
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanofibrous TiO2 produced using alternating field electrospinning of titanium alkoxide precursors: crystallization and phase development
Sarah L. Nealya,
Courtney Severinob,
W. Anthony Brayerb and
Andrei Stanishevsky *b
*b
aDepartment of Chemistry, University of Alabama at Birmingham, 901 14th Street South, Birmingham, AL 35294-1170, USA
bDepartment of Physics, University of Alabama at Birmingham, 1300 University Boulevard, Birmingham, AL 35294-1170, USA. E-mail: astan@uab.edu
First published on 13th February 2020
Abstract
High-yield, free-surface alternating field electrospinning (AFES) was effectively used in the fabrication of titanium oxide nanofibrous materials from the precursors based on titanium alkoxide and a blend of polyvinylpyrrolidone and hydroxypropyl cellulose. The alkoxide/polymer mass ratio in the precursor solution has significant effects on the precursor fiber production rate as well as the structure of resulting TiO2 nanofibers after thermal processing of precursor fibers at temperatures from 500 to 1000 °C. Within the range of tested process parameters, the best fiber production rate of ∼5.2 g h−1 was achieved, in terms of the mass of crystallized TiO2 nanofibers, with the precursor that corresponded to 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 TiO2/polymer mass ratio. TiO2 nanofibers produced by calcination at 500 °C for 3 h had 100–500 nm diameters and were composed of anatase (20–25 nm crystallite size) with rutile content 0.1–6.0 mol%, depending on the precursor composition. A considerable amount of anatase phase (up to 80 mol%) can be retained after thermal processing of TiO2 nanofibers at 750 °C for 3 h. A nanofibrous material composed of smooth and long, predominantly monocrystalline rutile, fibrous segments was produced at 1000 °C from the precursor with 2.5:1 TiO2/polymer mass ratio.
:1 TiO2/polymer mass ratio. TiO2 nanofibers produced by calcination at 500 °C for 3 h had 100–500 nm diameters and were composed of anatase (20–25 nm crystallite size) with rutile content 0.1–6.0 mol%, depending on the precursor composition. A considerable amount of anatase phase (up to 80 mol%) can be retained after thermal processing of TiO2 nanofibers at 750 °C for 3 h. A nanofibrous material composed of smooth and long, predominantly monocrystalline rutile, fibrous segments was produced at 1000 °C from the precursor with 2.5:1 TiO2/polymer mass ratio.
Introduction
Titanium dioxide (TiO2) nanomaterials have tunable semiconducting properties, interesting defect chemistry, large surface area and small particle size. TiO2 nanomaterials are increasingly used in photocatalysis, environmental remediation, sensor technology, photovoltaic structures, fuel cells, batteries, paints, plastic additives, self-cleaning surfaces, UV blockers in cosmetics, and many other applications.1–6 The anatase polymorph of TiO2 is the most commonly used crystalline form in such catalytic applications,7–10 but the more thermodynamically stable rutile polymorph11–13 and mixed-phase14,15 nano-titania also have their niche applications.There has been growing research interest in TiO2 nanofibers as one of the types of nano-titania materials.16–19 Several studies have indicated that TiO2 nanofibers can be increasingly more efficient in catalysis than TiO2 nanoparticles.20–23 For example, when compared to TiO2 nanoparticles, a 7-fold increase in hydrogen production has been achieved with mesoporous TiO2 nanofibers.20 Similarly, TiO2 nanofibers showed 10 times higher efficiency than nanoparticles in H2S oxidation.21 TiO2 nanofibers have also demonstrated a better photocatalytic activity in ethanol oxidation than that of commercial P25 TiO2 nanopowder.23 The catalytic performance of TiO2 nanofibers was attributed to the faster adsorption–desorption, efficient charge separation and recombination processes.20,22,23 Besides, scaffold-like porous structure of nanofibrous assemblies prevents aggregation, which is one of the serious limitations of nanoparticles. Further development and evaluation of TiO2-based nanofibrous catalysts needs to assess the capabilities for sizeable production of such materials with tunable structures and properties.
TiO2 nanofibers are mainly prepared by thermal processing of precursor fibers fabricated by direct current (DC) electrospinning.24–30 This simple method utilizes a static electric field to generate the propagating liquid precursor jets that form fibers upon drying. Usually, the precursor solution containing titanium alkoxide, polymer, and solvent in different molar ratios, is delivered through a capillary needle, which serves as the jet-generating electrode, and then solidified precursor fibers are collected on an oppositely charged electrode. Calcination of precursor fibers at temperatures in the range usually from 400 to 1000 °C results in the crystallization of TiO2 nanofibers with varying textural properties, phase composition, and fiber diameters.31,32 For example, Li and Xia24 obtained predominantly anatase crystalline phase TiO2 nanofibers with mean diameters in the range from 33 to 192 nm from a titanium propoxide (TTIP)/polyvinylpyrrolidone (PVP) acidic solution in ethanol. The precursor solution in those experiments was fed through a capillary needle with a flow rate between 0.1 and 0.5 mL h−1. The TTIP/PVP mass ratio was kept ∼3:1 (or ∼1:1 in terms of TiO2 mass) and PVP concentration varied, leading to the calculated yield of calcined TiO2 nanofibers in the range from 18 to 56 mg mL−1 after calcination of the as-spun precursor fibers at 500 °C. Tekmen et al.25 and Caratão et al.33 used similar precursors and electrospinning parameters, and found mixed anatase/rutile phase composition in TiO2 nanofibers calcined at 600 °C. It was also observed that the 200–900 nm diameter rutile fibers prepared from, in terms of TiO2 mass, 0.5:1 and 1:1 TiO2/PVP precursors,32 maintained their shape and polycrystallinity up to 1200 °C. In several other studies,25–28 the titanium alkoxide/PVP precursors were prepared with 0.2–1.4 TiO2/PVP mass ratios, warranting an expected yield of TiO2 nanofibers in the range of 15–50 mg mL−1. This precursor formulation also revealed comparable fiber diameters and calcination behavior, and the textural properties of fibers varied significantly. In another study, Li et al.16 used an acid-free titanium alkoxide-based precursor with, in terms of TiO2 mass, 2.5:1 TiO2/PVP mass ratio that corresponded to an expected yield of TiO2 nanofibers of 100 mg mL−1. The fibers calcined at up to 700 °C were apparently porous and rough but contained only anatase phase.
Onozuka et al.34 used a TTIP/polyvinylacetate precursor with N,N-dimethylformamide (DMF) as a solvent. A partially fused fibrous network composed of anatase TiO2 nanofibers with the diameters in the range from 150 to 700 nm has been observed after calcination at 500 °C. Choi35 utilized a TTIP/PMMA precursor with ∼0.17:1 TiO2/PMMA mass ratio and ∼20 mg mL−1 expected yield of TiO2 nanofibers. The calcined nanofibers revealed very small, 10–17 nm mean crystallite size, but the fibers had mostly rutile structure after calcination at only 450 °C, a relatively low temperature treatment for the resulting rutile polymorph.
