedure was applied for the Mn 3p3/2 signal, it can be reliably separated into three contributing peaks 641.0–641.4 eV, 642.5–642.9 eV and 644.1–646.1 eV, which may be assigned to Mn2+, Mn3+ and Mn4+, respectively.39 Because no crystalline MnO2 or Mn2O3 species were detected by XRD on all the catalysts calcined at 300 °C and 400 °C, MnO2 or Mn2O3 in the catalysts may be well dispersed on the catalyst surface in an amorphous state. It can be seen from Table 2 that the concentration of Mn4+ in Ce(5%)–Fe–Mn-400 °C was higher than that of other catalysts, and this catalyst presented higher performance for the catalysis of chlorobenzene. Accordingly, the higher ratio of Mn4+ on Ce–Fe–Mn catalysts might lead to more active sites, thereby enhancing chlorobenzene oxidation. The Fe 2p XPS spectra of the Ce–Fe–Mn catalysts is shown in Fig. 4B. Two main peaks of Fe 2p spectra correspond to Fe 2p3/2 and Fe 2p1/2. Peak fitting deconvolution of the Fe 2p3/2 peak indicates the presence of two different Fe species: Fe2+ (710.2–710.7 eV) and Fe3+ (712.2–712.8 eV).40 The Ce 3dXPS spectra of the Ce–Fe–Mn catalysts is shown in Fig. 4C. The peaks at u, u′′, u′′′, v, v′′, and v′′′ are attributed to Ce4+, and the peaks at u′ and v′ are considered to be Ce3+.41 As shown in Table 2, Ce was mainly in the +4 valence state and only a small fraction of the +3 valence state existed in Ce–Fe–Mn catalysts, which might be due to the lower content of lattice defect and oxygen vacancies.
 | ||
| Fig. 4 XPS spectra of the catalysts over the spectral regions of Mn 2p (A), Fe 2p (B), Ce 3d (C) and O 1s (D). | ||
| Catalysts | Ce (at%) | Fe (at%) | Mn (at%) | O (at%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Ce3+ | Ce4+ | Fe2+ | Fe3+ | Mn2+ | Mn3+ | Mn4+ | Oα | Oβ | |
| Ce(2%)–Fe–Mn-400 | 23.05 | 76.95 | 55.64 | 44.36 | 44.28 | 29.27 | 26.49 | 74.18 | 25.82 |
| Ce(5%)–Fe–Mn-400 | 12.50 | 87.50 | 48.71 | 51.29 | 31.79 | 34.48 | 33.74 | 37.33 | 62.67 |
| Ce(7.5%)–Fe–Mn-400 | 18.82 | 81.11 | 58.26 | 41.74 | 46.58 | 31.48 | 21.93 | 81.11 | 18.88 |
| Ce(10%)–Fe–Mn-400 | 30.72 | 65.66 | 58.57 | 41.42 | 43.52 | 37.95 | 18.53 | 80.72 | 19.98 |
| Ce(5%)–Fe–Mn-300 | 19.06 | 80.94 | 51.63 | 48.37 | 37.30 | 35.27 | 27.43 | 73.14 | 26.86 |
| Ce(5%)–Fe–Mn-500 | 12.18 | 87.82 | 53.44 | 46.56 | 40.90 | 33.11 | 25.99 | 74.65 | 25.35 |
The O 1s XPS spectra of the Ce–Fe–Mn catalysts is shown in Fig. 4D. The O 1s spectra showed two peaks: the surface lattice oxygen (denoted as Oα) at about 529.6 eV, and the chemisorbed oxygen species (denoted as Oβ).26 Between the two oxygen species, the chemisorbed oxygen was more active than the lattice oxygen.13 The surface chemisorbed oxygen has been reported to be in favor of catalytic oxidation of chlorobenzene.42 Therefore, Oβ plays an important role in the Ce–Fe–Mn catalysts during catalytic reaction. Table 2 shows that the ratio of chemisorbed oxygen to lattice oxygen was higher for Ce(5%)–Fe–Mn-400 than the others, which is consistent with the higher catalytic performance against chlorobenzene.
