DOI:
10.1039/C9RA10306F
(Paper)
RSC Adv., 2020,
10, 4243-4250
Cytotoxic alkaloids from the marine shellfish-associated fungus Aspergillus sp. XBB-4 induced by an amino acid-directed strategy†
Received
9th December 2019
, Accepted 20th January 2020
First published on 27th January 2020
Abstract
Eight different culture media were used to culture shellfish Panopea abbreviate associated fungus Aspergillus sp. XBB-4. In a glucose-peptone-yeast (GPY) culture medium supplied with amino acids, this fungus can produce chemodiversity metabolites. Four new alkaloids including three β-carboline alkaloids, aspercarbolines A–C (1–3) and one piperazinedione, asperdione A (13) along with nine known compounds were isolated. The structures were elucidated mainly based on the NMR, MS, ECD and X-ray single-crystal diffraction data. The possible biosynthetic pathways of aspercarbolines A–C (1–3) were proposed. All compounds (1–13) were evaluated for their cytotoxicity against six cancer cell lines, including human nasopharyngeal carcinoma cell lines CNE1, CNE2, HONE1 and SUNE1, and human hepatocellular carcinoma cell lines hepG2 and QGY7701.
Introduction
Currently, the demand for seafood across the world is increasing owing to a wide range of health benefits in it. For example, appropriate consumption of seafood can reduce the risk of developing coronary heart disease (CHD), high blood pressure, stroke, some cancers, rheumatoid arthritis and other inflammatory diseases.1 However, seafood allergy becomes a serious health problem at the same time as the data shows that up to 2.5% of the general population suffers worldwide.2 The occurrence of seafood allergy may be aroused due to two factors: some seafood especially shellfish are filter feeders and selectively accumulate microorganisms from surrounding waters. Then they are normally eaten whole, without removing the intestinal tract, and raw or just with a very mild heat treatment.3
Geoduck Panopea abbreviate is a kind of important economic shellfish and many research studies about it have been reported. For instance, P. C. Zaidman and E. Morsan studied the growth variability of geoduck in a metapopulation.4 M. Goman et al. tested the suitability of using geoduck shells to evaluate changes in both annual and seasonal sea surface temperature, since geoducks are long-lived and deposit well-defined annual growth lines within their shells each winter.5 Other studies were about the nutritional value of geoducks6 or how to make the artificial reproduction of them.7 Few studies were focused on the secondary metabolites from microbial communities which were associated with geoducks.
Recently, a fungal strain Aspergillus sp. XBB-4 was isolated from geoduck Panopea abbreviate in our laboratory. Our previous studies have shown that systematic alteration of easily accessible cultivation parameters could affect microorganisms' metabolism. For examples, five new triquinane-type sesquiterpenoids chondrosterins A–E were isolated from marine fungus Chondrostereum sp. cultured in PDB (potato dextrose broth) medium8 and 14 new alkaloids were obtained from marine fungus Scedosporium apiospermum F41-1 cultured in GPY (glucose peptone yeast) medium contained a variety of amino acids.9 Two new bisindole compounds, pseudboindoles A and B, together with 11 known indole alkaloids were efficiently isolated from the marine fungus Pseudallescheria boydii F44-1 which was cultured in GPY medium with an amino acid-directed strategy by Yuan et al.10 Therefore, in order to optimize the culture conditions of this fungal strain, eight different culture media were developed. Finally, according to the LC-MS spectra of their culture extracts, the fungal strain cultured in GPY + amino acids produced the alkaloids metabolites with chemodiversity. So the fungus Aspergillus sp. XBB-4 was cultured in GPY + amino acids medium on a large scale. Metabolites investigation resulted in the isolation and structural determination of 13 compounds, including three new β-carboline alkaloids, aspercarbolines A–C (1–3), one new piperazinedione, asperdione A (13) and nine known compounds (Fig. 1). Herein, the isolation, structure characterization, proposed biosynthetic pathway and cytotoxic activity of these compounds were reported.
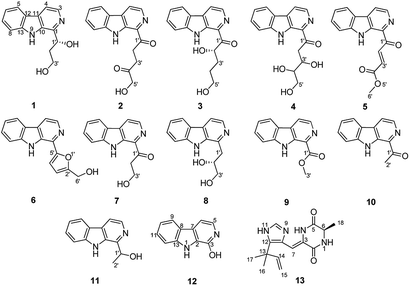 |
| | Fig. 1 Chemical structures of compounds 1–13. | |
Results and discussion
Structural elucidation
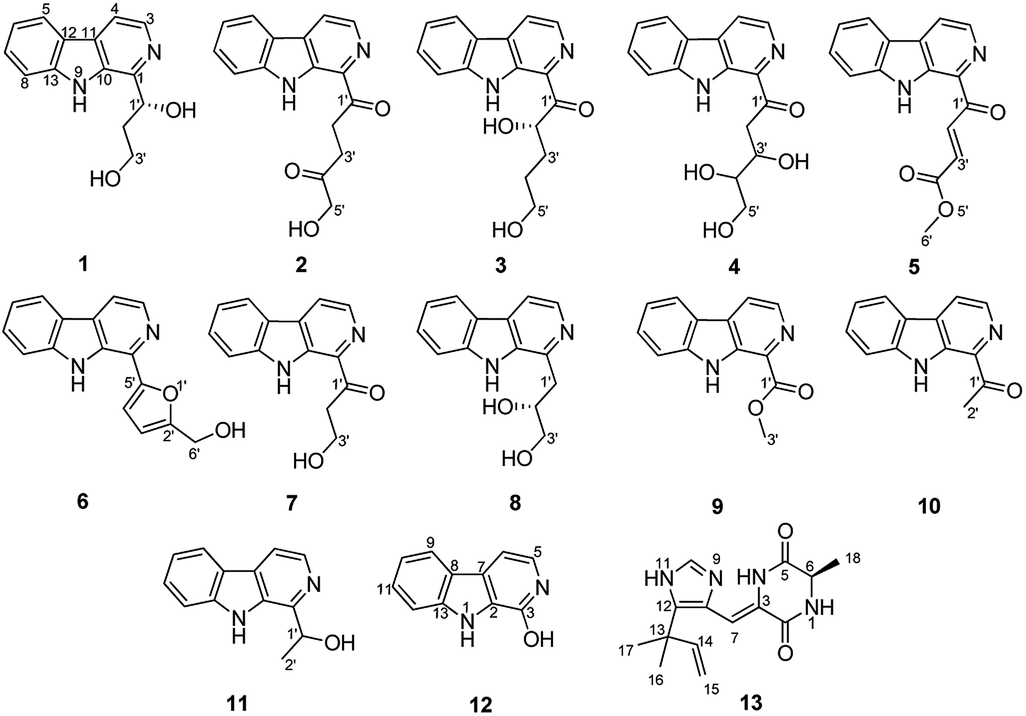
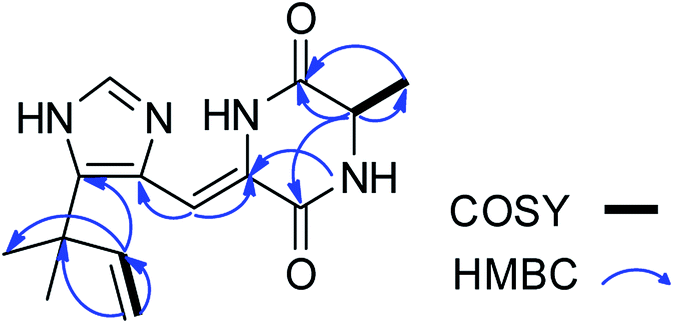