In most studies on TiO2 nanofibers fabrication, capillary needle DC electrospinning was used to spin precursor fibers at the precursor flow rates of less than 1 mL h−1, resulting in a productivity of TiO2 nanofibers most markedly below 100 mg h−1. The low process productivity has been a major limiting factor in practical applications of TiO2 nanofibers. A more productive, free-surface, “needleless” DC electrospinning has also been used for the fabrication of TiO2 nanofibers,36,37 but the precursor flow and fiber collection rates were not reported. Vahtrus et al.36 found that when the same precursor used in needle DC electrospinning was applied to free-surface electrospinning, the mean TiO2 fiber diameter was smaller after calcination. On the contrary, TiO2 nanofibers produced from capillary needle electrospun precursor fibers revealed more uniform microstructure and more predictable mechanical behavior. Shepa et al.37 used free-surface electrospinning of TTIP/PVP precursors with 0.05:1 to 0.63:1 TiO2/PVP mass ratios and expected yield of TiO2 nanofibers from 15 to 115 mg mL−1. The shape of calcined TiO2 nanofibers varied from ribbon-like, at low TTIP/PVP mass ratio and up to 90 mg mL−1 TiO2 yield, to cylindrical form at the highest used TTIP/PVP mass ratio and yield of TiO2. The fibers with either shape crystallized already at 350 °C with a mere 34% anatase phase, which was explained by low pH value of the precursor. All fibers transformed to rutile after calcination at 600 °C.
Titanium oxide nanofibers have also been prepared using solution blow spinning23 and centrifugal spinning38 techniques. Using solution blow spinning of a precursor with 1.9:1 TiO2/PVP mass ratio, Ghosh et al.23 achieved ∼0.95 g h−1 productivity in terms of the yield of TiO2 nanofibers, whereas approximately 7 g h−1 productivity was reached by Vasquez et al.38 using centrifugal spinning of a precursor with ∼0.83:1 TiO2/PVP mass ratio.
In this work, TiO2 nanofibers with different morphologies and phase compositions were fabricated from the titanium butoxide-based precursor fibers produced using alternating field electrospinning (AFES). This is a high-yield, free-surface electrospinning technique, which generates dense fibrous flows where the propagating fibers carry almost no electric charge.39 This unique attribute of AFES processing allows good flexibility in the precursor fibers collection and handling.40 Thus far, AFES has been successfully employed in the fabrication of several inorganic fibrous materials including amorphous silica,41 alumina,42 and zirconia43 nanofibers.
AFES requires apparently different chemical precursor compositions and process parameters than either capillary-needle or free-surface DC electrospinning to achieve electrospinnability and obtain fibers with similar characteristics.44 The goal of the present study was to explore the applicability of AFES for sizeable production of TiO2 nanofibers using alkoxide-based precursors, and establish the precursor–process–structure relationships for controlled synthesis of nanofibrous TiO2 with desired microstructures and crystalline phase compositions. The productivity of precursor fibers in terms of the yield of TiO2 nanofibers up to 5.2 g h−1 has been achieved with a fiber-generating dish-like electrode of 25 mm diameter. TiO2 nanofibers had 100–500 nm fiber diameters and different ratios of anatase/rutile crystalline phases depending on the precursor composition and calcination temperature. Unusual trends in titanium alkoxide-based precursor fibers generation during AFES and TiO2 nanofibers crystallization behavior have been observed and analyzed.
Experimental section
Preparation of TiO2 precursor solutions
Titanium(IV) n-butoxide (Ti(OBu)4, 99+%), polyvinylpyrrolidone (PVP, Mw = 1300000), hydroxypropyl cellulose (HPC, Mw = 100000) and glacial acetic acid (AA, Reagent grade) supplied by Alfa Aesar, and ethanol (200 Proof, Decon Labs, Inc) were used to prepare the TiO2 precursor solutions. In a typical procedure, the Ti(OBu)4 was combined with a mixture of ethanol and acetic acid in a dry box (25 ± 5% RH), and this precursor component was added to the 10 wt% solution of 1.0/1.0 mass ratio PVP/HPC polymer blend in ethanol. The molar ratio of Ti(OBu)4 to acetic acid was maintained as 1.0/1.0 in all experiments. The amount of ethanol in each precursor was adjusted to maintain ∼5 wt% polymer concentration. The Ti(OBu)4 to polymer mass ratio (Ti(OBu)4:PVP/HPC) varied to yield 0.5, 1.0, 1.5, 2.0, or 2.5 grams of TiO2 per one gram of polymer carrier. This corresponded to the projected yield of TiO2 nanofibers in the amount of 25, 50, 75, 100, and 125 mg per one mL of the precursor solution. The Ti(OBu)4/polymer precursors were labeled as ×0.5, ×1.0, ×1.5, ×2.0, and ×2.5, respectively. The precursors were stirred for up to 24 h using a magnetic stirrer and kept in sealed HDPE bottles at normal laboratory conditions (21 ± 1 °C and 42 ± 5% RH). All precursors were stable for at least six months.