The catalytic performances of the Fe–Mn and Ce–Fe–Mn catalysts were evaluated for the oxidation of chlorobenzene. The ratio of chlorobenzene conversion was investigated as a function of the temperature, 100–400 °C, and the results are shown in Fig. 5. As can be observed, pure Fe–Mn catalyst performed the lowest conversion with the ratio of chlorobenzene conversion at about 90% when temperature reached as high as 400 °C. Upon addition of Ce to the Fe–Mn catalyst, the ratio of chlorobenzene conversion remarkably increased up to a Ce content of 5%, and Ce(5%)–Fe–Mn was the most active among all catalysts achieving ∼99% chlorobenzene conversion at 250 °C. When the CeO2 content continuously increased to 7.5–10%, the catalytic activity started to decrease, even worse than that of Ce(2%)–Fe–Mn, which indicated that the optimum concentration of Ce is 5% for Ce–Fe–Mn catalysts to reach the largest chlorobenzene conversion. For Fe–Mn, the catalytic activity for chlorobenzene oxidation was obviously lower than those of Ce–Fe–Mn, showing that CeO2 addition offers significant promotion over Fe–Mn.
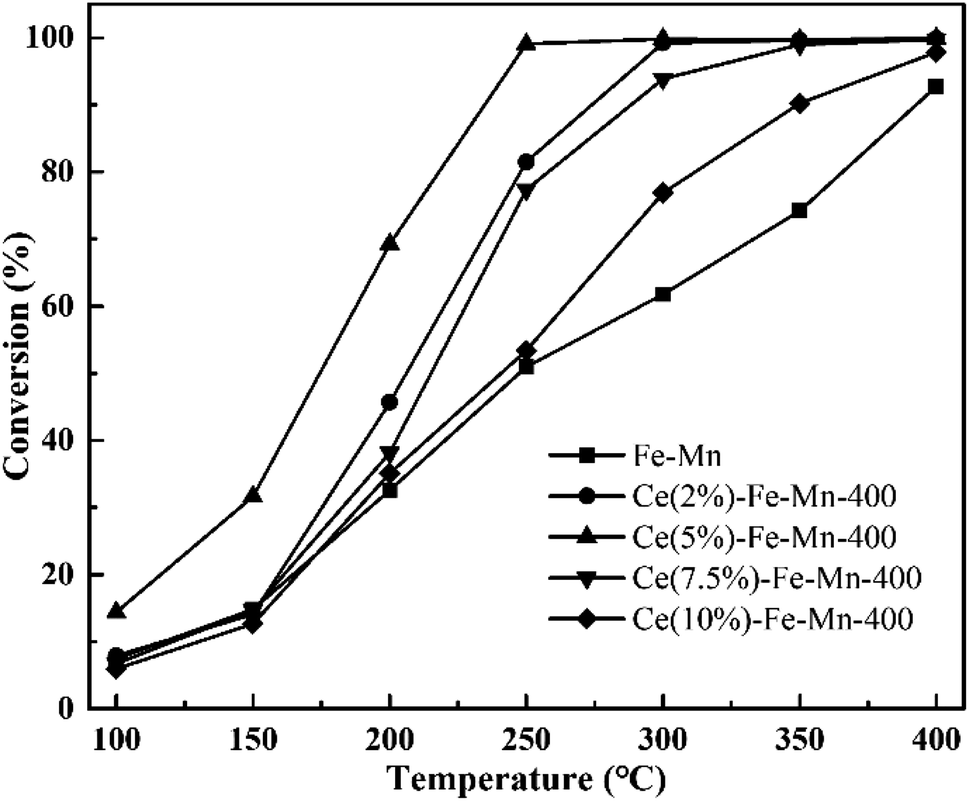 | ||
| Fig. 5 Catalytic performance of the Fe–Mn and Ce–Fe–Mn catalysts. Reaction conditions: 250 ppm CBz, 80% N2, 10% O2, and GHSV of 1500 h−1, 0.2 g catalysts. | ||
In order to further discuss the factors affecting the catalytic efficiency of chlorobenzene, the catalytic performances of Ce(5%)–Fe–Mn catalysts with different calcination temperatures were investigated. From Fig. 6, it is evident that there is no significant distinction in the ratio of chlorobenzene conversion between Ce(5%)–Fe–Mn-300 and Ce(5%)–Fe–Mn-400. This is because Ce(5%)–Fe–Mn-300 catalysts had a large BET surface area, while Ce(5%)–Fe–Mn-400 had