
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo3O4–Ag photocatalysts for the efficient degradation of methyl orange†
Hongmei Chen a,
Chenyang Xue*a,
Danfeng Cuia,
Maoxing Liub,
Yi Chena,
Yuankai Lia and
Wendong Zhang*a
a,
Chenyang Xue*a,
Danfeng Cuia,
Maoxing Liub,
Yi Chena,
Yuankai Lia and
Wendong Zhang*a
aScience and Technology on Electronic Test and Measurement Laboratory, North University of China, Taiyuan, Shanxi 030051, China. E-mail: xuechenyang@nuc.edu.cn; wdzhang@nuc.edu.cn
bDepartment of Mathematics, Qingdao University of Science and Technology, Qingdao, 266061, China
First published on 17th April 2020
Abstract
In this paper, a series of Co3O4–Ag photocatalysts with different Ag loadings were synthesized by facile hydrothermal and in situ photoreduction methods and fully characterized by XRD, SEM, TEM, FTIR spectroscopy, XPS, UV-vis and PL techniques. The catalysts were used for the degradation of methyl orange (MO). Compared with the pure Co3O4 catalyst, the Co3O4–Ag catalysts showed better activity; among these, the Co3O4–Ag-0.3 catalyst demonstrated the most efficient activity with 96.4% degradation efficiency after 30 h UV light irradiation and high degradation efficiency of 99.1% after 6 h visible light irradiation. According to the corresponding dynamics study under UV light irradiation, the photocatalytic efficiency of Co3O4–Ag-0.3 was 2.72 times higher than that of Co3O4 under identical reaction conditions. The excellent photocatalytic activity of Co3O4–Ag can be attributed to the synergistic effect of strong absorption under UV and visible light, reduced photoelectron and hole recombination rate, and decreased band gap due to Ag doping. Additionally, a possible reaction mechanism over the Co3O4–Ag photocatalysts was proposed and explained.
1. Introduction
Water pollution, which affects human health and the healthy development of the whole ecosystem, is becoming more and more serious with the rapid development of industries.1 To date, many types of treatments for water pollution have been developed by researchers,2,3 but the main drawbacks such as severe reaction conditions or complicated processes significantly limit their practical applications. In contrast, since previous research has reported that TiO2 can be used for water splitting and organic pollutant degradation,4,5 the use of semiconductors as photocatalysts has attracted increasing attention due to their perfect utilization of the clean and renewable solar energy.6–9 However, TiO2 can only absorb ultraviolet light due to its wide band gap (∼3.2 eV).10–14 Hence, various new types of semiconductors have been developed as photocatalysts to solve this problem.15–26 Among these, Co3O4 is a potential material for many applications because of its excellent electronic and magnetic properties.6,27,28 For example, it can be used as a material for supercapacitors due to its high theoretical capacitance (∼3560 F g−1).29–31 Additionally, it can be applied in various photocatalytic reactions because it is non-toxic, environmentally friendly and cost-efficient.32 Nevertheless, the Co3O4 nanoscale materials still suffer from some difficulties in real industrial applications. The photocatalytic efficiencies of simple Co3O4 nanomaterials are often low due to the fast recombination rate of photoelectrons and holes. Thus, it is really a challenge for researchers to design and explore new kinds of promising Co3O4 photocatalysts with higher efficiency. To date, various methods have been used to improve the catalytic activity of Co3O4 photocatalysts. For example, constructing p–n heterojunctions such as Co3O4/ZnO,33 Co3O4/Bi2WO6,34 Co3O4/TiO2,35 and Bi2O3/Co3O4 (ref. 36) is effective. Besides, doping noble metals into simple metal oxides can improve the photocatalytic activity.37 Among the noble metals, elemental Ag is widely used due to its lower cost. Its role in improving the catalytic activity mainly involves two aspects. First, Ag doping can separate the photogenerated carriers efficiently because of the formation of a Schottky barrier.38,39 Second, it can improve the response to visible light.40,41 The positive role of Ag as a cocatalyst has been reported in several catalytic systems, such as TiO2,41,42 SnO2,43 and Cu2O.44 However, to date, doping elemental Ag in the Co3O4 system for MO degradation has rarely been reported.Most of the previous studies have focused on Co3O4 materials and satisfactory progress has been made for pollutant degradation. However, there are still some problems for practical applications. First, the recovery and reuse of granular materials are difficult. Then, the product separation process after the reaction is complex and requires considerable energy. Finally, the mass loss of the materials is serious. On the contrary, materials coated on substrates have potential as they are easy to use and separate. However, the photocatalytic efficiencies of simple Co3O4 catalysts are still low. Hence, in this paper, a series of highly active Co3O4–Ag catalysts on Ni foam substrates were prepared for the degradation of MO. High activity with 96.4% degradation efficiency was achieved after 30 h UV light irradiation, and high degradation efficiency of 99.1% could be obtained after 6 h visible light irradiation over the Co3O4–Ag-0.3 photocatalysts. The impacts of the Ag additive on the structure, morphologies, and chemical properties of the photocatalysts were investigated thoroughly.