Fabrication of TiO2 nanofibers
An AFES apparatus capable of operating at alternating current (AC)–voltages up to 40 kV rms at 60 Hz was used to spin the precursor fibers.41–43,45 The Ti(OBu)4/polymer precursor solution was delivered at up to 180 mL h−1 flow rate through the base of a shallow dish-like electrode with the diameter varying from 10 to 25 mm (Fig. 1). The generated flow of precursor fibers propagated upward due to the “ionic wind” phenomena.40,46 The generation of fibrous flows was recorded at up to 900 fps using a Sony DSC-RX10M2 camera. The fibers were collected on a PTFE plastic mesh placed ∼50 cm above the electrode at an ambient temperature of 21 °C and relative humidity of 42%. A 5–7 cm thick, fluffy layer of the precursor fibers was removed from the collector and dried in a Heratherm Advanced Protocol oven (Thermo Fisher) at 120 °C in air to remove the residual solvent. The dried fibrous fluffy material was placed in a programmable furnace (Isotemp, Fisher Scientific), heated up at a rate of 3 °C min−1 in air, and calcined at 500 and 750 °C for 3 h, or sintered at 1000 °C for 2 hours.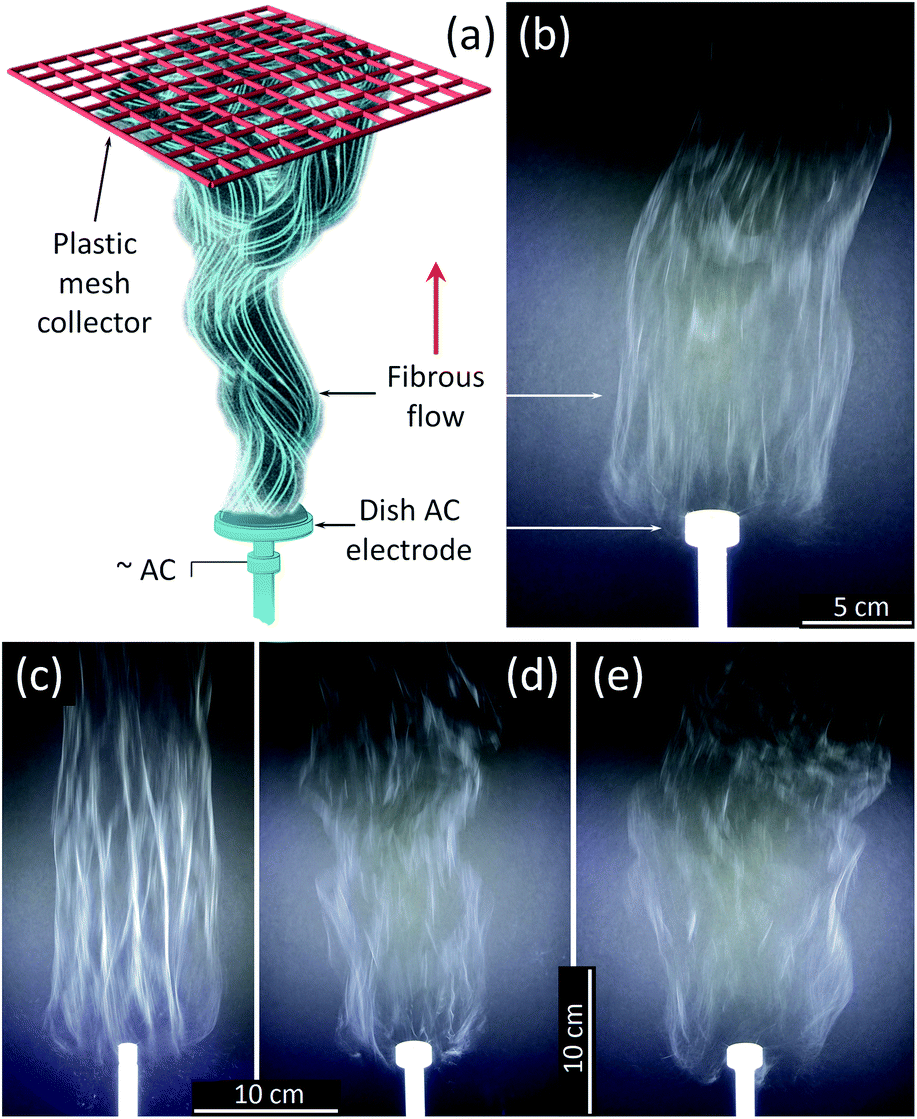 | ||
| Fig. 1 (a) AFES experiment schematics and photos of the flows of precursor fibers with ×1.5 TiO2/polymer yield mass ratio generated on (b) 25 mm and (c) 12.5 mm diameter electrode at 29.5 kV rms AC voltage; and with (d) ×0.5 and (e) ×2.5 TiO2/Polymer yield mass ratio generated using 25 mm diameter electrode at the same AC voltage. | ||
Characterization methods
A HAAKE RotoVisco 1 (Thermo Scientific) viscometer in a parallel plate configuration was used to determine the viscosity of precursor solutions. Apparent viscosity as a function of time was measured at steady shear rate of 1000 rpm over a period of 120 seconds. An FP30 conductivity meter (Mettler Toledo) was used to measure the electrical conductivity of the precursor solutions, and a PGX+ Goniometer (Testing Machines, Inc) was utilized to evaluate their surface tension from the shape of a pendant droplet. The tests were performed at 21 °C and repeated five times.Thermogravimetric Analysis (TGA) was performed on each as-spun Ti(OBu)4/polymer precursor material by using TG/DTA-SETSYS-16/18 (SETARAM, France) instrument. The samples of precursor fibers with 10 ± 3 mg mass were packed into the alumina crucibles, and heated at 10 °C min−1 in synthetic air at 50 mL min−1 gas flow rate. Thermogravimetric (TG), differential thermal analysis (DTA), and differential thermogravimetric (DTG) curves were recorded simultaneously at temperatures ranging from 20 to 900 °C.
The X-ray Diffraction (XRD) patterns were obtained using a PANalytical Empyrean X-ray diffractometer (copper Kα1 radiation, λ = 1.54059 Å, generator voltage 45 kV, and 40 mA current). The patterns were acquired using a 2θ scan in the range of 10–80° in a continuous scan mode with time per step 79.56 ms and scan step size 0.01313°. The diffraction optics included a hybrid monochromator with a 1/8° divergence and a 1/16° anti-scattering slits, and a parallel plate collimator on the diffracted beam path with a PIXcel1D detector. Crushed samples were pressed into zero-background silicon holders and rotated during the measurements to minimize the possibility of preferred orientation effects. Phase composition and crystallite size analyses were performed using the PANalytical High Score Plus software package and the International Centre for Diffraction Data (ICDD) powder diffraction file (PDF-2 ver. 2013) database.
The fiber diameter and surface morphology of TiO2 nanofibers were analyzed by scanning electron microscopy (SEM, FEI Quanta 650 FE-SEM with Bruker Flat Quad EDS detector). A 2–3 nm thick, AuPd layer was sputter coated onto the samples to reduce electric charging during the imaging. SEM imaging was done in secondary electron mode at an accelerating voltage of 15 kV and electron probe current of 2.5 μA. ImageJ image processing program was used to determine the fiber diameters and their size distribution.
Results and discussion
AFES of Ti(OBu)4/polymer precursors was first tested with pure PVP and HPC as the carrier polymers. There was only electrospraying observed with HPC alone, whereas the precursors with PVP spun well. However, this precursor produced fibers that collapsed and fused easily, unlike in the case of either capillary needle or free-surface DC-electrospinning.24–33,36,37 The fusion of AFES Ti(OBu)4/PVP fibers can be attributed to the high density of fibrous flow and increased amount of residual solvent in collected fibers. It has been determined that the PVP/HPC polymer blend with 1:1 mass ratio resulted in both good precursor spinnability and improved quality of collected fibrous material. It can be attributed to the hydrogen bonding between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of PVP and –OH groups of HPC, which led to good miscibility and optimized rheological properties of PVP/HPC blend for AFES. The parameters of tested precursors are given in Table 1.