more Mn4+ content, resulting in more chemisorption oxygen. Therefore, the ratio of chlorobenzene conversion with the two catalysts is basically the same after the synergistic effects. When the calcination temperature rises to 500 °C, the catalytic efficiency of chlorobenzene for Ce(5%)–Fe–Mn-500 obviously reduced, which was correlated with the production of Mn2O3, resulting in a decline of the oxygen mobility.24
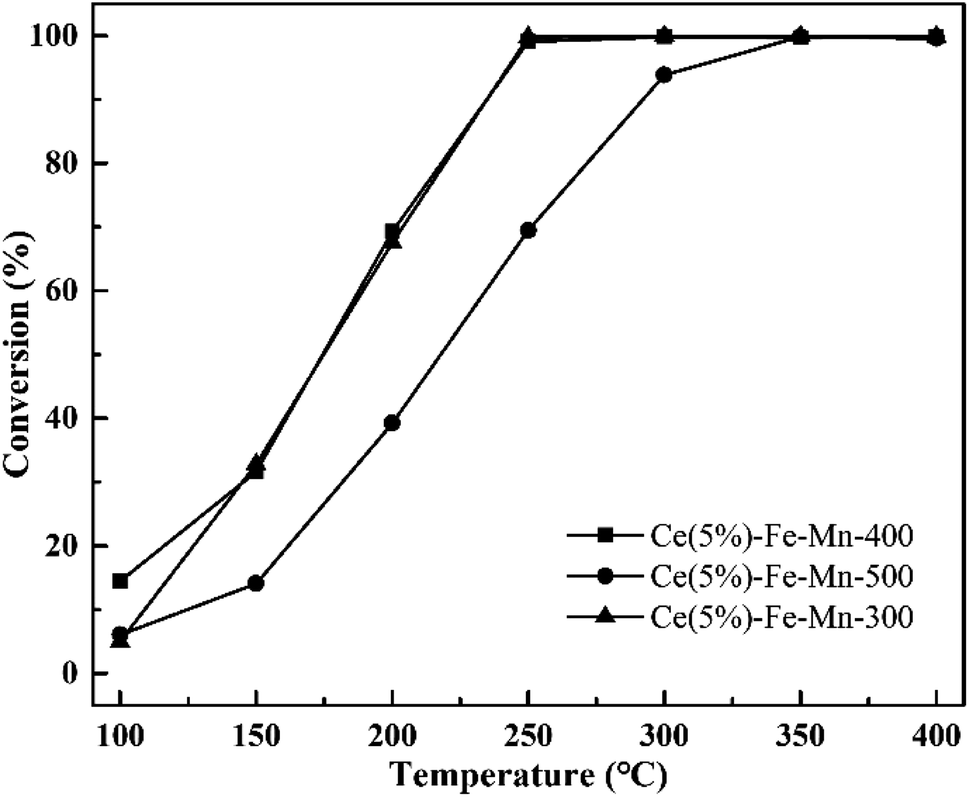 | ||
| Fig. 6 Catalytic performance of Ce–Fe–Mn catalysts with different calcination temperatures. | ||
The chlorobenzene oxidation reaction over Ce–Fe–Mn catalysts is mainly determined by two critical factors, one is the catalyst’s ability to trap surface chemisorbed oxygen groups which promotes oxygen transportability to enhance activity.43 Based on the H2-TPR results, it was shown that reaction temperature of the catalysts was associated to the amount of lattice oxygen of the catalysts. The Mn3O4 phase in Ce–Fe–Mn catalysts was more reducible at lower temperatures and could easily trap surface chemisorbed oxygen groups which increases the mobility of O atoms compared to the other catalysts. Another determining factor in catalytic oxidation reactions is the number of active surface sites.44 For Ce(5%)–Fe–Mn-400 and Ce(5%)–Fe–Mn-300 catalysts, owing to a large surface area and high dispersion of Mn, Fe, and Ce, a greater number of active surface sites were present for the adsorption of chlorobenzene molecules, resulting in the improvement of the catalytic activity. Moreover, the decrease in specific surface area caused by roasting is also one of the reasons why the activity decreases at higher temperatures, because the active surface sites for the adsorption of chlorobenzene molecules decreases.