Aspercarboline A (1) was isolated as a brown oil. Its molecular formula was determined to be C14H14N2O2 by HR-ESI-MS at m/z 243.11268 [M + H]+ (calcd for C14H15N2O2, 243.11280), having nine degrees of unsaturation (ESI Fig. S1†). The 13C NMR spectrum of 1 indicated the presences of 11 aromatic carbons and three aliphatic carbons, which could be classified to five quaternary carbons, seven methines and two methylenes according to the DEPT spectra (ESI Fig. S3–S5†). The diagnostic aryl proton signals at δH 8.21 (d, 8.0, H-5), 7.71 (d, 8.0, H-8), 7.54 (dd, 8.0, 1.0, H-7) and 7.25 (dd, 8.0, 1.0, H-6) along with the 1H–1H COSY correlations of H-5/H-6, H-6/H-7 and H-7/H-8 suggested a 1,2-disubstituted benzene ring in this molecule (Table 1 and ESI Fig. S2†). The 1H–1H COSY spectrum also indicated the presence of a pair of aromatic protons H-3 (δH 8.28, d, 5.0) and H-4 (δH 7.98, d, 5.0) (ESI Fig. S7†). The HMBC correlations from H-3 to C-1 (δC 148.3) and C-11 (δC 129.9); from H-4 to C-10 (δC 133.9), C-11 and C-12 (δC 122.0); from H-5 to C-7 (δC 128.9); from H-6 to C-8 (δC 113.0) and C-13 (δC 141.7); from H-7 to C-5 (δC 122.2), C-6 and C-8; from H-8 to C-6 and C-13 displayed a β-carboline fragment (Fig. 2 and ESI Fig. S8†). The 1H–1H COSY and HSQC spectra along with the molecular formula showed that 1 had two hydroxyl groups (δH 5.15, s; 4.03, s) (ESI Fig. S6†). The 1H–1H COSY cross-peaks of H-2′/H-1′, H-2′/H-3′ and the HMBC correlations from H-2′ to C-1′ and C-1, from H-3′ to C-2′ revealed the side chain –CH(OH)CH2CH2OH was connected to C-1. The absolute configuration of 1 was defined by comparison of the experimental and calculated ECD data. The calculated ECD curve of 1′R-1 was well matched with the experimental curve (Fig. 3). Therefore, the absolute configuration of 1 was defined as 1′R. The chemical structure of aspercarboline A (1) was unambiguously established, as shown in Fig. 1.
Table 1 1H and 13C NMR data for 1–3 (δ in ppm)
| No. |
1 (in acetone-d6)a |
2 (in CDCl3)b |
3 (in DMSO-d6)b |
| δC, type |
δH, mult. (J in Hz) |
δC, type |
δH, mult. (J in Hz) |
δC, type |
δH, mult. (J in Hz) |
| 1H and 13C NMR data were recorded at 500/125 MHz, respectively. 1H and 13C NMR data were recorded at 400/100 MHz, respectively. |
| 1 |
148.3, C |
|
135.6, C |
|
135.8, C |
|
| 3 |
138.0, CH |
8.28, d (5.0) |
138.4, CH |
8.54, d (4.8) |
137.3, CH |
8.49, d (4.8) |
| 4 |
114.2, CH |
7.98, d (5.0) |
119.5, CH |
8.17, d (4.8) |
119.2, CH |
8.41, d (4.8) |
| 5 |
122.2, CH |
8.21, d (8.0) |
122.0, CH |
8.15, d (8.0) |
121.7, CH |
8.29, d (8.0) |
| 6 |
120.3, CH |
7.25, dd (8.0, 1.0) |
121.0, CH |
7.34, dd (8.0, 1.2) |
120.1, CH |
7.29, dd (8.0, 1.2) |
| 7 |
128.9, CH |
7.54, dd (8.0, 1.0) |
129.5, CH |
7.60, dd (8.0, 1.2) |
128.8, CH |
7.58, dd (8.0, 1.2) |
| 8 |
113.0, CH |
7.71, d (8.0) |
112.1, CH |
7.57, d (8.0) |
113.0, CH |
7.80, d (8.0) |
| 9-NH |
|
10.63, s |
|
10.18, s |
|
11.90, s |
| 10 |
133.9, C |
|
135.3, C |
|
134.0, C |
|
| 11 |
129.9, C |
|
131.8, C |
|
130.9, C |
|
| 12 |
122.0, C |
|
120.8, C |
|
119.8, C |
|
| 13 |
141.7, C |
|
141.3, C |
|
141.8, C |
|
| 1′ |
73.3, CH |
5.37, dd (8.0, 4.0) |
202.8, C |
|
203.5, C |
|
| 2′ |
40.5, CH2 |
2.21, m; 2.11, m |
32.2, CH2 |
3.85, t (6.4) |
70.7, CH |
3.56, m |
| 3′ |
60.4, CH2 |
3.87, dt (10.5, 6.0); 3.80, dt (10.5, 6.0) |
32.3, CH2 |
2.89, t (6.4) |
28.0, CH2 |
1.96, m; 1.69, m |
| 4′ |
|
|
208.9, C |
|
33.6, CH2 |
3.42, m |
| 5′ |
|
|
68.5, CH2 |
4.47, s |
65.9, CH2 |
3.36, m |
| 1′-OH |
|
5.15, s |
|
|
|
|
| 2′-OH |
|
|
|
|
|
4.55, d (5.2) |
| 3′-OH |
|
4.03, s |
|
|
|
|
| 5′-OH |
|
|
|
3.13, s |
|
4.50, t (5.6) |
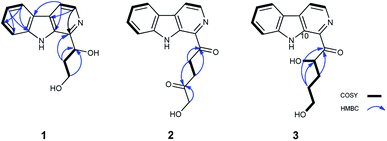 |
| | Fig. 2 1H–1H COSY (bold lines), main HMBC (blue arrows) correlations of compounds 1–3. | |
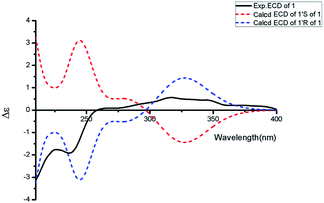 |
| | Fig. 3 Experimental ECD spectrum in MeOH and the calculated ECD spectra of compound 1. | |
Aspercarboline B (2) was obtained as a white powder and its molecular formula was established as C16H14N2O3 by HR-ESI-MS at m/z 283.10891 [M + H]+ (calcd for C16H15N2O3, 283.10772) requiring 11 degrees of unsaturation (ESI Fig. S9†). After careful comparison of the 1H and 13C NMR spectra, compound 2 was found to possess similar signals to compound 1 in the aromatic region, so compound 2 was quickly identified as a β-carboline alkaloid having different side chain (ESI Fig. S10 and S11†). The 13C NMR and DEPT spectra revealed two carbonyl carbons (δC 202.8 and 208.9) and three methylene groups (δC 68.5, 32.3 and 32.2) in the side chain (ESI Fig. S12 and S13†). From the HSQC spectrum and the 1H–1H COSY correlation between H-2′ (δH 3.85, t, 6.4) and H-3′ (δH 2.89, t, 6.4) revealed the presence of the fragment –CH2CH2– (ESI Fig. S14 and S15†). The remaining methylene group (δC 68.5, δH 4.47, s) was deduced to connect to the hydroxyl group (δH 3.13, s). The HMBC correlations of H-2′/C-1′ (δC 202.8), H-2′/C-4′ (δC 208.9), H-3′/C-1′, H-3′/C-4′, and H-5′/C-4′ elucidated the side chain as –C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)CH2CH2C(O)CH2OH (ESI Fig. S16†).