2. Experimental
2.1. Preparation of the photocatalysts
All chemicals were of analytical reagent (AR) grade and used as received without any further treatment.2.2. Characterizations
XRD patterns were obtained from 15° to 85° using a D2 Phaser desktop/max-RAX-ray diffractometer (Bruker, Germany) with Cu Kα radiation at 30 kV and 10 mA. FTIR spectra were measured over a NEXUS Thermo Nicolet IR-spectrometer. XPS was performed on an ESCALAB 250Xi spectrometer. SEM and HRTEM were performed on FEI Inspect F50 and FEI Inspect F30. Ultraviolet-visible (UV-vis) absorption spectra were collected on a DaoJin UV3600 spectrophotometer (Agilent Technologies, USA). The photoluminescence (PL) test was performed on a Raman microscope (Renishaw inVia).2.3. Photocatalytic activity evaluation
The catalytic activities of all the photocatalysts were evaluated for MO degradation under UV light or visible light (λ > 400 nm) and the concentration of the MO solutions for all the experiments was 6 mg L−1. The absorbance of the MO solutions was tested using a UNIC UV-2800A spectrophotometer at 465 nm. First, the Co3O4 and Co3O4–Ag photocatalysts were placed into the MO solutions (50 mL) for 30 min without irradiation and then, the degradation reaction was performed with ultraviolet light irradiation. During the course of the reaction, certain solutions were taken out and analyzed. For the trapping experiments of the active species for the photocatalytic degradation of MO under UV light, benzoquinone (BQ), isopropanol (IPA) and ethylenediaminetetraacetic acid (EDTA) were used as scavengers and the concentration was 0.001 mol L−1. The absorbances of the MO solutions were recorded immediately after irradiating for 18 h. According to the following formula, the degradation efficiency (D) can be estimated: D = [(C0 − Ct)/C0] × 100%.Here, C0 is the original absorbance and Ct is the absorbance after a certain sampling time of the MO solution.3. Results and discussion
3.1. Characterization of the photocatalysts
Fig. 1 displays the XRD patterns of the Co(OH)2, pure Co3O4 and Co3O4–Ag-0.3 photocatalysts. The obvious peaks at 2θ of 44.5°, 51.8°, and 76.4° corresponded to the (111), (200), and (220) crystal faces of Ni (JCPDS 04-0850) in all samples. The diffraction peaks at 2θ of 19.1°, 37.9°, 38.7° and 57.9° were assigned to the planes of the Co(OH)2 precursor (JCPDS 30-0443). In addition, the peaks positioned at 2θ = 19.0°, 36.9°, 38.5°, 55.7°, 59.4°, and 65.2° were assigned to the planes of Co3O4 (JCPDS 04-0850) for pure Co3O4 and Co3O4–Ag-0.3. However, no peaks of Ag species were observed because of its low mass in the Co3O4–Ag-0.3 sample.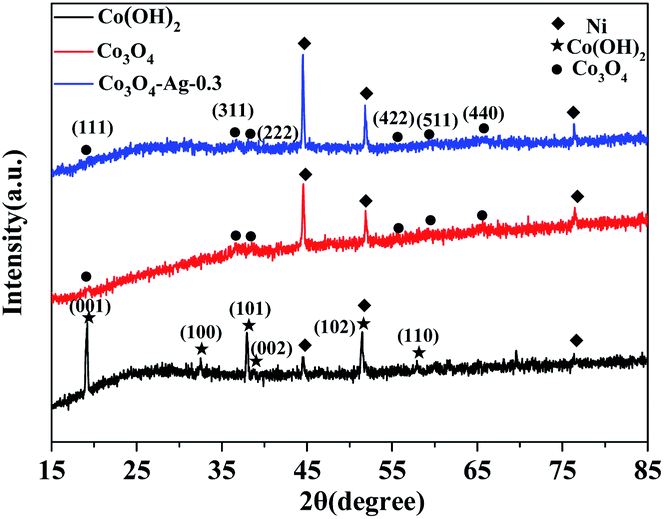 | ||
| Fig. 1 XRD patterns of Co(OH)2, pure Co3O4 and Co3O4–Ag-0.3. | ||
Fourier transform infrared spectroscopy (FTIR) was performed to analyse the surface properties of the catalysts. As depicted in Fig. 2, in the spectrum of Co(OH)2, the band at 1386 cm−1 belongs to the bending vibration of the surface adsorbed water, and the peak centered at 3642 cm−1 is relevant to the characteristic O–H stretching vibration.45 Besides, with regard to Co3O4, the absorption peaks at 667 and 570 cm−1 belong to the fingerprint stretching vibrations of the Co–O bonds, which can evidently signify the formation of Co3O4.46 These results verify the successful synthesis of the Co(OH)2 precursor and Co3O4, which is consistent with the XRD data.