O groups of PVP and –OH groups of HPC, which led to good miscibility and optimized rheological properties of PVP/HPC blend for AFES. The parameters of tested precursors are given in Table 1.
| Yield TiO2/polymer ratio, mg mg−1 | Viscosity, mPa s | Electrical conductivity, μS cm−1 | Surface tension, mN m−1 |
|---|---|---|---|
| ×0.5 | 112 ± 10 | 23.4 ± 0.3 | 17.82 ± 0.08 |
| ×1.0 | 118 ± 10 | 45.9 ± 0.3 | 18.71 ± 0.07 |
| ×1.5 | 123 ± 8 | 176.5 ± 0.5 | 19.48 ± 0.06 |
| ×2.0 | 121 ± 7 | 95.5 ± 0.3 | 20.36 ± 0.07 |
| ×2.5 | 122 ± 9 | 65 ± 0.5 | 21.15 ± 0.05 |
All tested Ti(OBu)4/polymer precursor compositions with 1:1 PVP/HPC polymer carrier produced precursor fibers in the range of AC voltages from 12 to 40 kV rms. The lower voltage limit was determined mostly by the corona inception voltage threshold,46 and it fluctuated between 12 and 15 kV depending on the electrode diameter and precursor composition. The best process performance was achieved at ∼29.5 ± 2.5 kV rms AC-voltage when using a dish-like electrode with a 25 mm diameter (Fig. 1a and b). Smaller electrode size produced strong and uniform flows but usually led to an increase of fiber bundles (Fig. 1c). The larger electrodes demonstrated noticeable differences in fiber generation and fibrous flow propagation depending on the precursor composition. Such differences, though at a lesser scale, were observed with the 25 mm diameter electrode and appeared as the variations in the flow width and some intermittent turbulence in the flow behavior (Fig. 1d and e). These differences can be reduced by the adjustment of AC–voltage and the precursor delivery rate to account for small variations in the viscosity and electrical conductivity of the precursors.
The amount of Ti(OBu)4 in the precursor solution also had a strong effect on the flow and fiber collection rates (Fig. 2). The rate at which the liquid precursor has to be supplied to the electrode, to support the stable generation of fibrous flow, decreased from ∼2.0 mL min−1 to ∼0.45 mL min−1 when the mass ratio of Ti(OBu)4 to polymer (and the corresponding amount of TiO2) in the precursor increased five times. This resulted in significant variation in the process productivity with a maximum yield of fibrous material reached at 1.5:1.0 TiO2/polymer mass ratio in precursor fibers. If the collected fibrous material would be simultaneously converted to titanium oxide, this would correspond to the productivity of TiO2 nanofibers of ∼5.2 g h−1. The observed reduction in the flow rate does not follow the changes in viscosity and electrical conductivity of precursor solutions. All precursors had similar viscosity and surface changes, whereas electrical conductivity varied in the range from 20 to 180 μS cm−1, with a maximum conductivity of 180 μS cm−1 observed in the precursor with 1.5:1.0 TiO2/polymer mass ratio (Table 1). It has been suggested that the observed reduction in the fiber generation rate is primarily related to the increasing density of precursor solution and its effect on the mass/momentum transfer in generated fibrous flow, rather than to the changes of viscosity. The apparent similarity in the dependences of electrical conductivity of precursor solutions and TiO2 yield is coincidental.
 | ||
| Fig. 2 Precursor flow rate and the projected productivity of TiO2 nanofibers in continuous process vs. the TiO2/polymer yield mass ratio in the precursor solution. | ||