O)CH2CH2C(O)CH2OH (ESI Fig. S16†).
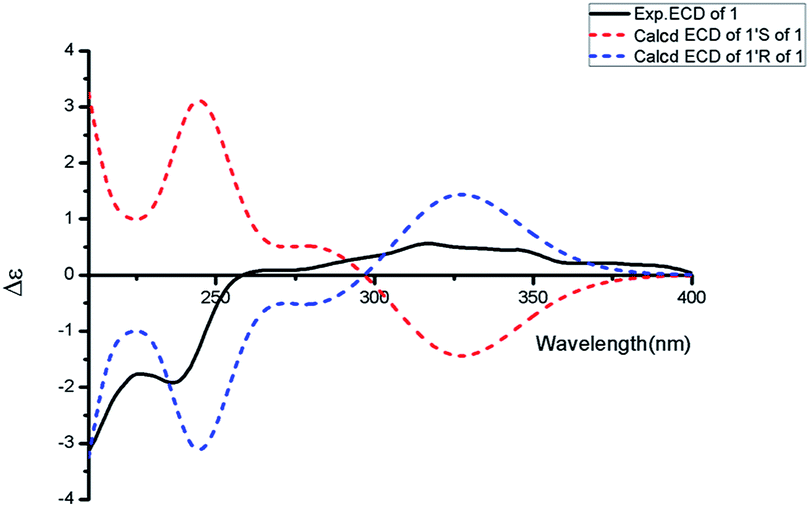
Aspercarboline C (3) was isolated as a white powder. The molecular formula was identified as C16H16N2O3 based on HR-ESI-MS peak at m/z 285.12302 [M + H]+ (calcd for C16H17N2O3, 285.12337) (ESI Fig. S17†), indicating 10 degrees of unsaturation. Its similar 1H and 13C NMR spectra signals to compound 1 in the aromatic region, suggested a β-carboline alkaloid as well (ESI Fig. S18 and S19†). Based on the analysis of the 13C NMR and DEPT spectra, three methylenes, one methine and one quaternary carbon were contained at the side chain (ESI Fig. S20†). The quaternary carbon at δC 203.5 (C-1′) revealed a ketone carbonyl. From the 1H–1H COSY correlations of H-2′ (δH 3.56)/H-3′ (δH 1.69, 1.96)/H-4′ (δH 3.42)/H-5′ (δH 3.36), H-2′/2′-OH (δH 4.55) and H-5′ (δH 3.36)/5′-OH (δH 4.50), the fragment of –CH(OH)CH2CH2CH2OH was established (Fig. 2 and ESI Fig. S22†). Moreover, the HMBC correlations (ESI Fig. S23†) from H-3′ to C-1′ and H-4′ to C-1′ demonstrated that the carbonyl group (C-1′) was linked with C-1 (δC 135.8) and C-2′ (δC 70.7) to connect the long side chain and the β-carboline skeleton. The absolute configuration of 3 was defined as 2′S because the calculated ECD curve of 2′S-3 was almost consistent with the experimental one (Fig. 4). Finally, the structure of 3 was established as shown in Fig. 1.
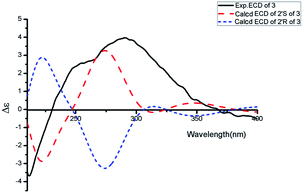 |
| | Fig. 4 Experimental ECD spectrum in MeOH and the calculated ECD spectra of compound 3. | |
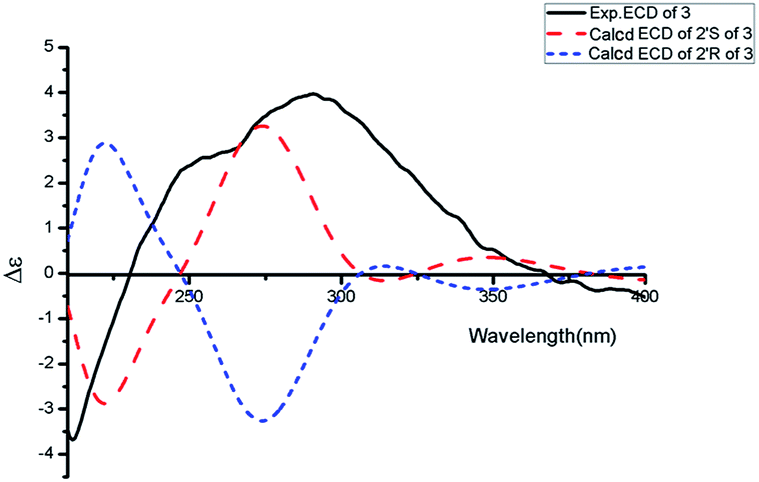
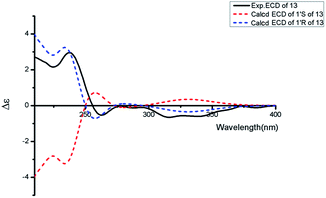
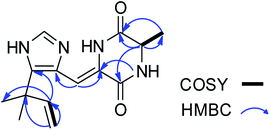
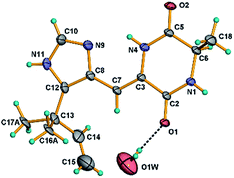
Asperdione A (13) was obtained as colorless crystals with the molecular formula C14H18N4O2 determined by the HR-ESI-MS peak at m/z 275.15079 [M + H]+ (calcd for C14H19N4O2, 275.15025), indicating the presence of 4 degrees of unsaturation (ESI Fig. S24†). In the 1H NMR spectrum of 13, there was a peak (δH 1.51) (Table 2) in the upfield whose integral was 6.00 (ESI Fig. S25†). It suggested that 13 might have two same methyl groups which was confirmed by the observation of a peak (δC 28.1) in the 13C NMR spectrum whose signal was almost double strong as the other peaks (ESI Fig. S26†). From the 13C NMR and DEPT spectra, C-2 (δC 160.2) and C-5 (δC 166.1) could be defined as two carbonyl groups (ESI Fig. S27 and S28†). The 1H–1H COSY cross peak of H-6 (δH 4.26, q, 7.2, 1H)/H-18 (δH 1.57, d, 7.2, 3H) and C-6 (δC 51.9) deduced a –CHCH3 fragment was attached to –NH group (ESI Fig. S30†). The HMBC correlations from H-18 to C-5 (δC 166.1) and C-6; H-6 to C-2, C-5 and C-18 (δC 21.5); H-7 to C-2, C-3 (δC 132.4), C-8 (δC 124.3) and C-12; and H-4 to C-3, C-8 and C-12 demonstrated the presence of piperazinedione skeleton (Fig. 6 and ESI Fig. S31†). The 1H–1H COSY correlation of H-14 (δH 6.04) and H-15 (δH 5.17 and 5.20) along with the HMBC correlations from H-16 and H-17 to C-12, C-13, C-14 and C-15; H-15 to C-12, C-13 and C-14; and H-14 to C-12, C-13, C-16 and C-17 presented the fragments of –C–C(CH3)2CHCH2. Thus, the planar structure of 13 was elucidated (Fig. 1). The absolute configuration of 13 was defined by comparison of the experimental and calculated ECD data. The calculated ECD curve of 6R-13 was well matched with the experimental curve (Fig. 5). In addition of the X-ray single crystal diffraction analysis, the absolute configuration of 13 was defined as 6R (Fig. 7). Compound 13 is the configuration isomer of a known compound, 3-[[5-(1,1-dimethyl-2-propen-1-yl)-1H-imidazol-4-yl]methylene]-6-methyl-(6S)-2,5-piperazinedione which was synthesized by E. Couladouros, et al. in 2005 (ref. 11) and was isolated as a natural product for the first time by P. Phainuphong et al. in 2018.12