 | ||
| Fig. 2 Fourier transform infrared spectra of Co(OH)2 and pure Co3O4. | ||
Fig. 3 shows the representative SEM and TEM images of Co3O4–Ag-0.3. As seen in Fig. 3(a–c), Co3O4–Ag-0.3 displays well-developed and defined three-dimensional (3D) “budded flower” morphology. The synthesized Co3O4–Ag-0.3 crystals were evenly distributed on the Ni foam substrate and the diameter of the “budded flowers” was about 5.7 μm. Moreover, Fig. 3(d and e) show the images of the nanoparticles of the Co3O4–Ag-0.3 catalyst obtained via TEM. As illustrated in Fig. 3(f), the lattice spacings of 0.214 and 0.205 nm correspond to the (222) and (311) planes of the Co3O4 crystals. Additionally, the lattice spacing of 0.24 nm was assigned to the (200) crystal facet of Ag crystals.47
 | ||
| Fig. 3 SEM (a–c) and TEM images (d–f) of Co3O4–Ag-0.3. | ||
XPS of Co3O4–Ag-0.3 was carried out to explore the surface composition and chemical state of the photocatalyst. Fig. 4(a) reveals the presence of Ni, Co, Ag and O in the photocatalyst. The Co 2p spectrum in Fig. 4(b) shows two strong peaks at around 794.86 eV and 779.86 eV. The former peak is attributed to Co 2p1/2, while the latter belongs to Co 2p3/2.48,49 The presence of Co3O4 can be further verified by the O 1s XPS peak at 529.66 eV in Fig. 4(d).50 Fig. 4(c) presents Ag 3d peaks at 373.26 eV and 367.81 eV and they are assigned to Ag 3d3/2 and Ag 3d5/2, respectively,47 indicating the successful deposition of Ag on the Co3O4 photocatalyst. Hence the successful formation of Co3O4–Ag-0.3 can be inferred, which is in line with the XRD and TEM results.
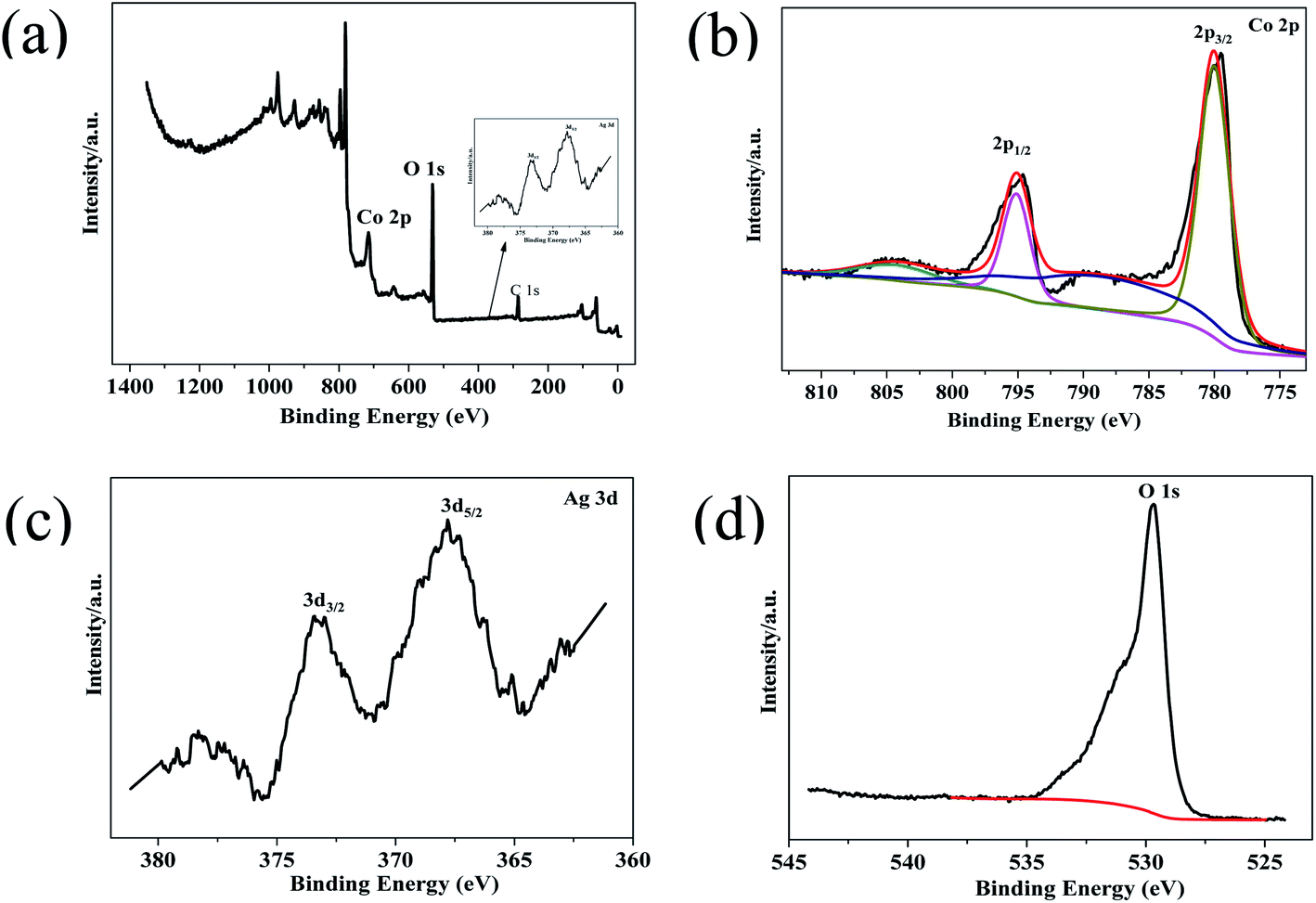 | ||
| Fig. 4 XPS survey spectrum of Co3O4–Ag-0.3 (a), and high-resolution spectra for (b) Co 2p, (c) Ag 3d, and (d) O 1s. | ||
Additionally, the optical properties of the Co3O4 and Co3O4–Ag-0.3 photocatalysts were evaluated by UV-vis spectroscopy (Fig. 5). The Co3O4 and Co3O4–Ag-0.3 photocatalysts exhibited strong absorption under UV and visible light in the ranges of 200–450 and 500–750 nm, respectively.51,52 The former band belonged to O2−–Co2+ and the latter was attributed to the O2–Co3+ charge transfer.53 As presented in Fig. 6, Egs are 1.50 eV and 2.04 eV for Co3O4, while they are 1.42 eV and 1.95 eV for the Co3O4–Ag-0.3 photocatalyst according to the absorption intensities, which are consistent with previous reports.54,55 The change in Egs for Co3O4 and Co3O4–Ag-0.3 may be attributed to the synergy of two aspects. One is the quantum confinement effect in nanomaterials56 and the other is the dielectric confinement effect, which is similar to that observed for Cu2O@Ag.44 From Fig. 5 and 6, it can be revealed that both the UV and visible light absorptions of the composite samples are strengthened, while Egs become narrower with the addition of Ag compared to that of the pure Co3O4 catalyst; this is because Ag doping can restrain the recombination of electrons and holes to endow Co3O4–Ag-0.3 with stronger light-harvesting ability to improve the catalytic activity.