Thermogravimetric analysis of as spun precursor fibers indicated that the residual solvent is removed upon heating to 130 °C, and the main decomposition of intermediates and polymer occurs between 250 and 500 °C (Fig. 3a). The decrease in the mass loss corresponded to increasing amount of Ti(OBu)4 in the precursor and expected yield of TiO2. However, the mass loss was higher than that predicted from as-spun precursor fiber composition based on the mass of TiO2 and polymer (Fig. 3b). It occurred due the formation of Ti(OH)4 as a main product of a hydrolysis reaction resulting, to some extent, from absorption of atmospheric moisture by as-spun fibers. The anticipated reaction scheme during the formation of precursor fibers is shown in Fig. 4.47,48 The reaction of Ti(OBu)4 with acetic acid results in a TiAOB(OBu)C(OAc)D complexes, which are further hydrolyzed to titanium hydroxide, Ti(OH)4. The hydrolysis of Ti(OBu)4 occurred as water formed in a partial reaction of acetic acid and ethanol, and water absorbed from the atmosphere during the spinning process. The decomposition of Ti(OH)4 and polymer matrix occur mainly between 150–500 °C. Additional mass loss (∼1–2%) between 500 and 1000 °C was associated with removal of residual carbon.
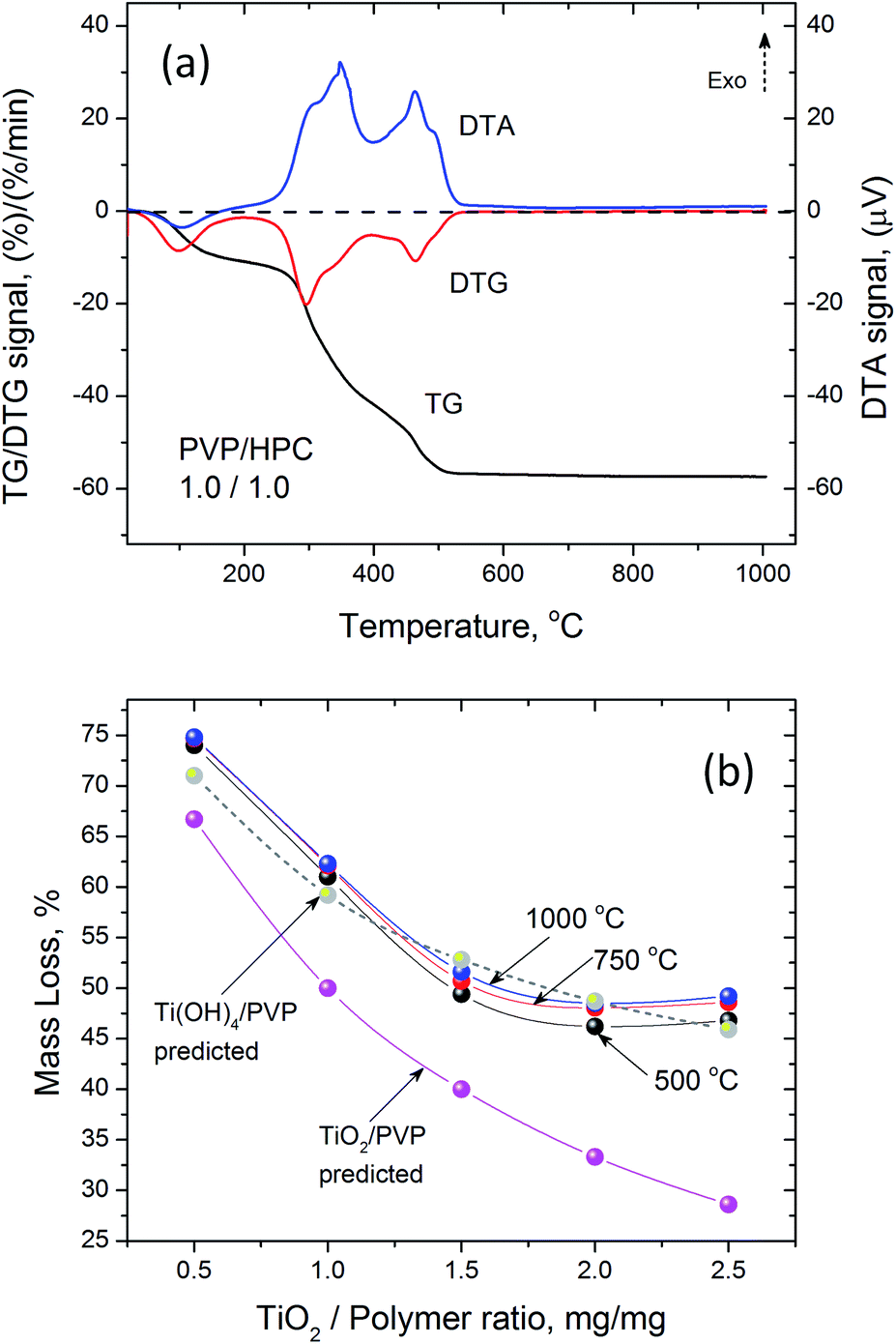 | ||
| Fig. 3 (a) Typical DTA/TG/DTG curves of the decomposition of Ti(OBu)4/polymer precursor nanofibers with ×1.0 TiO2/polymer yield mass ratio and 1:1 PVP/HPC mass ratio; (b) mass loss of precursor fibers with different TiO2/polymer yield mass ratios during calcination after initial stabilization at 120 °C. | ||
 | ||
| Fig. 4 A simplified Ti(OBu)4 precursor reaction scheme during the AFES of Ti(OH)4/(PVP:HPC) precursor fibers. | ||
The thermal processing at 500 °C has been frequently used to produce anatase TiO2 nanofibers from electrospun precursors.24,28,34,36 SEM analysis of AFES fibers after calcination at 500 °C for 3 hours revealed a random fibrous network (Fig. 5a) with the TiO2 fiber surface morphologies varying from somewhat rough (Fig. 5b) to relatively smooth (Fig. 5d) with the increasing content of TiO2/polymer mass ratio in as spun precursor fibers. The fiber diameters were in the (200–400) ± 100 nm range but the changes in the diameter did not follow exactly the increasing content of TiO2 (Fig. 5e). Furthermore, the average diameter of fibers decreased somewhat at low and increased at high TiO2 content when precursor fibers were calcined at 750 and 1000 °C without changes in general trend. Such behavior can be associated with different fiber porosity and carbon residue content (2.8–5.1) ± 0.4 wt% according to EDS after calcination at 500 °C. These two parameters can affect the rate of further changes in the fibers morphology and microstructure, and the fibers final form, depending on the initial fiber diameter, temperature and processing time. The changes in residual carbon content correlated with the mass loss during thermal processing at 750 and 1000 °C (Fig. 3b). There was no carbon detected in TiO2 nanofibers prepared from the precursor with 0.5:1.0 TiO2/polymer mass ratio whereas ∼0.2 ± 0.06 wt% of carbon was present in nanofibers prepared from precursor with 2.5:1.0 TiO2/polymer mass ratio after calcination at 750 °C for 3 h.
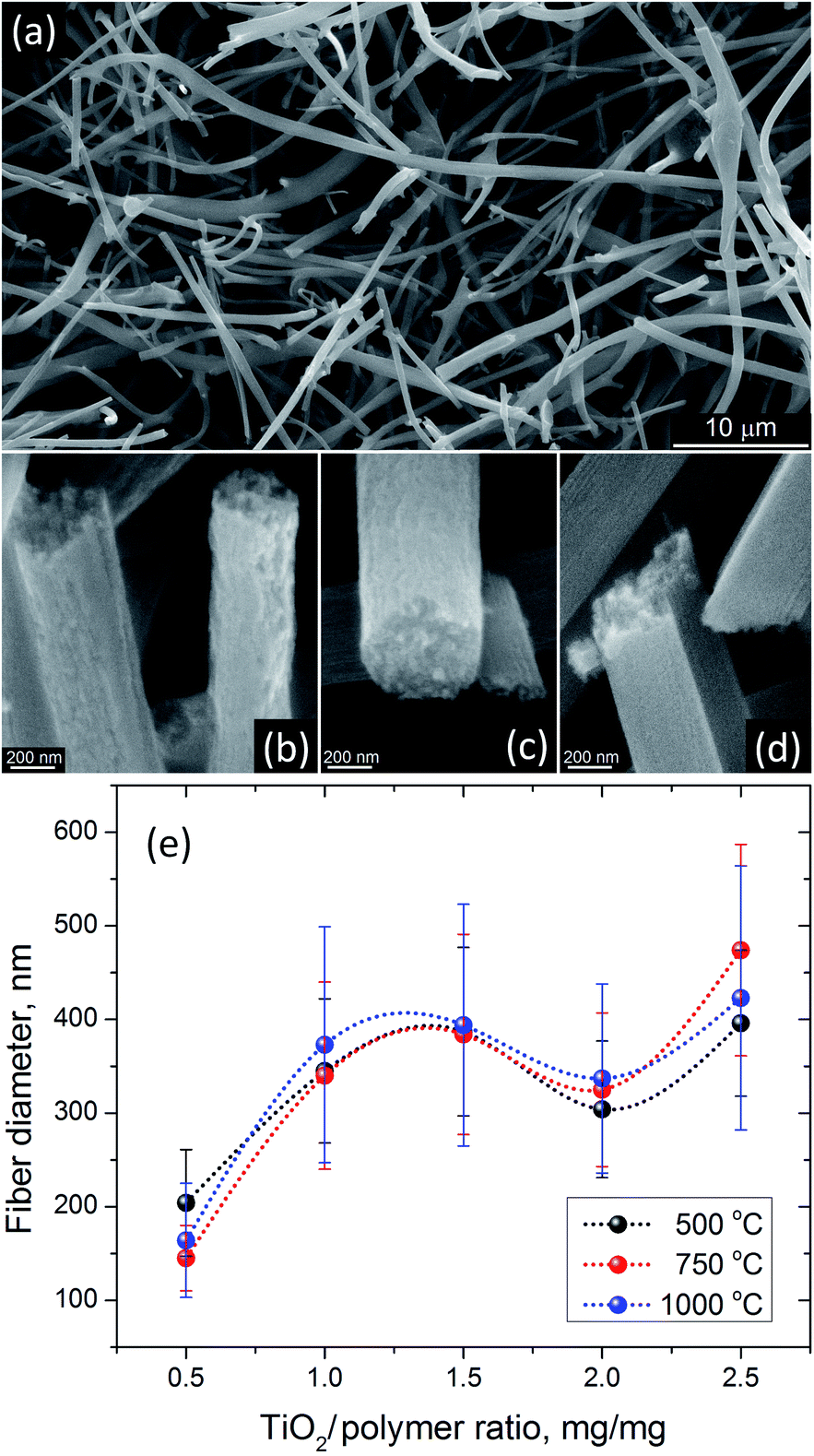 | ||
| Fig. 5 SEM images of (a) typical appearance of TiO2 nanofibers after calcination at 500 °C for 3 h, and their surface morphology when prepared from the precursors with (b) ×0.5, (c) ×1.5 and (d) ×2.5 TiO2/polymer yield mass ratios. (e) TiO2 nanofiber diameter after calcination at 500, 750 and 1000 °C as a function of TiO2/polymer yield mass ratio in the precursor solution. Dotted lines show general trend. | ||
The XRD and SEM analyses helped to achieve better understanding of the observed macroscopic phenomena during the crystallization of nanofibrous TiO2 from AFES precursor fibers. The XRD patterns, changes in crystallite sizes and rutile content are shown in Fig. 6. The position and intensity of XRD peaks assigned to anatase and rutile phases corresponded to those in ICDD files #00-064-0863 and #01-070-7347, respectively. The results of XRD patterns analysis are summarized in Table 2.