Table 2 1H (600 MHz) and 13C NMR (150 MHz) data for 13 in CDCl3 (δ in ppm)
| No. |
δC, type |
δH, mult. (J in Hz) |
| 1 |
NH |
9.13, s |
| 2 |
160.2, CO |
|
| 3 |
132.4, C |
|
| 4 |
NH |
7.55, s |
| 5 |
166.1, CO |
|
| 6 |
51.9, CH |
4.26, q (7.2) |
| 7 |
105.4, CH |
6.93, s |
| 8 |
124.3, C |
|
| 9 |
N |
|
| 10 |
132.5, CH |
6.02, brs |
| 11 |
NH |
11.95, s |
| 12 |
136.8, C |
|
| 13 |
37.8, C |
|
| 14 |
144.8, CH |
6.04, dd (17.4, 10.8) |
| 15 |
113.6, CH2 |
5.20, d (17.4); 5.17, d (10.8) |
| 16 |
28.1, CH3 |
1.51, s |
| 17 |
28.1, CH3 |
1.51, s |
| 18 |
21.5, CH3 |
1.57, d (7.2) |
 |
| | Fig. 5 Experimental ECD spectrum in MeOH and the calculated ECD spectra of compound 13. | |
 |
| | Fig. 6 1H–1H COSY (bold lines), main HMBC (blue arrows) correlations of compound 13. | |
 |
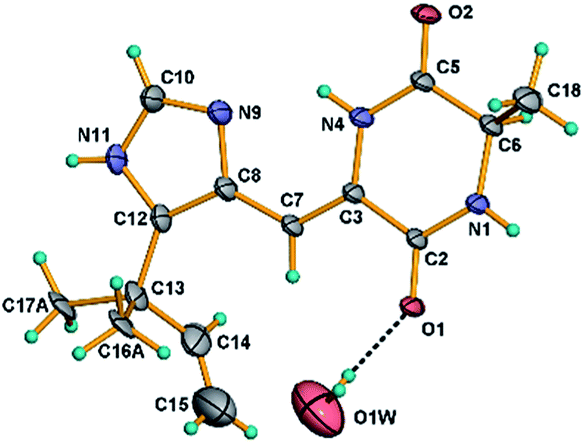
| | Fig. 7 ORTEP diagram for the single-crystal X-ray structure of compound 13. | |
The other compounds were identified as 1-(1-oxo-3,4,5-trihydroxy-1-pentyl)-β-carboline (4),13 4-(9H-β-carbolin-1-yl)-4-oxobut-2-enoic acid methyl ester (5),14 perlolyrin (6),15 1-(9H-β-carbolin-1-yl)-3-hydroxy-propan-1-one (7),16 cordysinin E (8),17 1-methoxycarbonyl-β-carboline (9),18 1-acetyl-β-carboline (10),19 cordysinin C (11),20 and 3-hydroxy-β-carboline (12)21 based on their NMR and MS data (ESI Fig. S32–S50†) and by comparing with those previously reported in the literatures.
Proposed biosynthetic pathway
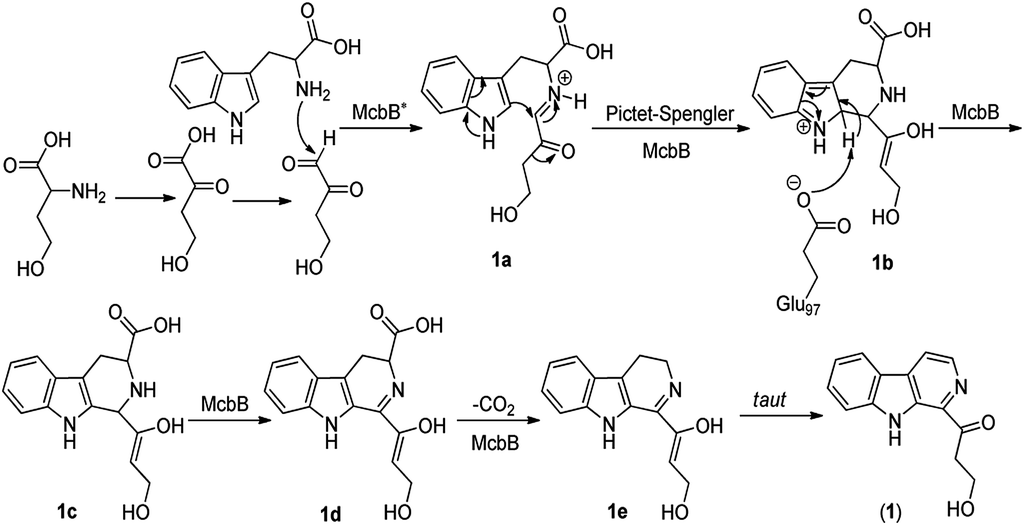
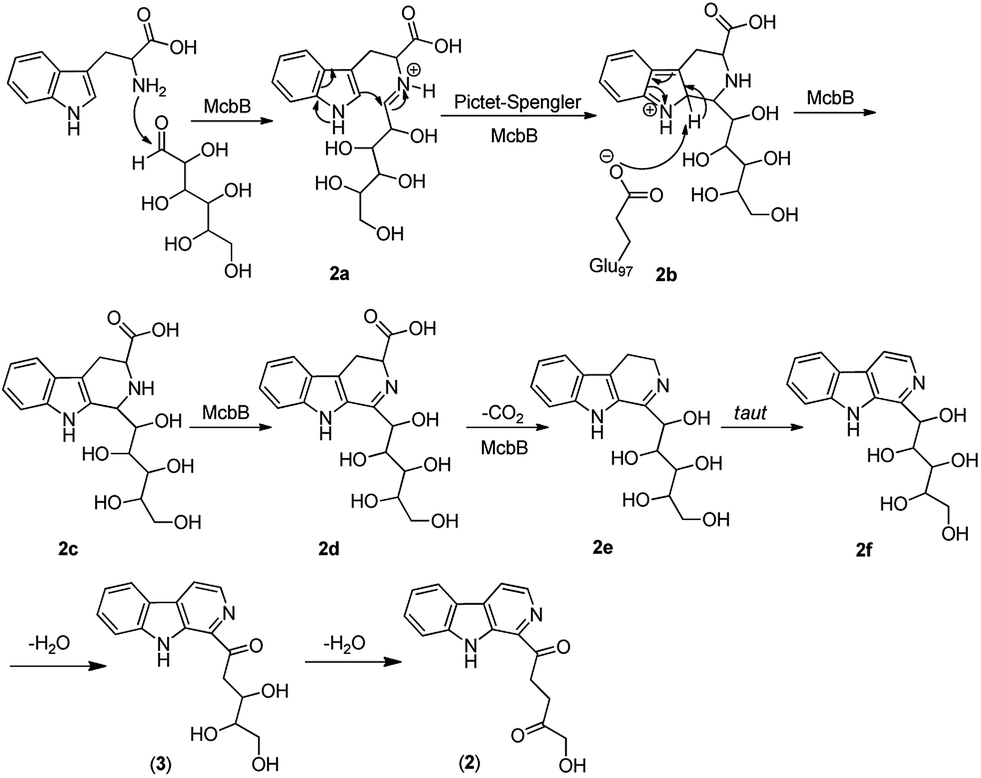
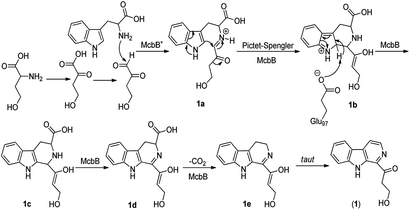
Although it was proved that the origin of the indole part of β-carboline alkaloids was from tryptophan, the other counterparts can be derived from different components.22 Many researches demonstrated that the enzymatic Pictet–Spengler reaction, catalyzed by Pictet–Spenglerase, was one of the characteristics of β-carboline alkaloid biosynthesis.23–25 Feeding experiments with 5-F-Trp and 13C labeled glucose and acetate revealed that the synthesis of β-carboline involved a Pictet–Spengler cyclization, a decarboxylation and C-ring oxidation process.26,27 Combining of the published literatures and basic organic synthesis mechanism, the biosynthetic pathway of compounds 1–3 were proposed. For compound 1, McbB (McbB is an enzyme which was demonstrated to be absolutely essential for β-carboline core construction26) catalyzes two substrates to obtain compound 1a which is utilized in the Pictet–Spengler cyclization to afford 1b. The negatively charged Glu97 is used as the active site to complete the C-ring aromatization and then via oxidation to product 1d. 1e is obtained by decarboxylation from 1d and followed oxidation lead to the product of compound 1 (Fig. 8). Similarly, the hypothetical biosynthetic pathway of compounds 2 and 3 is given as showed in Fig. 9. Compound 2f is produced after Pictet–Spengler cyclization, C-ring aromatization, oxidation and decarboxylation. Subsequent elimination of one H2O from the carbohydrate-derived side chain of 2f produces compound 3. Finally, subsequent elimination of one more H2O from compound 3 obtains compound 2.
 |
| | Fig. 8 Proposed biosynthetic pathway of compound 1. | |
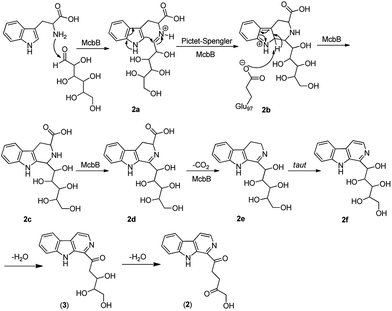 |
| | Fig. 9 Proposed biosynthetic pathways of compounds 2 and 3. | |
Biological activity