According to the analysis above, the possible mechanism for chlorobenzene catalytic oxidation by the Ce–Fe–Mn catalysts is shown in Fig. 7. In the chlorobenzene catalytic combustion, C–Cl bonds are weaker than C–H bonds, and hence, more prone to attack by nucleophiles.45 The first step in the oxidation of chlorobenzene is the C–Cl molecular bonds of chlorobenzene are destroyed, and the free Cl− ions are adsorbed to the metal cations. On the other hand, during the catalytic oxidation, chlorobenzene molecules are adsorbed on the surface of Ce–Fe–Mn catalysts and oxidized by the active oxygen into H2O and CO2.46 The free Cl− ions produce HCl, and react with chemisorbed oxygen to form Cl2 simultaneously.38 The chlorobenzene oxidation reaction is shown eqn (1):
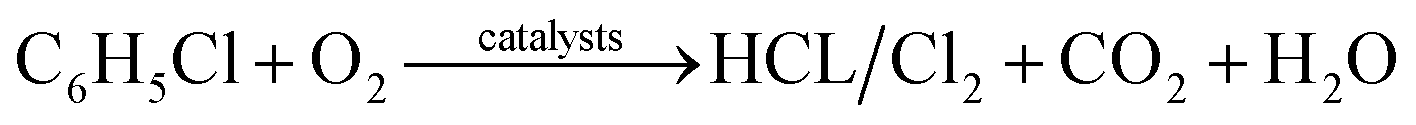 | (1) |
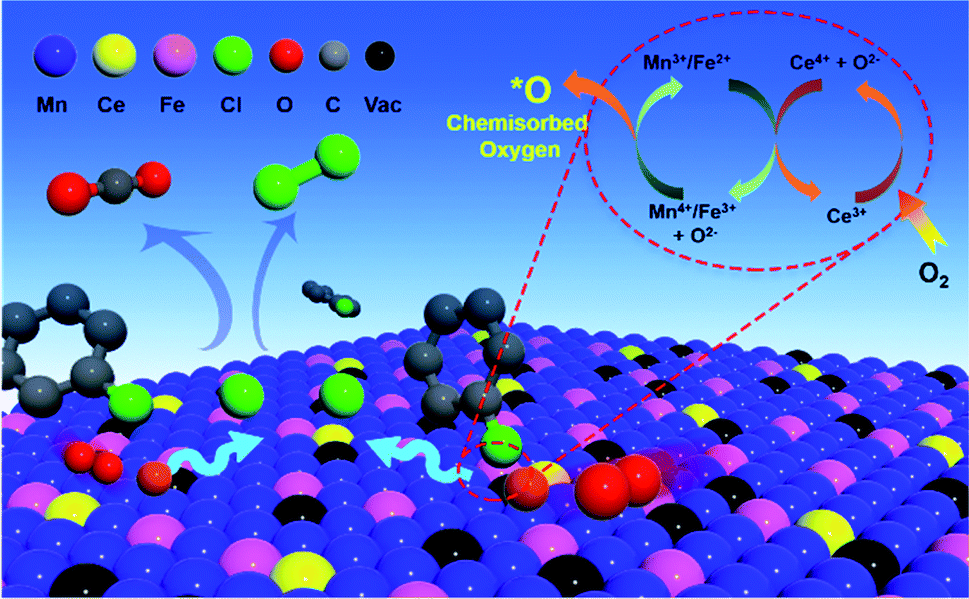 | ||
| Fig. 7 Synergistic mechanism of Ce–Fe–Mn catalysts. | ||
Cl species need be removed as Cl2 or HCl from the surface of catalysts so as to avoid deactivating the catalysts.47 In this study, the removal of Cl species was promoted by elevating the temperature (e.g. to 250 °C) and synergistic mechanisms on the Ce–Fe–Mn catalysts. It may be deduced that the removal of Cl species is a slow step so that it determines the rate of chlorobenzene catalytic oxidation. According to a previous report, the reduction of Mn3O4 and Fe2O3 releases additional O atoms attached to the catalysts’ surface to become surface chemisorbed oxygen.41 The redox cycle of Fe–Mn catalysts is shown as eqn (2):
| Mn4+ + Fe2+ → Mn3+ + Fe3+ | (2) |
Additionally, Ce plays a co-catalytic role in the Ce–Fe–Mn catalysts. Ce4+ has strong oxidizing properties, which can oxidize Mnx+ and Fex+. At the same time, Ce3+ is oxidized to CeO2 by O2 in the air16 (eqn (3)–(5)).