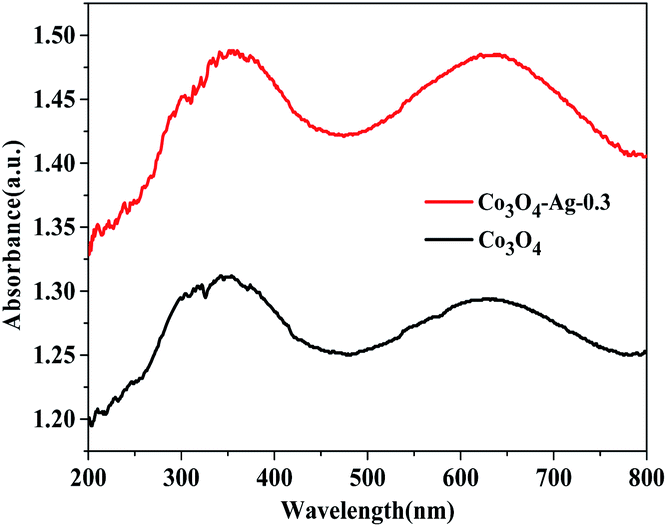 | ||
| Fig. 5 UV-vis absorption spectra of Co3O4 and Co3O4–Ag-0.3. | ||
 | ||
| Fig. 6 Plots of (αhν)2 versus photon energy of Co3O4 and Co3O4–Ag-0.3. | ||
The PL spectra of Co3O4 and Co3O4–Ag-0.3 were obtained at room temperature. Fig. 7 indicates that the excitation wavelength of the PL spectra for the pure Co3O4 and Co3O4–Ag-0.3 photocatalysts is about 647 nm. However, the Co3O4–Ag-0.3 composite photocatalyst showed a weaker emission intensity compared with the pure Co3O4 photocatalyst, indicating that the recombination of the photogenerated charge carriers could be inhibited more efficiently over Co3O4–Ag-0.3 than over pure Co3O4 because of the formation of a Schottky barrier. It is well known that electrons can easily transfer from one material with a lower work function to the other with a higher work function.57 Thus, the migration of holes from Co3O4 to Ag particles was accelerated by the Schottky barrier because of the different work functions of Ag (4.26 eV)58 and Co3O4 (6.2 eV),59 which could inhibit the photogenerated charge carrier recombination and improve the photocatalytic activity.
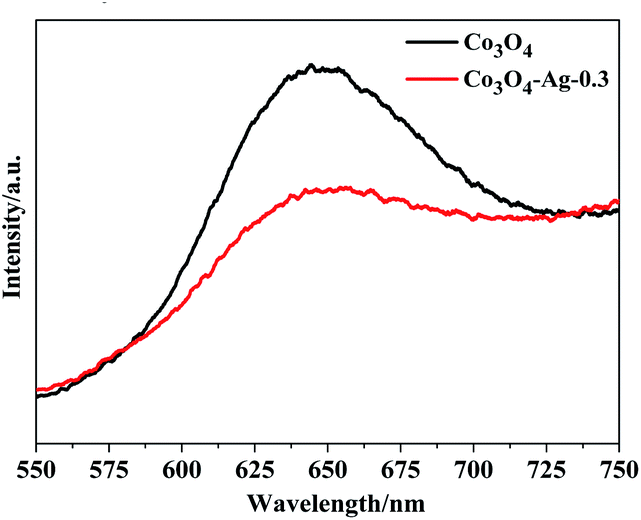 | ||
| Fig. 7 PL spectra of Co3O4 and Co3O4–Ag-0.3. | ||
3.2. Catalytic performance of the photocatalysts
Fig. 8(a and b) present the relative concentrations of MO solutions at different irradiation times and Fig. S1† shows the corresponding UV-vis spectra of MO over the Co3O4–Ag photocatalysts with different Ag loadings. It could be seen that the degradation efficiency for MO was only 6.4% without any photocatalyst. However, MO degraded greatly in the presence of photocatalysts as the irradiation time increased. The degradation efficiency of pure Co3O4 for MO was 71.2% after 30 h irradiation. In order to understand the role of different Ag loadings precisely, a series of photocatalysts including Co3O4–Ag-0.05, Co3O4–Ag-0.15, Co3O4–Ag-0.45, and Co3O4–Ag-0.6 were synthesized and their degradation efficiency was tested. The degradation activity increased from 89.5% to 96.4% on increasing the loading of Ag from 0.05% to 0.3% after 30 h irradiation. Nevertheless, the degradation efficiency decreased from 96.4% to 79.7% on further increasing the loading of Ag from 0.3% to 0.6%, respectively. Consequently, the photocatalytic activity increased with the loading of Ag compared with the