Compounds 1–13 were evaluated for their cytotoxicity against six cancer cell lines, including human nasopharyngeal carcinoma cell lines CNE1, CNE2, HONE1 and SUNE1, human hepatocellular carcinoma cell lines hepG2 and QGY7701 (Table 3). Results showed that compound 3 exhibited a moderate inhibitory effect against all six cell lines except hepG2 with IC50 values of 16.29, 20.58, 20.11, 45.31, 50.85 and 28.97 μM. Compound 4 showed moderate cytotoxicity against CNE1, CNE2, HONE1 and QGY7701 with the IC50 values from 20.64 to 43.43 μM. Compound 6 presented a strong inhibitory effect against CNE2 with an IC50 value of 8.69 μM and moderate inhibitory effects against the other five cell lines. Compounds 7, 10 and 11 displayed moderate cytotoxicity against QGY7701 with the mean IC50 values of 30.40, 35.49 and 49.27 μM respectively while 12 exhibited a strong cytotoxicity to hepG2 and QGY7701. Compound 13 showed potent cytotoxicity against these cancer cell lines with the IC50 values ranged from 10.72 to 22.00 μM. In contrast, compounds 1, 2, 5, 8 and 9 were apparently inactive in this assay (IC50 > 50 μM). Hirsutanol A was used as a positive control.28–30
Table 3 Cytotoxicities of compounds 1–13 against six cancer cell lines, IC50 (μM)a
| Compounds |
CNE1 |
CNE2 |
HONE1 |
SUNE1 |
HepG2 |
QGY7701 |
| “−” means “>100”. |
| 1 |
95.70 |
— |
77.61 |
85.94 |
— |
— |
| 2 |
— |
— |
84.29 |
94.05 |
— |
— |
| 3 |
16.29 |
20.58 |
20.11 |
45.31 |
50.85 |
28.97 |
| 4 |
43.31 |
38.53 |
42.85 |
64.95 |
65.03 |
20.64 |
| 5 |
60.42 |
64.04 |
74.58 |
70.72 |
— |
51.93 |
| 6 |
22.34 |
8.69 |
31.35 |
23.44 |
49.79 |
32.76 |
| 7 |
— |
— |
— |
— |
76.53 |
30.40 |
| 8 |
— |
— |
— |
— |
— |
— |
| 9 |
— |
— |
80.28 |
— |
— |
59.40 |
| 10 |
74.67 |
— |
97.39 |
— |
60.13 |
35.49 |
| 11 |
62.57 |
— |
85.13 |
— |
— |
49.27 |
| 12 |
40.58 |
— |
71.16 |
— |
16.93 |
8.82 |
| 13 |
22.00 |
18.93 |
21.61 |
16.93 |
12.33 |
10.72 |
| Hirsutanol A |
10.08 |
12.72 |
17.40 |
3.50 |
10.11 |
21.12 |
Experimental
General experimental procedures
The 1D and 2D NMR spectra were obtained on Bruker AV-400, AV-500 and AV-600 spectrometers (Bruker Bio Spin AG, Industriestrasse 26, Fällanden, Switzerland). HR-ESI-MS data were acquired on a Thermo Fisher LTQ Orbitrap Elite high-resolution mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). LC-MS data were collected on a Thermo Fisher system (Thermo Fisher Scientific Inc., Waltham, MA, USA) with an Ultimate 3000 RS pump, auto-sampler, diode array detector and column compartment, and coupled with an ISQ EC mass spectrometer. CD spectra were measured on a JASCO J-1700 circular dichroism spectrometer (JASCO International Co. Ltd., Tokyo, Japan). A Shimadzu UV-Vis-NIR spectrophotometer (Shimadzu Corporation, Nakagyo-ku, Kyoto, Japan) was used to obtain UV data. IR spectra were collected on a PerkinElmer Frontier FT-IR spectrophotometer (PerkinElmer Inc., Waltham, MA, USA). Optical rotations were measured using an Anton Paar (MCP 300) polarimeter at 25 °C. X-ray crystallographic analysis was performed on an Agilent Gemini Ultra diffractometer (Cu Kα radiation). Column chromatography was performed with silica gel (SiO2, 200–300 mesh, Qingdao Marine Chemical Inc., Qingdao, China) and Sephadex LH-20 (GE Healthcare Bio-Sciences, Uppsala, Sweden). Preparative HPLC was performed on a Shimadzu LC-20AT HPLC pump (Shimadzu Corporation, Nakagyo-ku, Kyoto, Japan) and equipped with a SPD-20A dual λ absorption detector (Shimadzu Corporation, Nakagyo-ku, Kyoto, Japan) and a Capcell-Pak C18 UG80 HPLC column (250 mm × 20 mm, Shiseido Co., Ltd., Minato-ku, Tokyo, Japan) added with a Spolar HPLC packed column (250 mm × 4.6 mm, Shiseido Co., Ltd., Minato-ku, Tokyo, Japan).