| Mn3+ + Ce4+ + O2− → Mn4+ + Ce3+ | (3) |
| Fe2+ + Ce4+ + O2− → Fe3+ + Ce3+ | (4) |
| Ce3+ + O2 → Ce4+ + O2− | (5) |
Therefore, the interaction of Ce, Fe and Mn can accelerate the conversion of O2 into surface chemisorbed oxygen and improve the content of surface chemisorbed oxygen. Since the adsorption of surface O atoms on the oxygen vacancies of catalysts is the rate determining step for the HCl oxidation reaction, the surface chemisorbed oxygen is critical for the so-called Deacon process48 (eqn (6)).
| 2HCl + O → H2O + Cl2 | (6) |
Therefore, the higher the amount of surface chemisorbed oxygen in Ce–Fe–Mn catalysts, the easier the removal of Cl from the catalyst surface, resulting in the improvement of the reaction efficiency. This implies that the synergistic mechanism between Ce–Fe–Mn is combined to promote the chlorobenzene oxidation mutually.
4 Conclusions
In summary, Ce–Fe–Mn catalysts were synthesized by an oxalic acid assisted co-precipitation method, and the influence of ceria addition to Fe–Mn with different calcination temperatures upon the catalytic oxidation of chlorobenzene was examined. By comparison, it was found that catalysts containing CeO2 exhibited high surface area and amounts of surface-active oxygen, leading to the enhancement of the catalytic activity of chlorobenzene. In addition, calcination temperatures clearly affected the activity of the prepared catalysts. When calcined at temperatures of 300 °C and 400 °C, low reduction temperatures of Ce–Fe–Mn catalysts could increase the mobility of oxygen. Therefore, the synergistic mechanism between Ce–Fe–Mn was further enhanced, more chemisorbed oxygen was produced, and so the catalytic activity of chlorobenzene was improved. The decrease in the activity of Ce–Fe–Mn catalysts calcined at a high temperature can be attributed to the decrease in the oxygen mobility and surface area. For Ce(5%)–Fe–Mn-400 catalyst, the higher surface area could lead to the increase in the number of oxygen vacancies, more of the Mn3O4 phase could produce more chemisored oxygen, thus leading to a good catalytic oxidation of chlorobenzene. This investigation may offer a promising strategy for designing efficient catalysts for the destruction of chlorobenzene.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51674002, 21673001, 21773114, 21972065) and the Key Project of the National Nature Science Foundation of China (U1660206).References
- H.-m. Long, Q. Shi, H.-l. Zhang, R.-f. Wei, T.-j. Chun and J.-x. Li, Application status and comparison of dioxin removal technologies for iron ore sintering process, J. Iron Steel Res. Int., 2018, 25, 357–365 CrossRef.
- X. Gao, B. Ji and Q. Huang, Thermal dechlorination of heavily PCB-contaminated soils from a sealed site of PCB-containing electrical equipment, Environ. Sci. Pollut. Res. Int., 2016, 23, 15544–15550 CrossRef CAS PubMed.