 | ||
| Fig. 6 XRD patterns of TiO2 nanofibers prepared from the precursors with different TiO2/polymer yield mass ratios after calcination at (a) 500 °C and (b) 750 °C. Symbols ● – indicate the peaks from anatase and ♦ – from rutile; (c) rutile content and (d) anatase and rutile crystallite size in TiO2 nanofibers after calcination at 500 °C and 750 °C vs. the precursor composition. Dotted lines show general trend. | ||
| Yield TiO2/polymer ratio, mg mg−1 | 500 °C | 750 °C | ||||||
|---|---|---|---|---|---|---|---|---|
| Anatase | Rutile | Anatase | Rutile | |||||
| Size, nm | mol% | Size, nm | mol% | Size, nm | mol% | Size, nm | mol% | |
| ×0.5 | 22.9 | 97.5 | 18.6 | 2.5 | 34.5 | 5.1 | 41.1 | 94.9 |
| ×1.0 | 22.4 | 98.5 | — | 1.5 | 47.1 | 36.0 | 58.8 | 64.0 |
| ×1.5 | 25.9 | 99.5 | — | 0.5 | 42.6 | 64.2 | 53.4 | 35.8 |
| ×2.0 | 22.2 | 94.0 | 19.8 | 6.0 | 40.9 | 43.6 | 48.4 | 56.4 |
| ×2.5 | 29.7 | 99.9 | — | 0.1 | 44.5 | 84.1 | 65.4 | 15.9 |
As expected, calcination at 500 °C resulted in the formation of mainly nanocrystalline anatase phase of TiO2 with 0.1–6.0 mol% of rutile (Fig. 6a and c) according to Rietveld analysis.49 The average size of anatase crystallites was in the range of 22–30 nm (Fig. 6d and Table 2). The content of rutile phase (Fig. 6c) was less than 0.1 mol% in the sample with the largest size of anatase crystallites (at 2.5:1 TiO2/polymer mass ratio), whereas the sample with the smallest size of anatase crystallites contained about 6 mol% of rutile (at 2.0:1 TiO2/polymer mass ratio). The size of rutile crystallites was close to that of anatase, although it was not possible to retrieve the data for some compositions due to the low content of rutile phase. This result coincides with experimental and theoretical data on the effect of small particle size on anatase to rutile transformation.50–52 The changes in crystallinity and phase composition during calcination at 750 °C did not precisely follow the parameters of initial structure (Fig. 6c, d and Table 2). For example, although the amount of rutile phase was still less than 20 mol% in the sample with 2.5:1 TiO2/polymer mass ratio, the average size of rutile crystallites was the largest with respect to other TiO2/polymer mass ratio compositions (Fig. 6c and d). The sample with 0.5:1 TiO2/polymer mass ratio exhibited almost 95 mol% of rutile phase and the smallest size (35–40 nm) of both anatase and rutile crystallites. The size of the rutile crystallites was larger than that of anatase for all TiO2/polymer ratios. It was suggested that the growth of crystallites and the rate of anatase to rutile conversion depended, under other equal conditions, on the textural properties of TiO2 nanofibers (e.g., volume, size and shape of micro/meso pores) and the amount of residual carbon after calcination at 500 °C. These factors affect the diffusion pathway and the changes in free energy of the system,53 and their contribution depends on the initial composition of precursor fibers and annealing strategy. All samples revealed only rutile TiO2 phase (ICDD file #01-070-7347) after calcination and sintering at 1000 °C. It was not possible to reliably determine the crystallite size in these samples from XRD data.
SEM images of nanofibrous TiO2 calcined at 750 °C showed increasing differences in the fiber morphology (Fig. 7a, c and e), depending on the precursor composition and average fiber diameter. The apparent grain size in TiO2 nanofibers prepared from 0.5:1 TiO2/polymer mass ratio precursor (Fig. 7a) was close to the fiber diameter despite the smallest size of TiO2 crystallites determined from XRD data. The resulting grain size in TiO2 nanofibers prepared from 2:1 TiO2/polymer mass ratio precursor was similar (not shown in this figure) to that of nanofibers from 0.5:1 precursor although those nanofibers had as much as twice of larger diameters. The samples calcined from 1:1 (not shown) and 1.5:1 precursor fibers were rather similar, noticeably denser and had the visibly smallest grain size (Fig. 7c). Nanofibrous TiO2 prepared from 2.5:1 precursor had a slightly larger grain size and rougher morphology than the materials from 1:1 and 1.5:1 precursor fibers, which was especially visible in the fiber cross-sections (Fig. 7e). These results seem to continue the trend observed in the fiber shape development and formation of cylindrical, denser TiO2 nanofibers in free-surface DC-electrospun precursor fibers with higher than 0.5:1 TiO2/polymer ratio.37
 | ||
| Fig. 7 SEM images of TiO2 nanofibers prepared from the precursors with (a and b) ×0.5, (c and d) ×1.5, and (e and f) ×2.5 TiO2/polymer yield mass ratios after (a, c and e) calcination at 750 °C and (b, d and f) sintering at 1000 °C. Corresponding higher magnification images are shown in insets. | ||
Further calcination and sintering at 1000 °C led to more significant differences in the final TiO2 nanofiber morphology and rutile phase grain size, depending on the initial composition of the precursor fibers (Fig. 7b, d and f). TiO2 nanofibers with the greatest amount of rutile, formed at 750 °C (96 mol% for 0.5:1 TiO2/polymer ratio), demonstrated a slight increase in fiber diameters caused by the continuing axial shrinkage of fibers resulting from grain growth. This fibrous material was brittle and SEM imaging revealed that many nanofibers broke into small pieces (Fig. 7b). All this can be attributed to the initial microarchitecture of precursor fibers with 0.5:1 composition, which had noticeable interfiber fusion and bundling due to the larger amount of polymer and residual solvent in as spun fibers. On the opposite end, TiO2 nanofibers prepared from 2.5:1 precursor showed some decrease in fiber diameter, with the formation of a significant fraction of long, smooth nanofibers without distinct grain morphology (Fig. 7f). It was suggested that such smooth nanofibers were composed of elongated monocrystalline rutile segments. The fiber morphology is very similar to that observed in monocrystalline Al2O3 nanofibers sintered at 1300 °C.54,55 The variations in SEM contrast (dark spots) on nanofiber surface (inset in Fig. 7f) appear, most probably, due to the presence of large voids formed during the grain coalescence. The formation of long monocrystalline rutile segments in TiO2 nanofibers can be caused by the transformation of rather “spherical” anatase grains to larger, elongated and preferentially oriented rutile crystallites, followed by spontaneous coalescence of those monocrystalline rutile segments.54 The change from “spherical” to elongated crystallite shape explains the decrease in the fiber diameter as well as the preserved continuity of nanofibers due to lower residual stress. TiO2 nanofibers prepared from 1.5:1 precursor showed transitional behavior (Fig. 7d). In this sample, clear difference in the crystallization of thicker and thinner nanofibers was observed. Again, like after the calcination at 750 °C, TiO2 nanofibers from 2:1 precursor were more similar to those prepared from 0.5:1 precursor after calcination and sintering at 1000 °C. Nanofibrous TiO2 made of 1:1 precursor had microarchitecture exactly between those prepared from the precursors with 0.5:1 and 1.5:1 TiO2/polymer mass ratios.