activity of pure Co3O4, and Co3O4–Ag-0.3 showed the most excellent degradation efficiency (96.4%). Furthermore, the kinetics of MO degradation for all catalysts were investigated according to the experimental results. Fig. S2† shows the linear relationship between ln(C0/C) and the irradiation time for all the samples. The slope of the catalysts increased from 0.0401 to 0.1091 as the loading of Ag increased from 0.05% to 0.3%, while the slope decreased from 0.1091 to 0.052 when the Ag doping increased from 0.3% to 0.6%, respectively, indicating that the photocatalytic activity of Co3O4–Ag-0.3 was 2.72 times that of pure Co3O4 at identical reaction conditions. Additionally, we investigated the photocatalytic degradation of MO over the Co3O4–Ag-0.3 catalyst under visible light. As shown in Fig. 8(c), the blank experiment indicates that the concentration of MO only slightly decreases without adding catalysts, implying that the photodegradation can be ignored. The degradation efficiency for MO was 99.1% over the Co3O4–Ag-0.3 catalyst after 6 h visible light irradiation, which was greatly higher than the degradation efficiency (40.5%) over the same catalyst under UV light irradiation, indicating the excellent visible light-harvesting activity.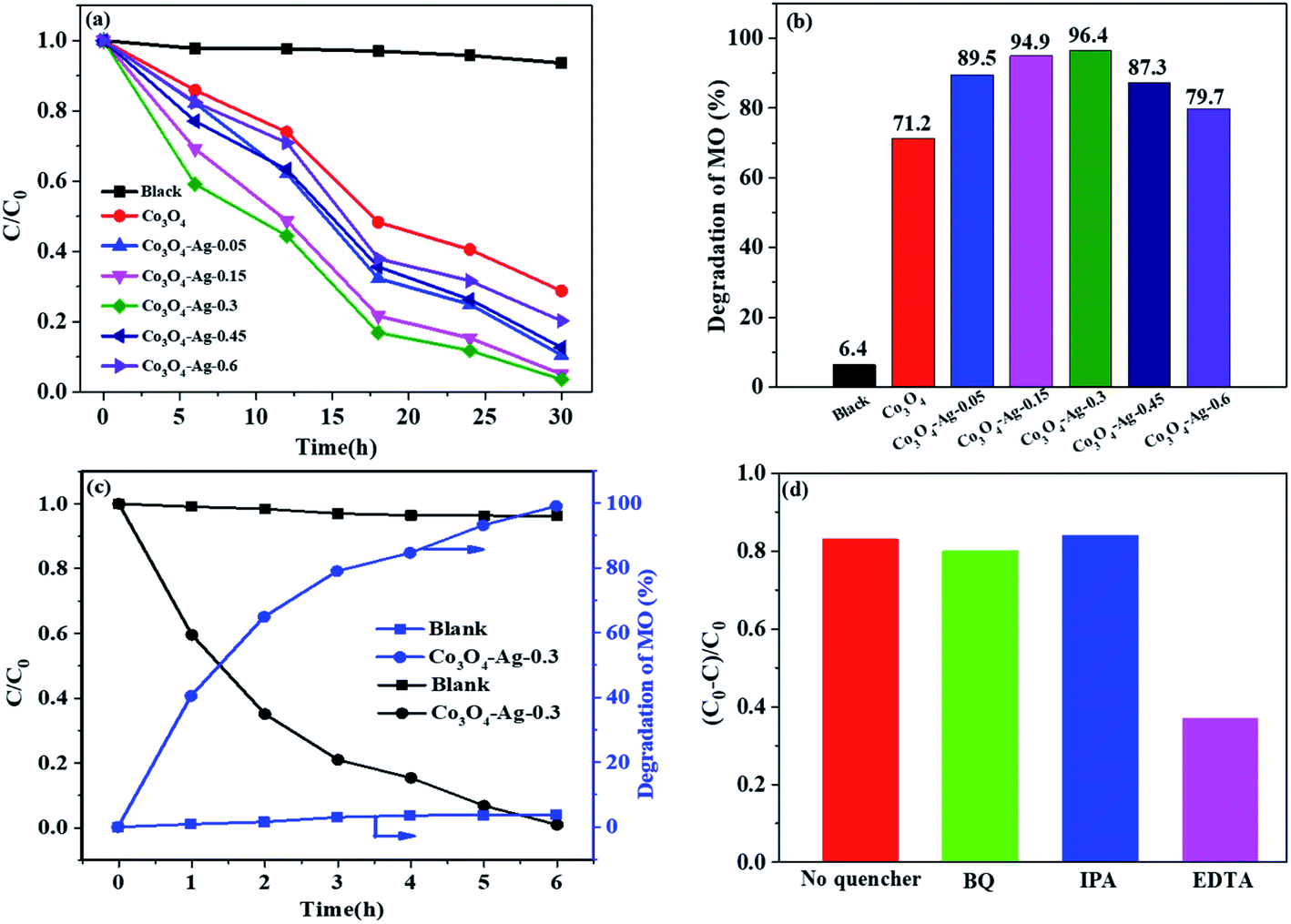 | ||
| Fig. 8 Degradation rate (a) and degradation efficiency of MO in 30 h (b) over the series of catalysts under UV light. Photocatalytic degradation of MO with or without Co3O4–Ag-0.3 under visible light (c). Trapping experiments of the active species during the photocatalytic degradation of MO with Co3O4–Ag-0.3 under UV radiation (d). | ||
The superior degradation performance of the Co3O4–Ag catalysts was related to the introduction of Ag. First, as seen in Fig. 3, the structure of Co3O4–Ag is uniform and regular. On the other hand, as revealed in Fig. 5 and 6, the absorption intensities for UV and visible light for the Co3O4–Ag catalysts significantly increase, while Egs become narrower with the addition of Ag compared with that for the pure Co3O4 catalyst. Moreover, from the PL spectra in Fig. 7, we can infer that the doping of Ag inhibits the recombination of the photogenerated charge carriers more efficiently than that for pure Co3O4 because of the formation of the Schottky barrier, and this is another reason for the excellent catalytic activity of the Co3O4–Ag catalysts.