- X. Gao, W. Wang and X. Liu, Low-temperature dechlorination of hexachlorobenzene on solid supports and the pathway hypothesis, Chemosphere, 2008, 71, 1093–1099 CrossRef CAS PubMed.
- R. E. Bailey, D. van Wijk and P. C. Thomas, Sources and prevalence of pentachlorobenzene in the environment, Chemosphere, 2009, 75, 555–564 CrossRef CAS PubMed.
- J. Chen, D. Zhong, H. Hou, C. Li, J. Yang, H. Zhou, L. Hu and L. Wang, Ferrite as an effective catalyst for HCB removal in soil: characterization and catalytic performance, Chem. Eng. J., 2016, 294, 246–253 CrossRef CAS.
- B. K. Gullett, D. F. Natschke and K. R. Bruce, Thermal Treatment of 1,2,3,4-Tetrachlorodibenzo-p-dioxin by Reaction with Ca-Based Sorbents at 23–300 °C, Environ. Sci. Technol., 1997, 31, 1855–1862 CrossRef CAS.
- A. Fadli, C. Briois, C. Baillet and J. P. Sawerysyn, Experimental study on the thermal oxidation of chlorobenzene at 575–825 °C, Chemosphere, 1999, 38, 2835–2848 CrossRef CAS.
- B. Higgins, M. J. Thomson, D. Lucas, C. P. Koshland and R. F. Sawyer, An experimental and numerical study of the thermal oxidation of chlorobenzene, Chemosphere, 2001, 42, 703–717 CrossRef CAS PubMed.
- R. Q. Long and R. T. Yang, Carbon nanotubes as superior sorbent for dioxin removal, J. Am. Chem. Soc., 2001, 123, 2058–2059 CrossRef CAS PubMed.
- M. B. Chang, K. H. Chi and G. P. Chang-Chien, Evaluation of PCDD/F congener distributions in MWI flue gas treated with SCR catalysts, Chemosphere, 2004, 55, 1457–1467 CrossRef CAS PubMed.
- C. Du, S. Lu, Q. Wang, A. G. Buekens, M. Ni and D. P. Debecker, A review on catalytic oxidation of chloroaromatics from flue gas, Chem. Eng. J., 2018, 334, 519–544 CrossRef CAS.
- C. He, Y. Yu, J. Shi, Q. Shen, J. Chen and H. Liu, Mesostructured Cu–Mn–Ce–O composites with homogeneous bulk composition for chlorobenzene removal: catalytic performance and microactivation course, Mater. Chem. Phys., 2015, 157, 87–100 CrossRef CAS.
- Z. Wang, G. Shen, J. Li, H. Liu, Q. Wang and Y. Chen, Catalytic removal of benzene over CeO2–MnOx composite oxides prepared by hydrothermal method, Appl. Catal., B, 2013, 138–139, 253–259 CrossRef CAS.
- P. Sun, W. Wang, X. Weng, X. Dai and Z. Wu, Alkali Potassium Induced HCl/CO2 Selectivity Enhancement and Chlorination Reaction Inhibition for Catalytic Oxidation of Chloroaromatics, Environ. Sci. Technol., 2018, 52, 6438–6447 CrossRef CAS PubMed.
- P. Sun, W. Wang, X. Dai, X. Weng and Z. Wu, Mechanism study on catalytic oxidation of chlorobenzene over MnxCe1−xO2/H-ZSM5 catalysts under dry and humid conditions, Appl. Catal., B, 2016, 198, 389–397 CrossRef CAS.
- X. Liu, L. Chen, T. Zhu and R. Ning, Catalytic oxidation of chlorobenzene over noble metals (Pd, Pt, Ru, Rh) and the distributions of polychlorinated by-products, J. Hazard. Mater., 2019, 363, 90–98 CrossRef CAS PubMed.
- W. L. Wang, Q. Meng, Y. Xue, X. Weng, P. Sun and Z. Wu, Lanthanide perovskite catalysts for oxidation of chloroaromatics: secondary pollution and modifications, J. Catal., 2018, 366, 213–222 CrossRef CAS.