SEM images in Fig. 8 summarize the main features of TiO2 nanofibers crystallization during the thermal processing of AFES precursor fibers at 500, 750, and 1000 °C. Initially, nanocrystalline TiO2 nanofibers with anatase structure form at 500 °C from initial Ti(OH)4-based solidified gel network (Fig. 8a). The anatase crystallite size, amount of carbon residue, and fiber porosity can affect the diffusion processes, anatase to rutile conversion rate, and grain growth as temperature increases (Fig. 8b). These factors strongly influence the grain size and morphology of the resulting rutile TiO2 nanofibers after calcination and sintering at 1000 °C. In particular, the formation of elongated rutile grains (Fig. 8c) and their coalescence into even longer, apparently monocrystalline rutile segments in TiO2 nanofibers can be related to preservation of the anatase phase at higher temperatures.
 | ||
| Fig. 8 Representative SEM images showing the surface morphology and crystallinity changes in TiO2 nanofibers prepared from ×1.5 TiO2/polymer yield mass ratio precursor after calcination at (a) 500 °C and (b) 750 °C, and (c) sintering at 1000 °C. | ||
Conclusions
The results of this study demonstrated that a high-yield, free-surface alternating field electrospinning (AFES) process can be used to efficiently generate the fibrous TiO2 precursor materials from the solutions based on titanium butoxide and a blend of PVP and HPC polymers. Thermal processing of such precursor fibers in the range of temperatures between 500 and 1000 °C produces nanocrystalline TiO2 nanofibers with different morphology, diameters and composition of crystalline phases, depending on the mass ratio of alkoxide and polymer in the precursor. The precursor fibers generation rate drops directly proportionally to the increase of alkoxide/polymer mass ratio, regardless of voltage, electrode diameter, and rheological properties of precursors. Within the tested alkoxide/polymer mass ratios, the best fiber production rate was achieved with the precursor that corresponded to 1.5:1 TiO2/polymer mass ratio. In terms of crystallized nanofibrous TiO2, the process productivity was ∼5.2 g h−1 of titania nanofibers, when the single 25 mm diameter electrode and precursor flow rate of 1.1 mL min−1 were used. TiO2 nanofibers produced from this precursor had rutile phase content less than 0.5 mol% after calcination at 500 °C for 3 h. However, the lowest content of rutile was found in TiO2 nanofibers calcined from precursor fibers with 2.5:1 TiO2/polymer mass ratio. That material also retained the largest amount of anatase phase after calcination at 750 °C. It was demonstrated that the mass ratio of alkoxide and polymer in the precursor plays a critical role in the crystallization of anatase and rutile structures in AFES derived titania nanofibers. It has been suggested that the preservation of significant amount of anatase phase in TiO2 nanofibers at relatively high temperature (at least 750 °C) may be a prerequisite for the formation of nanofibrous material composed predominantly of long monocrystalline rutile segments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the National Science Foundation (NSF) [grant number DMR-1708600]. The authors thank Dr Karolina Chalupka, Lodz University of Technology, for the help with TGA measurements. SN and CS thank the support from the NSF International Research Experience for Students (IRES) Program [grant number OISE-1558268].References
- T. R. Li, Y. W. Li, Z. Y. Bai, X. C. Peng, Z. Q. Zhong and T. He, Appl. Mech. Mater., 2013, 295–298, 2227 Search PubMed.
- Y. Wang, Y. He, Q. Lai and M. Fan, J. Environ. Sci., 2014, 26, 2139 CrossRef PubMed.
- D. P. MacWan, P. N. Dave and S. Chaturvedi, J. Mater. Sci., 2011, 46, 3669 CrossRef.
- T. D. Malevu, B. S. Mwankemwa, S. V. Motloung, K. G. Tshabalala and R. O. Ocaya, Phys. E, 2019, 106, 127 CrossRef.
- J. H. Lee, J. W. Kim, J. M. Kim and S. J. Choung, Appl. Chem. Eng., 2013, 24, 231 Search PubMed.
- X. P. Wu, Key Eng. Mater., 2017, 727, 314 Search PubMed.
- A. J. Gardecka, C. Bishop, D. Lee, S. Corby, I. P. Parkin, A. Kafizas and S. Krumdieck, Appl. Catal., B, 2018, 224, 904 CrossRef.
- J. Zhang, B. Wu, L. Huang, P. Liu, X. Wang, Z. Lu, G. Xu, E. Zhang, H. Wang, Z. Kong, J. Xi and Z. Ji, J. Alloys Compd., 2016, 661, 441 CrossRef.
- M. Ghadiry, M. Gholami, L. C. Kong, C. W. Yi, H. Ahmad and Y. Alias, Sensors, 2015, 16, 39 CrossRef PubMed.
- D. Wu, S. Zhang, S. Jiang, J. He and K. Jiang, J. Alloys Compd., 2015, 624, 94 CrossRef.
- S. Gatla, N. Madaan, J. Radnik, V. N. Kalevaru, M. M. Pohl, B. Lücke, A. Martin, U. Bentrup and A. Brückner, J. Catal., 2013, 297, 256 CrossRef.
- C. A. Walenta, S. L. Kollmannsberger, C. Courtois, R. N. Pereira, M. Stutzmann, M. Tschurl and U. Heiz, Phys. Chem. Chem. Phys., 2019, 21, 1491 RSC.
- Z. Liu, V. Sriram and J. Y. Lee, Appl. Catal., B, 2017, 207, 143 CrossRef.
- J. C. Bear, V. Gomez, N. S. Kefallinos, J. D. McGettrick, A. R. Barron and C. W. Dunnill, J. Colloid Interface Sci., 2015, 460, 29 CrossRef PubMed.
- A. Kim, D. P. Debecker, F. Devred, V. Dubois, C. Sanchez and C. Sassoye, Appl. Catal., B, 2018, 220, 615 CrossRef.
- J. Li, H. Qiao, Y. Du, C. Chen, X. Li, J. Cui, D. Kumar and Q. Wei, Sci. World J., 2012, 2012, 154939 Search PubMed.