To explain the possible photocatalytic mechanism, trapping experiments of the active species during the photocatalytic degradation of MO over the Co3O4–Ag-0.3 catalysts with or without scavengers were performed under UV light. As shown in Fig. 8(d), three scavengers (BQ, IPA, and EDTA) have been adopted. An 83.5% MO degradation efficiency could be obtained over the pure Co3O4–Ag-0.3 catalyst. It is well known that BQ and IPA are suitable scavengers of superoxide radicals (·O2−) and hydroxyl radicals (·OH), respectively.60,61 When BQ and IPA were introduced, the experimental results were almost unchanged, indicating that the superoxide radicals (·O2−) and hydroxyl radicals (·OH) were not the main active species for MO degradation. However, the degradation efficiency decreased greatly to 36.8% in the presence of EDTA, which is the scavenger of holes. Hence, the photogenerated holes (h+) were concluded to be the main active species for the photocatalytic degradation of MO.
Based on all the above-mentioned results and the trapping experiments of the active species during the photocatalytic degradation of MO, a reaction mechanism was proposed. When Ag was introduced into the Co3O4 catalyst, a Schottky junction structure was successfully formed (Fig. 9(a)), which could promote the migration of photo-generated holes from Co3O4 to the Ag metal, leaving electrons in the CB of Co3O4 because the work function of Co3O4 (Ws) was higher than that of the Ag element (Wm),58,59 as shown in Fig. 9(b). Therefore, the doping of Ag in Co3O4 can decrease the recombination rate of the electron–hole pairs and promote the charge separation simultaneously to generate more free holes, which are the main active species for MO degradation, so as to increase the photocatalytic activity effectively.
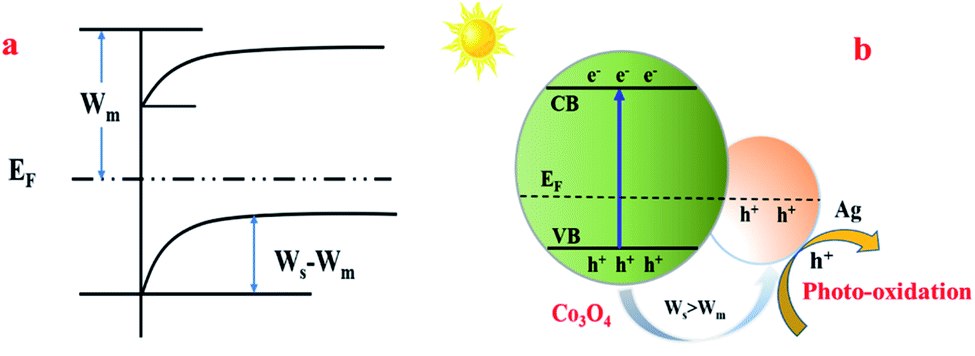 | ||
| Fig. 9 The proposed reaction mechanism of MO degradation over the Co3O4–Ag photocatalysts. Ws: the work function of Co3O4; Wm: the work function of Ag; EF: the balanced Fermi level. | ||
4. Conclusions
In conclusion, the doping of Ag into Co3O4 played a critical role in improving the MO degradation efficiency for the Co3O4–Ag photocatalysts using Ni foams as substrates because of the synergistic effect of the strengthened response to UV and visible light, narrower energy gap, and weaker PL intensity. Efficient activity with 96.4% MO degradation efficiency after 30 h UV light irradiation was achieved over the Co3O4–Ag-0.3 catalyst, and the photocatalytic activity of Co3O4–Ag-0.3 was 2.72 times that of pure Co3O4. Meanwhile, an excellent MO degradation efficiency of 99.1% over the Co3O4–Ag-0.3 catalyst was obtained after 6 h visible light irradiation. Additionally, a possible photocatalytic reaction mechanism was proposed based on the experimental results. Hence, the Co3O4–Ag catalyst covered on the Ni foam is a promising photocatalytic material for pollutant degradation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial supports from the School Foundation for North University of China (Grant No. 110246), Shanxi Science Foundation of China (Grant No. 201801D221197), Shanxi Scholarship Council of China (Grant No. 2017-094) and Shanxi ‘1331 project’ Key Subject Construction (1331KSC).References
- Y. Chen, J. Peng, H. Xiao, H. Peng, L. Bu, Z. Pan, Y. He, F. Chen, X. Wang and S. Li, Appl. Surf. Sci., 2017, 420, 773–781 CrossRef CAS.