- Y. Lu, Q. Dai and X. Wang, Catalytic combustion of chlorobenzene on modified LaMnO3 catalysts, Catal. Commun., 2014, 54, 114–117 CrossRef CAS.
- N. Zhou, B. He, X. Wang and Z. Hu, Preparation and characterization of Au@TiO2 core-shell hollow nanoparticles with CO oxidation performance, J. Nanopart. Res., 2014, 16(11), 2676 CrossRef.
- F. G. Durán, B. P. Barbero, L. E. Cadús, C. Rojas, M. A. Centeno and J. A. Odriozola, Manganese and iron oxides as combustion catalysts of volatile organic compounds, Appl. Catal., B, 2009, 92, 194–201 CrossRef.
- T. Odoom-Wubah, Q. Li, I. Adilov, J. Huang and Q. Li, Towards efficient Pd/Mn3O4 catalyst with enhanced acidic sites and low temperature reducibility for Benzene abatement, Mol. Catal., 2019, 477, 110558 CrossRef CAS.
- J. Li, L. Li, W. Cheng, F. Wu, X. Lu and Z. Li, Controlled synthesis of diverse manganese oxide-based catalysts for complete oxidation of toluene and carbon monoxide, Chem. Eng. J., 2014, 244, 59–67 CrossRef CAS.
- H. Xu, N. Yan, Z. Qu, W. Liu, J. Mei, W. Huang and S. Zhao, Gaseous Heterogeneous Catalytic Reactions over Mn-Based Oxides for Environmental Applications: A Critical Review, Environ. Sci. Technol., 2017, 51, 8879–8892 CrossRef CAS PubMed.
- S. C. Kim and W. G. Shim, Catalytic combustion of VOCs over a series of manganese oxide catalysts, Appl. Catal., B, 2010, 98, 180–185 CrossRef CAS.
- L. J. France, Q. Yang, W. Li, Z. Chen, J. Guang, D. Guo, L. Wang and X. Li, Ceria modified FeMnO-enhanced performance and sulphur resistance for low-temperature SCR of NOx, Appl. Catal., B, 2017, 206, 203–215 CrossRef CAS.
- P. Venkataswamy, D. Jampaiah, F. Lin, I. Alxneit and B. M. Reddy, Structural properties of alumina supported Ce–Mn solid solutions and their markedly enhanced catalytic activity for CO oxidation, Appl. Surf. Sci., 2015, 349, 299–309 CrossRef CAS.
- D. A. Aguilera, A. Perez, R. Molina and S. Moreno, Cu–Mn and Co–Mn catalysts synthesized from hydrotalcites and their use in the oxidation of VOCs, Appl. Catal., B, 2011, 104, 144–150 CrossRef CAS.
- M. M. Barroso Quiroga, B. P. Barbero and L. E. Cadus, Synthesis of a catalyst of Mn–Fe–O by mechano-chemical reaction, Appl. Catal., A, 2014, 474, 26–33 CrossRef CAS.
- J. Chen, X. Chen, X. Chen, W. Xu, Z. Xu, H. Jia and J. Chen, Homogeneous introduction of CeOy into MnOx-based catalyst for oxidation of aromatic VOCs, Appl. Catal., B, 2018, 224, 825–835 CrossRef CAS.
- J. Kan, L. Deng, B. Li, Q. Huang, S. Zhu, S. Shen and Y. Chen, Performance of co-doped Mn–Ce catalysts supported on cordierite for low concentration chlorobenzene oxidation, Appl. Catal., A, 2017, 530, 21–29 CrossRef CAS.
- Z. Wu, B. Jiang and Y. Liu, Effect of transition metals addition on the catalyst of manganese/titania for low-temperature selective catalytic reduction of nitric oxide with ammonia, Appl. Catal., B, 2008, 79, 347–355 CrossRef CAS.
- Z. Fan, J. W. Shi, C. Gao, G. Gao, B. Wang and C. Niu, Rationally Designed Porous MnOx–FeOx Nanoneedles for Low-Temperature Selective Catalytic Reduction of NOx by NH3, ACS Appl. Mater. Interfaces, 2017, 9, 16117–16127 CrossRef CAS PubMed.