Fungal material
Aspergillus sp. XBB-4 was isolated from the inner tissue of geoduck Panopea abbreviate which was collected from the South China Sea. The sequence data of this fungal strain have been submitted to GenBank with accession number MK863524. This fungal strain was stored at School of Chemistry, Sun Yat-sen University, Guangzhou 510275, China.
Optimization of culture conditions
The culture conditions could be classified to two kinds, liquid culture media and solid culture media. The main component of liquid culture media was water while the solid one was rice. Considering that salt might affect the osmotic pressure of cells and alter the metabolism, and the supplement with amino acids might induce marine fungi to produce diverse alkaloids, the key cultivation parameter alteration was whether having salt and amino acids or not. Detail composition of each nutrient source in the different culture media can see the ESI Table S1.† The LC-MS prescreening of their culture extracts indicated that fungus Aspergillus sp. XBB-4 could produce most diverse metabolites in culture medium D (GPY + amino acids) composed of glucose 15 g, peptone 10 g, yeast extract 2 g, L-lysine 2 g, phenylalanine 2 g, L-threonine 2 g, L-tryptophan 2 g, L-methionine 2 g, nicotinamide 1 g, folic acid 1 g, and sea salt 30 g in 1 L water (ESI Fig. S51†). Therefore, culture medium D was selected to ferment this fungus for scale-up production.
Culture method
Aspergillus sp. XBB-4 was cultured in liquid medium D (GPY + amino acids). Fungal mycelia were cut and transferred aseptically to 1000 mL conical flasks each containing 400 mL sterilized liquid medium. The fungus was static incubated at 25 °C for 30 days.
Extraction and isolation
The mycelia and liquid culture (100 L) were separated by cheesecloth. The culture broth was extracted successively three times with EtOAc (60 L) and concentrated by low-temperature rotary evaporation to obtain a crude extract (45.1 g).
The crude extract was subjected to a silica gel chromatography (column diameter: 8 cm, length: 70 cm, silica gel: 500 g) with a gradient of petroleum ether–EtOAc (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0–0:10, v/v) followed by EtOAc–MeOH (10:0–0:10, v/v) to afford 40 fractions (Fr. 1–Fr. 40). Fr. 29 was purified by silica gel column in a step gradient elution with EtOAc–MeOH (10:0–0:10, v/v) to get 10 sub-fractions (Fr. 29-1–Fr. 29-10). Fr. 29-6 was further purified by reversed-phase silica gel column chromatography using MeOH–H2O (50:50–100:0, v/v) gradient elution to obtain Fr. 29-6-1–Fr. 29-6-4. Then Fr. 29-6-3 was further purified by RP-HPLC using MeOH–H2O (60:40, v/v) as the eluent to obtain compound 1 (4.9 mg). Compound 8 (2.6 mg) was obtained from Fr. 29-6-4 by Sephadex LH-20 gel column chromatography and RP-HPLC eluted with MeOH–H2O (60:40, v/v). Compounds 2 (1.9 mg) and 9 (2.3 mg) were gained as the result of the purification of Fr. 29-2 through RP-HPLC with a solvent system methanol–water (70:30, v/v). Compound 3 (8.9 mg) was obtained by the natural precipitation of component Fr. 24. HPLC purification of Fr. 29-3 with a solvent system MeOH–H2O (70:30, v/v) gave compound 4 (6.5 mg). Fr. 30 was purified by RP HPLC with an eluent of MeOH–H2O (70:30, v/v) to afford compound 5 (3.0 mg). Fr. 20 was separated by RP-HPLC eluted with MeOH–H2O (70:30, v/v) to obtain sub-fractions Fr. 20-1–Fr. 20-3, and then Fr. 20-2 was further separated by RP-HPLC in MeOH–H2O (70:30, v/v) to afford compound 6 (58.0 mg). Similarly, compound 11 (5.0 mg) was isolated by the further RP-HPLC separation of Fr. 20-3. Fr. 14 and Fr. 15 was merged after 1H NMR prescreening, and then were purified with HPLC using MeOH–H2O (75:25, v/v) as eluent to obtain compound 7 (6.2 mg). Compound 10 (11.3 mg) was gained as white powder from the precipitation of Fr. 4. Fr. 21 was subjected to a reversed-phase silica gel column chromatography using a step gradient of MeOH–H2O (50:50–100:0, v/v) as eluent to furnish four sub-fractions Fr. 21-1–21-4, and then Fr. 21-3 was purified by Sephadex LH-20 gel column chromatography eluted with 100% methanol to give compound 12 (2.9 mg). Fr. 25 was purified on RP-HPLC using MeOH–H2O (80:20, v/v) as eluent to obtain compound 13 (7.2 mg).
:0–0:10, v/v) followed by EtOAc–MeOH (10:0–0:10, v/v) to afford 40 fractions (Fr. 1–Fr. 40). Fr. 29 was purified by silica gel column in a step gradient elution with EtOAc–MeOH (10:0–0:10, v/v) to get 10 sub-fractions (Fr. 29-1–Fr. 29-10). Fr. 29-6 was further purified by reversed-phase silica gel column chromatography using MeOH–H2O (50:50–100:0, v/v) gradient elution to obtain Fr. 29-6-1–Fr. 29-6-4. Then Fr. 29-6-3 was further purified by RP-HPLC using MeOH–H2O (60:40, v/v) as the eluent to obtain compound 1 (4.9 mg). Compound 8 (2.6 mg) was obtained from Fr. 29-6-4 by Sephadex LH-20 gel column chromatography and RP-HPLC eluted with MeOH–H2O (60:40, v/v). Compounds 2 (1.9 mg) and 9 (2.3 mg) were gained as the result of the purification of Fr. 29-2 through RP-HPLC with a solvent system methanol–water (70:30, v/v). Compound 3 (8.9 mg) was obtained by the natural precipitation of component Fr. 24. HPLC purification of Fr. 29-3 with a solvent system MeOH–H2O (70:30, v/v) gave compound 4 (6.5 mg). Fr. 30 was purified by RP HPLC with an eluent of MeOH–H2O (70:30, v/v) to afford compound 5 (3.0 mg). Fr. 20 was separated by RP-HPLC eluted with MeOH–H2O (70:30, v/v) to obtain sub-fractions Fr. 20-1–Fr. 20-3, and then Fr. 20-2 was further separated by RP-HPLC in MeOH–H2O (70:30, v/v) to afford compound 6 (58.0 mg). Similarly, compound 11 (5.0 mg) was isolated by the further RP-HPLC separation of Fr. 20-3. Fr. 14 and Fr. 15 was merged after 1H NMR prescreening, and then were purified with HPLC using MeOH–H2O (75:25, v/v) as eluent to obtain compound 7 (6.2 mg). Compound 10 (11.3 mg) was gained as white powder from the precipitation of Fr. 4. Fr. 21 was subjected to a reversed-phase silica gel column chromatography using a step gradient of MeOH–H2O (50:50–100:0, v/v) as eluent to furnish four sub-fractions Fr. 21-1–21-4, and then Fr. 21-3 was purified by Sephadex LH-20 gel column chromatography eluted with 100% methanol to give compound 12 (2.9 mg). Fr. 25 was purified on RP-HPLC using MeOH–H2O (80:20, v/v) as eluent to obtain compound 13 (7.2 mg).