- J. Gómez-Pastora, S. Dominguez, E. Bringas, M. J. Rivero, I. Ortiz and D. D. Dionysiou, Chem. Eng. J., 2017, 310, 407–427 CrossRef.
- M. Chong, B. Jin, C. W. K. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- S. N. Frank and A. J. Bard, J. Am. Chem. Soc., 1997, 99, 303–304 CrossRef.
- C. Han, L. Ge, C. Chen, Y. Li, X. Xiao, Y. Zhang and L. Guo, Appl. Catal., B, 2014, 147, 546–553 CrossRef CAS.
- F. Jing, R. Liang, J. Xiong, R. Chen, S. Zhang, Y. Li and L. Wu, Appl. Catal., B, 2017, 206, 9–15 CrossRef CAS.
- Y. Sun, G. Wang and K. Yan, J. Hazard. Mater., 2011, 36, 15502–15508 CAS.
- P. Adriana, L. Vincent, S. Kevin, G. Michael and T. Elijah, Nature, 2011, 10, 456–461 CrossRef PubMed.
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef CAS.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- C. D. Valentin, G. Pacchioni and A. Selloni, J. Phys. Chem. C, 2009, 133, 20543–20552 CrossRef.
- M. Zou, L. Feng, A. S. Ganeshraja, F. Xiong and M. Yang, Solid State Sci., 2016, 60, 1–10 CrossRef CAS.
- Z. Lian, W. Wang, S. Xiao, X. Li, Y. Cui, D. Zhang, G. Li and H. Li, Sci. Rep., 2015, 5, 10461–10470 CrossRef CAS PubMed.
- K. Nakashima, M. Kera, I. Fujii and S. Wada, Ceram. Int., 2013, 39, 3231–3234 CrossRef CAS.
- K. Domem, A. Kudo and T. Onishi, J. Phys. Chem., 1986, 90, 292–295 CrossRef.
- M. Cantarella, A. D. Mauro, A. Gulino, L. Spitaleri, G. Nicotra, V. Privitera and G. Impellizzeri, Appl. Catal., B, 2018, 238, 509–517 CrossRef CAS.
- G. Wang, Z. Li, M. Li, Y. Feng, W. Li, S. Lv and J. Liao, Ceram. Int., 2018, 44, 1291–1295 CrossRef CAS.
- S. Tokunaga, H. Kato and A. Kudo, Chem. Mater., 2001, 13, 4624–4628 CrossRef CAS.
- F. F. Abdi and R. V. D. Krol, J. Phys. Chem. C, 2012, 116, 9398–9404 CrossRef CAS.
- W. J. Jo, J. W. Jang, K. J. Kong, H. J. Kang, J. Y. Kim, H. Jun, K. P. S. Parmar and J. S. Lee, Angew. Chem., Int. Ed., 2012, 51, 3147–3151 CrossRef CAS PubMed.
- H. Ma, K. Teng, Y. Fu, Y. Song, Y. Wang and X. Dong, Energy Environ. Sci., 2011, 4, 3067–3074 RSC.
- J. S. Jang, S. H. Choi, H. G. Kim and J. S. Lee, J. Phys. Chem. C, 2008, 112, 17200–17205 CrossRef CAS.
- X. Jiang, L. Wang, F. Yu, Y. Nie, Q. Xing, X. Liu, Y. Pei, J. Zou and We. Dai, ACS Sustainable Chem. Eng., 2018, 6, 12695–12705 CrossRef CAS.
- S. Song, C. Lu, X. Wu, S. Jiang, C. Sun and Z. Le, Appl. Catal., B, 2018, 227, 145–152 CrossRef CAS.
- J. Cao, B. D. Luo, H. L. Lin and S. F. Chen, J. Hazard. Mater., 2011, 190, 700–706 CrossRef CAS PubMed.
- F. Teng, M. Chen, N. Li, X. Hua, J. Wang, Q. Zhang, Y. Wang, D. D. Meng and G. Li, RSC Adv., 2013, 3, 743–751 RSC.
- Y. Luo, J. Luo, W. Zhou, X. Qi, H. Zhang, D. Y. W. Yu, C. Li, H. Fan and T. Yu, J. Mater. Chem. A, 2013, 1, 273–281 RSC.
- W. Liu, X. Li, M. Zhu and X. He, J. Power Sources, 2015, 282, 179–186 CrossRef CAS.
- X. Zheng, Z. Han, W. Yang, F. Qu, B. Liu and X. Wu, Dalton Trans., 2016, 45, 16850–16858 RSC.
- J. Wang, X. Zhang, Q. Wei, H. Lv, Y. Tian, Z. Tong, X. Liu, J. Hao, H. Qu, J. Zhao, Y. Li and L. Mai, Nano Energy, 2016, 19, 222–233 CrossRef CAS.
- X. Zhang, P. Guo, Q. Pan, K. Shi and G. Zhang, J. Alloys Compd., 2017, 727, 514–521 CrossRef CAS.
- H. Xu, M. Shi, C. Liang, S. Wang, C. Xia, C. Xue, Z. Hai and S. Zhuiykov, Nanoscale Res. Lett., 2018, 13, 195–210 CrossRef PubMed.