- K. Zhuang, Y.-p. Zhang, T.-j. Huang, B. Lu and K. Shen, Sulfur-poisoning and thermal reduction regeneration of holmium-modified Fe–Mn/TiO2 catalyst for low-temperature SCR, J. Fuel Chem. Technol., 2017, 45, 1356–1364 CrossRef CAS.
- X. Guo, J. Jia, H. Dong, Q. Wang, T. Xu, B. Fu, R. Ran, P. Liang, X. Huang and X. Zhang, Hydrothermal synthesis of Fe Mn bimetallic nanocatalysts as high-efficiency cathode catalysts for microbial fuel cells, J. Power Sources, 2019, 414, 444–452 CrossRef CAS.
- H. Xiao, J. Wu, X. Wang, J. Wang, S. Mo, M. Fu, L. Chen and D. Ye, Ozone-enhanced deep catalytic oxidation of toluene over a platinum–ceria-supported BEA zeolite catalyst, Mol. Catal., 2018, 460, 7–15 CrossRef CAS.
- S. C. Kim and W. G. Shim, Influence of physicochemical treatments on iron-based spent catalyst for catalytic oxidation of toluene, J. Hazard. Mater., 2008, 154, 310–316 CrossRef PubMed.
- W. Weimin, Y. Yongnian and Z. Jiayu, Redox behavior of trimanganese tetraoxide catalysts, Recl. Trav. Chim. Pays-Bas, 2010, 114, 22–25 CrossRef.
- F. He, Y. Chen, P. Zhao and S. Liu, Effect of calcination temperature on the structure and performance of CeOx–MnOx/TiO2 nanoparticles for the catalytic combustion of chlorobenzene, J. Nanopart. Res., 2016, 18(5), 119 CrossRef.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. Smart, Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- L. Xing, Y. Xu and Q. Zhong, Mn and Fe Modified Fly Ash As a Superior Catalyst for Elemental Mercury Capture under Air Conditions, Energy Fuels, 2012, 26, 4903–4909 CrossRef CAS.
- F. Kong, J. Qiu, H. Liu, R. Zhao and Z. Ai, Catalytic oxidation of gas-phase elemental mercury by nano-Fe2O3, J. Environ. Sci., 2011, 23, 699–704 CrossRef CAS.
- F. Liu, H. He, Y. Ding and C. Zhang, Effect of manganese substitution on the structure and activity of iron titanate catalyst for the selective catalytic reduction of NO with NH3, Appl. Catal., B, 2009, 93, 194–204 CrossRef CAS.
- C. E. Hetrick, J. Lichtenberger and M. D. Amiridis, Catalytic oxidation of chlorophenol over V2O5/TiO2 catalysts, Appl. Catal., B, 2008, 77, 255–263 CrossRef CAS.
- H. Pan, Y. Jian, C. Chen, C. He, Z. Hao, Z. Shen and H. Liu, Sphere-Shaped Mn3O4 Catalyst with Remarkable Low-Temperature Activity for Methyl-Ethyl-Ketone Combustion, Environ. Sci. Technol., 2017, 51, 6288–6297 CrossRef CAS PubMed.
- J. Lichtenberger, Catalytic oxidation of chlorinated benzenes over V2O5/TiO2 catalysts, J. Catal., 2004, 223, 296–308 CrossRef CAS.
- Q. Dai, X. Wang and G. Lu, Low-temperature catalytic combustion of trichloroethylene over cerium oxide and catalyst deactivation, Appl. Catal., B, 2008, 81, 192–202 CrossRef CAS.
- X. Wang, Q. Kang and D. Li, Low-temperature catalytic combustion of chlorobenzene over MnOx–CeO2 mixed oxide catalysts, Catal. Commun., 2008, 9, 2158–2162 CrossRef CAS.
- H. Huang, Q. Dai and X. Wang, Morphology effect of Ru/CeO2 catalysts for the catalytic combustion of chlorobenzene, Appl. Catal., B, 2014, 158–159, 96–105 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10489e |
| This journal is © The Royal Society of Chemistry 2020 |