Aspercarboline A (1). Brown oil; [α]25D = +1.5 (c 0.65, MeOH). UV (MeOH) λmax nm (logε): 235 (3.16). ECD (0.08 mM, MeOH) λmax (Δε) 317 (+0.57), 210 (−3.14) nm. IR νmax: 3326, 2970, 2934, 1626, 1537, 1448, 1405, 1252, 1084, 999 cm−1. 1H and 13C NMR data see Table 1. HR-ESI-MS at m/z 243.11268 [M + H]+ (calcd for C14H15O2N2, 243.11280).
Aspercarboline B (2). White powder; [α]25D = +60.0 (c 0.25, MeOH). UV (MeOH) λmax nm (logε): 216 (3.12). IR νmax: 3390, 2961, 2918, 1716, 1667, 1624, 1323, 1266, 1206, 1064, 1018, 805, 741 cm−1. 1H and 13C NMR data see Table 1. HR-ESI-MS at m/z 283.10891 [M + H]+ (calcd for C16H15N2O3, 283.10772).
Aspercarboline C (3). White powder; [α]25D = +84.6 (c 0.20, MeOH). UV (MeOH) λmax nm (logε): 235 (3.12). ECD (0.07 mM, MeOH) λmax (Δε) 290 (+3.99), 212 (−3.71) nm. IR νmax: 3320, 1672, 1624, 1432, 1323, 1203, 1124, 1011, 937, 839, 738 cm−1. 1H and 13C NMR data see Table 1. HR-ESI-MS peak at m/z 285.12302 [M + H]+ (calcd for C16H17N2O3, 285.12337).
Asperdione A (13). Colorless crystals; [α]25D = +10.0 (c 0.60, MeOH). UV (MeOH) λmax nm (logε): 318 (4.51). ECD (0.07 mM, MeOH) λmax (Δε) 238 (+2.96), 316 (−0.65) nm. IR νmax: 3299, 2973, 2938, 1662, 1630, 1434, 1384, 1337, 1305, 1280, 1198, 1034, 949, 902, 788 cm−1. 1H and 13C NMR data see Table 2. HR-ESI-MS peak at m/z 275.15079 [M + H]+ (calcd for C14H19N4O2, 275.15025).
Absolute configurations of compounds 1, 3 and 13
The absolute configurations of compounds 1, 3 and 13 were determined by experimental and calculated ECD spectra. The calculated ECD spectra of compounds 1, 3 and 13 were obtained using the TDDFT method at the PBE1PBE/6-311++G (d, p) level in methanol. Additionally, the ECD spectra were generated from dipole-length dipolar and rotational strengths using a Gaussian band shape with a 0.3 eV exponential half-width and elaborated using the SpecDis program.
X-ray crystal data of compound 13
Crystals of 13 were obtained as colorless crystals from a MeOH solution. Crystal X-ray diffraction data for 13 were collected on a Bruker SMART APEX CCD using graphite-monochromated Cu Kα radiation (λ = 1.54178 Å) at room temperature. Its structure was solved by direct methods (SHELXS-97), expanded using difference Fourier techniques, and a = 27.726(2) Å, b = 14.7975(12) Å, c = 7.8558(11) Å, V = 3223.0(6) Å3, Z = 64, d = 1.419 g cm−3. The data were processed using CrysAlis. The thermal ellipsoids are plotted at 30% probability level. The water molecule O1W resides on a 2-fold axis. The carbon atoms C-16 and C-17 exhibit two-fold disorder and only the most probable orientations are shown in the figure for clarity. Crystallographic data of 13 has been deposited with the Cambridge Crystallographic Data Centre (CCDC number 1969177).
Cytotoxicity assay
Human nasopharyngeal carcinoma cell lines CNE1, CNE2, SUNE1 and HONE1 and hepatocellular carcinoma cell lines HepG2 and QGY7701 were generously provided by Professor Mu-Sheng Zeng (Cancer Center, Sun Yat-sen University) and conserved in State Key Laboratory of Oncology in South China, Cancer Center, Sun Yat-sen University, Guangzhou, P. R. China. The culture medium was DMEM (Gibco) with 10% fetal bovine serum (FBS) (Gibco). All of the cells were authenticated using short tandem repeat profiling, tested for Mycoplasma contamination, and cultured for less than 2 months.
The in vitro cytotoxicities of 1–13 were determined by means of the colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. The tested human cancer cell lines at a density of 1.5 × 107 cells per L and were seeded in 96-well plates. Compounds at different concentrations (1.5625–100 μM) were added to the cultures and then 0.5 mg mL−1 MTT was added to the culture medium after 72 h. After incubated for 4 h at 37 °C, the supernatant of the plates was removed. The formazan crystals were dissolved in DMSO (150 μL), and the absorbance was measured at 570 nm by using a microplate reader (Bio-Rad, Hercules, CA, USA), and the data were analyzed with the CalcuSystem software package.
Conclusions
In summary, 13 alkaloids including three new β-carboline alkaloids aspercarbolines A–C (1–3), one new piperazinedione, asperdione A (13) and nine known alkaloid compounds were obtained from a geoduck Panopea abbreviate associated fungus Aspergillus sp. XBB-4 by amino acid-directed strategy. The optimization of culture conditions presented the importance of amino acids and salt in inducing the production of alkaloids especially β-carboline alkaloids from fungus. Aspercarboline C (3) and asperdione A (13) exhibited significant cytotoxic activity against human nasopharyngeal carcinoma cell lines CNE1, CNE2, HONE1 and SUNE1, human hepatocellular carcinoma cell lines hepG2 and QGY7701. The biosynthetic pathways of compounds 1–3 were also proposed. This work provides a feasible way to explore the potential of fungal biosynthesis which can enrich natural product compound library and provide lead compounds for potential drugs development.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financed by the National Natural Science Foundation of China (No. 81872795), Guangdong Provincial Science and Technology Research Program (No. 2016A020222004 and 2019B070702003), Natural Science Foundation of Guangdong Province (No. 2018A030313157), and the National Science and Technology Major Project for New Drug Innovation and Development (No. 2017ZX09305010).
References
- E. K. Lund, Food Chem., 2013, 140, 413–420 CrossRef CAS PubMed.
- L. L. Fu, C. Wang, Y. Zhu and Y. B. Wang, Trends Food Sci. Technol., 2019, 88, 80–92 CrossRef CAS.
- W. M. Laura, C. R. Malco, P. K. Joseph, L. Mark, F. P. Margaret, S. Mary and L. K. Alan, Innovative Food Sci. Emerging Technol., 2005, 6, 257–270 CrossRef.
- P. C. Zaidman and E. Morsan, Fish. Res., 2015, 172, 423–431 CrossRef.
- M. Goman, B. L. Ingram and A. Strom, Quat. Int., 2008, 188, 117–125 CrossRef.
- W. Liu, C. M. Pearce, R. S. McKinley and I. P. Forster, Aquaculture, 2016, 452, 326–341 CrossRef CAS.
- M. K. Liu, C. B. Wang, L. F. Kong and Q. Li, Mar. Sci., 2013, 37, 103–106 Search PubMed.
- H. J. Li, Y. L. Xie, Z. L. Xie, Y. Chen, C. K. Lam and W. J. Lan, Mar. Drugs, 2012, 10, 627–638 CrossRef CAS PubMed.
- L. H. Huang, Y. X. Chen, J. C. Yu, J. Yuan, H. J. Li, W. Z. Ma, R. K. Watanapokasin, C. Hu, S. I. Niaz, D. P. Yang and W. J. Lan, Org. Lett., 2017, 19, 4888–4891 CrossRef CAS PubMed.
- M. X. Yuan, Y. Qiu, Y. Q. Ran, G. K. Feng, R. Deng, X. F. Zhu, W. J. Lan and H. J. Li, Mar. Drugs, 2019, 17, 77 CrossRef CAS PubMed.
- E. Couladouros, A. Magos and A. Strongilos, Greek Patent, GR 1004602 B1 20040618, 2004.
- P. Phainuphong, V. Rukachaisirikul, S. Saithong, S. Phongpaichit, J. Sakayaroj, C. Srimaroeng, A. Ontawong, A. Duangjai, P. Muangnil and C. Muanprasat, Bioorg. Med. Chem., 2018, 26, 4502–4508 CrossRef CAS PubMed.
- N. Aimi, M. Kitajima, N. Oya, W. Nitta, H. Takayama, S. Sakai, I. Kostenyuk, Y. Gleba, S. Endress and J. Stockigt, Chem. Pharm. Bull., 1996, 44, 1637–1639 CrossRef CAS.
- H. Zhao, Q. L. Wang, S. B. Hou and G. Chen, J. Nat. Prod., 2019, 33, 2359–2362 CAS.
- Y. Li, M. Zhao and K. L. Parkin, J. Agric. Food Chem., 2011, 59, 2332–2340 CrossRef CAS PubMed.
- S. Mohamed, S. Dirk, A. S. Khaled, H. Elisabeth, G. W. Iris, W. D. Irene and L. Hartmut, Rev. Latinoam. Quim., 2007, 35, 58–67 Search PubMed.
- M. L. Yang, P. C. Kuo, T. L. Hwang and T. S. Wu, J. Nat. Prod., 2011, 74, 1996–2000 CrossRef CAS PubMed.
- N. Yang, Y. H. Shi, A. Z. Xiong, Y. Zhou, L. H. Gu, R. Wang, L. Yang and Z. T. Wang, J. Sep. Sci., 2018, 41, 3014–3021 CrossRef CAS PubMed.
- D. S. Lee, S. H. Eom, S. Y. Jeong, H. J. Shin, J. Y. Je, E. W. Lee, Y. H. Chung, Y. M. Kim, C. K. Kang and M. S. Lee, Environ. Toxicol. Pharmacol., 2013, 35, 171–177 CrossRef CAS PubMed.
- Y. L. Cui, N. Shen, J. Dang, L. J. Mei, Y. D. Tao and Z. G. Liu, J. Sep. Sci., 2017, 40, 2895–2905 CrossRef CAS PubMed.
- W. H. Jiao, H. Gao, C. Y. Li, G. X. Zhou, S. Kitanaka, A. Ohmurae and X. S. Yao, Magn. Reson. Chem., 2010, 48, 490–495 CAS.
- B. Proksa, D. Uhrin, M. Sturdikova and J. Fuska, Acta Biotechnol., 1990, 10, 337–340 CrossRef CAS.
- S. Diem and M. Herderich, J. Agric. Food Chem., 2001, 49, 5473–5478 CrossRef CAS PubMed.
- J. J. Maresh, L. A. Giddings, A. Friedrich, E. A. Loris, S. Panjikar, B. L. Trout, J. Stockigt, B. Peters and S. E. O'Connor, J. Am. Chem. Soc., 2008, 130, 710–723 CrossRef CAS PubMed.
- Q. Chen, S. F. Zhang and Y. C. Xie, J. Biotechnol., 2018, 281, 137–143 CrossRef CAS PubMed.
- Q. Chen, C. T. Ji, Y. X. Song, H. B. Huang, J. Y. Ma, X. P. Tian and J. H. Ju, Angew. Chem., Int. Ed., 2013, 52, 9980–9984 CrossRef CAS PubMed.
- W. Yu. Zhao, W. Y. Zhou, J. J. Chen, G. D. Yao, B. Lin, X. B. Wang, X. X. Huang and S. J. Song, Phytochemistry, 2019, 159, 39–45 CrossRef CAS PubMed.
- H. J. Li, W. J. Lan, C. K. Lam, F. Yang and X. F. Zhu, Hirsutane sesquiterpenoids from the marine-derived fungus Chondrostereum sp., Chem. Biodivers., 2011, 8, 317–324 CrossRef CAS PubMed.
- F. Yang, W. D. Chen, R. Deng, H. Zhang, J. Tang, K. W. Wu, D. D. Li, G. K. Feng, W. J. Lan, H. J. Li, X. F. Zhu and A. Hirsutanol, A novel sesquiterpene compound from fungus Chondrostereum sp., induces apoptosis and inhibits tumor growth through mitochondrial-independent ROS production: hirsutanol A inhibits tumor growth through ROS production, J. Transl. Med., 2013, 11, 32 CrossRef CAS PubMed.
- F. Yang, W. D. Chen, R. Deng, D. D. Li, K. W. Wu, G. K. Feng, H. J. Li and X. F. Zhu, Hirsutanol A induces apoptosis and autophagy via reactive oxygen species accumulation in breast cancer MCF-7 cells, J. Pharmacol. Sci., 2012, 119, 214–220 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: HRESIMS, 1D and 2D NMR spectra of compounds 1–3 and 13; 1D NMR spectra of compounds 4–12; ECD calculation of compounds 1, 3 and 13; crystallographic data of compound 13; composition of each nutrient source in eight culture media and HPLC spectra of extracts from eight culture media. CCDC 1969177. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra10306f |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Chi-Keung Lama,
Gong-Kan Fengd,
Rong Dengd,
Xiao-Feng Zhud,
Hou-Jin Li
c,
Chi-Keung Lama,
Gong-Kan Fengd,
Rong Dengd,
Xiao-Feng Zhud,
Hou-Jin Li