- Q. Xiao, J. Zhang, C. Xiao and X. Tan, Catal. Commun., 2008, 9, 1247–1253 CrossRef CAS.
- W. Ahmad, T. Noor and M. Zeeshan, Catal. Commun., 2017, 89, 19–24 CrossRef CAS.
- S. H. Hsieh, G. J. Lee, C. Y. Chen, J. Chen, S. H. Ma, T. L. Horng, K. Chen and J. J. Wu, Top. Catal., 2013, 56, 623–629 CrossRef CAS.
- X. Liu, J. Iocozzia, Y. Wang, X. Cui, Y. Chen, S. Zhao, Z. Li and Z. Lin, Energy Environ. Sci., 2017, 10, 402–434 RSC.
- Q. Lu, Z. Lu, Y. Lu, L. Lv, Y. Ning, H. Yu, Y. Hou and Y. Yin, Nano Lett., 2013, 13, 5698–5702 CrossRef CAS PubMed.
- M. R. Khan, T. W. Chuan, A. Yousuf, M. N. K. Chowdhurya and C. K. Cheng, Catal. Sci. Technol., 2015, 5, 2522–2531 RSC.
- P. Mazierski, A. Malankowska, M. Kobylanski, M. Diak, M. Kozak, M. J. Winiarski, T. Klimczuk, W. Lisowski, G. Nowaczyk and A. Z. Medynska, ACS Catal., 2017, 7, 2753–2764 CrossRef CAS.
- Y. Cao, Z. Xing, Z. Li, X. Wu, M. Hu, X. Yan, Q. Zhu, S. Yang and W. Zhou, J. Hazard. Mater., 2018, 343, 181–190 CrossRef CAS PubMed.
- T. Sakata, T. Kawai and K. Hashimoto, J. Phys. Chem., 1984, 88, 2344–2350 CrossRef CAS.
- X. Wan, R. Ma, S. L. Tie and S. Lan, Mater. Sci. Semicond. Process., 2014, 27, 748–757 CrossRef CAS.
- L. Jian, D. Cai, G. Su, D. Lin, M. Lin, J. Li, J. Liu, X. Wan, S. Tie and S. Lan, Appl. Catal., A, 2016, 512, 74–84 CrossRef CAS.
- Y. Li and Y. Wu, Chem. Mater., 2010, 22, 5537–5542 CrossRef CAS.
- S. K. Jesudoss, J. J. Vijaya, P. I. Rajan, K. Kaviyarasu, M. Sivachidambaram, L. J. Kennedy, H. A. Al-Lohedan, R. Jothiramalingam and M. A. Munusamy, Photochem. Photobiol. Sci., 2017, 16, 766–778 RSC.
- H. Chen, J. Tan, J. Cui, X. Yang, H. Zheng, Y. Zhu and Y. Li, Mol. Catal., 2017, 433, 346–353 CrossRef CAS.
- H. Sun, X. Sun, T. Hu, M. Yu, F. Lu and J. Lian, J. Phys. Chem. C, 2014, 118, 2263–2272 CrossRef CAS.
- Q. Liao, N. Li, S. Jin, G. Yang and C. Wang, J. Am. Chem. Soc., 2015, 9, 5310–5317 CAS.
- Z. Shang, M. Sun, S. Chang, X. Che, X. Cao, L. Wang, Y. Guo, W. Zhan, Y. Guo and G. Lu, Appl. Catal., B, 2017, 209, 33–44 CrossRef CAS.
- M. Pudukudy and Z. Yaakob, Chem. Pap., 2014, 68, 1087–1096 CAS.
- S. Vijayakumar, A. Kiruthika Ponnalagi, S. Nagamuthu and G. Muralidharan, Electrochim. Acta, 2013, 106, 500–505 CrossRef CAS.
- T. He, D. Chen, X. Jiao, Y. Wang and Y. Duan, Chem. Mater., 2005, 17, 4023–4030 CrossRef CAS.
- D. Barreca, C. Massignan, S. Daolio, M. Fabrizio, C. Piccirillo, L. Armelao and E. Tondello, Chem. Mater., 2001, 13, 588–593 CrossRef CAS.
- A. Gulino, P. Dapporto, P. Rossi and I. Fragala, Chem. Mater., 2003, 15, 3748–3752 CrossRef CAS.
- M. Zhou, J. Yu, S. Liu, P. Zhai and L. Jiang, J. Hazard. Mater., 2008, 154, 1141–1148 CrossRef CAS PubMed.
- T. T. Y. Tan, C. K. Yip, D. Beydoun and R. Amal, Chem. Eng. J., 2003, 95, 179–186 CrossRef CAS.
- L. Chen, Y. Cui, S. Shi, H. Luo and Y. Gao, Appl. Surf. Sci., 2018, 450, 318–327 CrossRef CAS.
- M. Liu, J. Liu, Z. Li and F. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 7052–7060 CrossRef CAS PubMed.
- X. P. Qiu, J. S. Yu, H. M. Xu, W. X. Chen, W. Hu, H. Y. Bai and G. L. Chen, Appl. Surf. Sci., 2016, 362, 498–505 CrossRef CAS.
- X. P. Qiu, J. S. Yu, H. M. Xu, W. X. Chen, W. Hu and G. L. Chen, Appl. Surf. Sci., 2016, 382, 249–259 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10437b |
| This journal is © The Royal Society of Chemistry 2020 |