- C. Ligon, K. Latimer, Z. D. Hood, K. D. Gilroy and K. Senevirathne, RSC Adv., 2018, 8, 32865 RSC.
- S. Madhugiri, B. Sun, P. G. Smirniotis, J. P. Ferraris and K. J. Balkus, Microporous Mesoporous Mater., 2004, 69, 77 CrossRef.
- J. Zhang, Y. Cai, X. Hou, X. Song, P. Lv, H. Zhou and Q. Wei, Beilstein J. Nanotechnol., 2017, 8, 1297 CrossRef PubMed.
- S. K. Choi, S. Kim, S. K. Lim and H. Park, J. Phys. Chem., 2010, 114, 16475 Search PubMed.
- N. Shahzad and R. W. Azfar, Environ. Sci. Pollut. Res., 2017, 24, 1133 CrossRef.
- B. Su, J. Xin, J. Li, N. Dong, K. Shao, Q. Wang and Z. Lei, Micro Nano Lett., 2016, 11, 323 CrossRef.
- M. Ghosh, M. Lohrasbi, S. S. C. Chuang and S. C. Jana, ChemCatChem, 2016, 8, 2525 CrossRef.
- D. Li and Y. Xia, Nano Lett., 2003, 3, 555 CrossRef.
- C. Tekmen, A. Suslu and U. Cocen, Mater. Lett., 2008, 62, 4470 CrossRef.
- J. Y. Park and S. S. Kim, Met. Mater. Int., 2009, 15, 95 CrossRef.
- M. L. Hu, M. H. Fang, C. Tang, T. Yang, Z. H. Huang, Y. G. Liu, X. W. Wu and X. Min, Nanoscale Res. Lett., 2013, 8, 1 CrossRef PubMed.
- J. Zhang, X. Hou, Z. Pang, Y. Cai, H. Zhou, P. Lv and Q. Wei, Ceram. Int., 2017, 43, 15911 CrossRef.
- O. Secundino-Sánchez, J. Diaz-Reyes, J. Aguila-López and J. F. Sánchez-Ramírez, J. Mol. Struct., 2019, 1194, 163 CrossRef.
- O. Solcova, T. Balkan, Z. Guler, M. Morozova, P. Dytrych and A. Sezai Sarac, Sci. Adv. Mater., 2014, 6, 2618 CrossRef.
- P. Aghasiloo, M. Yousefzadeh, M. Latifi and R. Jose, J. Alloys Compd., 2019, 790, 257 CrossRef.
- O. Landau and A. Rothschild, J. Electroceram., 2015, 35, 148 CrossRef.
- B. Caratão, E. Carneiro, P. Sá, B. Almeida and S. Carvalho, J. Nanotechnol., 2014, 2014, 472132 Search PubMed.
- K. Onozuka, B. Ding, Y. Tsuge, T. Naka, M. Yamazaki, S. Sugi, S. Ohno, M. Yoshikawa and S. Shiratori, Nanotechnol, 2006, 17, 1026 CrossRef PubMed.
- D. Y. Choi, C. H. Hwang, J. W. Lee, I. H. Lee, I. H. Oh and J. Y. Park, Mater. Lett., 2013, 106, 41 CrossRef.
- M. Vahtrus, A. Šutka, S. Vlassov, A. Šutka, B. Polyakov, R. Saar, L. Dorogin and R. Lõhmus, Mater. Charact., 2015, 100, 98 CrossRef.
- I. Shepa, E. Mudra, M. Vojtko, O. Milkovic, Z. Dankova, V. Antal, A. Annušová, E. Majková and J. Dusza, Results Phys., 2019, 13, 102243 CrossRef.
- H. Vasquez, H. Gutierrez, K. Lozano and G. Leal, J. Eng. Fibers Fabr., 2015, 10, 129 Search PubMed.
- P. Pokorny, E. Koštáková, F. Sanetmik, P. Mikes, J. Chvojka, T. Kalous, M. Bilek, K. Pejchar, J. Valtera and D. Lukaš, Phys. Chem. Chem. Phys., 2014, 16, 26816 RSC.
- J. Valtera, T. Kalous, P. Pokorny, O. Batka, M. Bilek, J. Chvojka, P. Mikes, E. Kuzelova Kostakova, J. Spankova, J. Beran, A. Stanishevsky and D. Lukáš, Sci. Rep., 2019, 9, 1801 CrossRef.
- A. Stanishevsky and J. Tchernov, J. Non-Cryst. Solids, 2019, 525, 119653 CrossRef.
- A. Stanishevsky, W. A. Brayer, P. Pokorny, T. Kalous and D. Lukáš, Ceram. Int., 2016, 42, 17154 CrossRef.
- A. Stanishevsky, R. Yager, J. Tomaszewska, M. Binczarski, W. Maniukiewicz, I. Witońska and D. Lukáš, Ceram. Int., 2019, 45, 18672 CrossRef CAS.
- A. Stanishevsky, J. Wetuski, M. Walock, I. Stanishevskaya, H. Yockell-Lelièvre, E. Košťáková and D. Lukaš, RSC Adv., 2015, 5, 69534 RSC.
- K. Paulett, W. A. Brayer, K. Hatch, T. Kalous, J. Sewell, T. Liavitskaya, S. Vyazovkin, F. Liu, D. Lukáš and A. Stanishevsky, J. Appl. Polym. Sci., 2018, 135, 45772 CrossRef.
- A. M. Drews, L. Cademartiri, G. M. Whitesides and K. J. M. Bishop, J. Appl. Phys., 2012, 114, 143302 CrossRef.
- F. X. Perrin, V. Nguyen and J. L. Vernet, J. Sol-Gel Sci. Technol., 2003, 28, 205 CrossRef CAS.
- R. Parra, M. S. Góes, M. S. Castro, E. Longo, P. R. Bueno and J. A. Varela, Chem. Mater., 2008, 20, 143–150 CrossRef CAS.
- M. Salari, M. Rezaee, A. T. Chidembo, K. Konstantinov and H. K. Liu, J. Nanosci. Nanotechnol., 2012, 12, 4724 CrossRef CAS.
- A. A. Gribb and J. F. Banfield, Am. Mineral., 1997, 82, 717 CAS.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855 CrossRef CAS.
- W. Li, C. Ni, H. Lin, C. P. Huang and S. Ismat Shah, J. Appl. Phys., 2004, 96, 6663 CrossRef CAS.
- K. N. P. Kumar, K. Keizer and A. J. Burggraaf, J. Mater. Chem., 1993, 3, 1141 RSC.
- P. C. Yu, R. J. Yang, Y. Y. Tsai, W. Sigmund and F. S. Yen, J. Eur. Ceram. Soc., 2011, 31, 723 CrossRef CAS.
- M. Vahtrus, M. Umalas, B. Polyakov, L. Dorogin, R. Saar, M. Tamme, K. Saal, R. Lõhmus and S. Vlassov, Mater. Charact., 2015, 107, 119 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |