
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSaccharomonosporine A inspiration; synthesis of potent analogues as potential PIM kinase inhibitors†
Asmaa M. AboulMagd a,
Hossam M. Hassanbc,
Ahmed M. Sayedc,
Usama Ramadan Abdelmohsen*de and
Hamdy M. Abdel-Rahman*af
a,
Hossam M. Hassanbc,
Ahmed M. Sayedc,
Usama Ramadan Abdelmohsen*de and
Hamdy M. Abdel-Rahman*af
aPharmaceutical Chemistry Department, Faculty of Pharmacy, Nahda University, Beni Suef, Egypt. E-mail: hamdy.abdelrahman@nub.edu.eg
bPharmacognosy Department, Faculty of Pharmacy, Beni-Suef University, Beni-Suef, Egypt
cPharmacognosy Department, Faculty of Pharmacy, Nahda University, Beni-Suef, Egypt
dPharmacognosy Department, Faculty of Pharmacy, Minia University, Minia, Egypt. E-mail: usama.ramadan@mu.edu.eg
eDepartment of Pharmacognosy, Faculty of Pharmacy, Deraya University, 7 Universities Zone, 61111 New Minia City, Egypt
fMedicinal Chemistry Department, Faculty of Pharmacy, Assiut University, Assiut 71526, Egypt
First published on 13th February 2020
Abstract
Saccharomonosporine A was recently reported as a natural anti-cancer agent working through inhibition of a Proviral integration site for Moloney murine leukemia virus-1 (PIM-1) kinase. Structural bioisosteres of this natural product were synthesized and tested against PIM kinase enzymes. They showed potent inhibitory activity against all the known PIM kinases (PIM-1, 2 and 3) with IC50 values ranging from 0.22 to 2.46 μM. Compound 5 was the most potent pan-inhibitor with IC50 values of 0.37, 0.41, and 0.3 μM, against PIM-1, 2, 3 respectively. Compounds 4–6 were tested for their cytotoxic activities against 3 cell lines: H1650, HT-29, and HL-60. Compound 5 exhibited significant cytotoxic activity against human colon adenocarcinoma HT-29 and the human promyelocytic leukemia HL-60, with IC50 μM values of 1.4 and 1.7 respectively. Molecular docking and homology modeling studies were carried out to confirm the affinity of these synthesized compounds to the three different PIM kinases. Additionally, a number of in silico predictions, ADME/Tox, were adopted to evaluate their drug-likeness.
1. Introduction
Marine natural product bioprospecting has established a significant number of drug candidates. Cytarabine is one of the already marketed marine natural agents. However, most of them are still in preclinical or early clinical development.1 Renieramycin M extracted from the blue sponge Xestospongia sp. is the first marine anticancer drug approved by the European Union and by the Food and Drug Administration (FDA) for the treatment of human colon, lung and brain cancer.2 To date, the synthetic analogue of halichondrin B, eribulin mesylate, which is isolated from the Porifera Halichondria okadai is the only antitumor agent derived from marine sponges and was approved by the FDA and European Medicines Agency (EMA) in 2011.3 Furthermore, the recent discovery of numerous taxonomically unique marine-derived actinomycetes,3,4 was associated with frequent isolation of novel bioactive secondary metabolites, and still offers a promising source for the discovery of further unusual natural products with diverse biological activity.5,6 Many of these secondary metabolites are clinically useful anticancer drugs, such as anthracyclines represented in daunomycin, and doxorubicin, bleomycin and actinomycin D derived from peptides pentostatin regarded as antimetabolites, in addition to carzinophilin, and mitomycins.7–9 In 2019, ULDF5 was a new indole derived compound isolated from the metabolic extracts of the marine actinomycetes that is structurally similar to staurosporine and it seemed to exhibit cytotoxic activity against HeLa cell line with IC50 0.075 μg mL−1.10PIM (Provirus Integration site for Moloney murine leukemia virus) kinases are a family of constitutively active serine/threonine kinases that promote cell survival.11 This family comprise of three different isoforms (PIM-1, PIM-2 and PIM-3) that share 60–70% sequence identity in their kinase domains and have very closely related ATP binding site.12–14
PIM kinases are able to suppress apoptosis via direct phosphorylation and inhibition of several pro-apoptotic proteins such as BAD, MAP3K5, FOXO3, so that, they can act as oncogenic survival factors.15 In addition, PIM kinases control several signaling pathways that are responsible for cancer progression, proliferation and drug resistance.16,17 PIM-1 and PIM-2 have been revealed to be over expressed in several hematological and solid tumors,18–20 while PIM-3 is highly explicit in pancreatic cancer and prostate cancer. Meanwhile, several adenocarcinomas have been shown elevated levels of all PIM kinases.21 Thus, one promising strategy for cancer drug development suggested being the inhibition function of the overlapping and compensatory nature of PIM-1, PIM-2, and PIM-3. Many PIM inhibitors particularly PIM-1 inhibitors have been reported and some of them are already in clinical trials.22 The development of selective PIM-2 or PIM-3 inhibitors is challenging due to the high affinity of both PIM-2 and PIM-3 for ATP, and thus, only a small number of PIM-2 kinase inhibitors have been reported,23–25 and to our knowledge, there are no selective PIM-3 inhibitors that have been described before. Recently, three reports have described the synthesis of inhibitors for all three PIM kinases.26–28
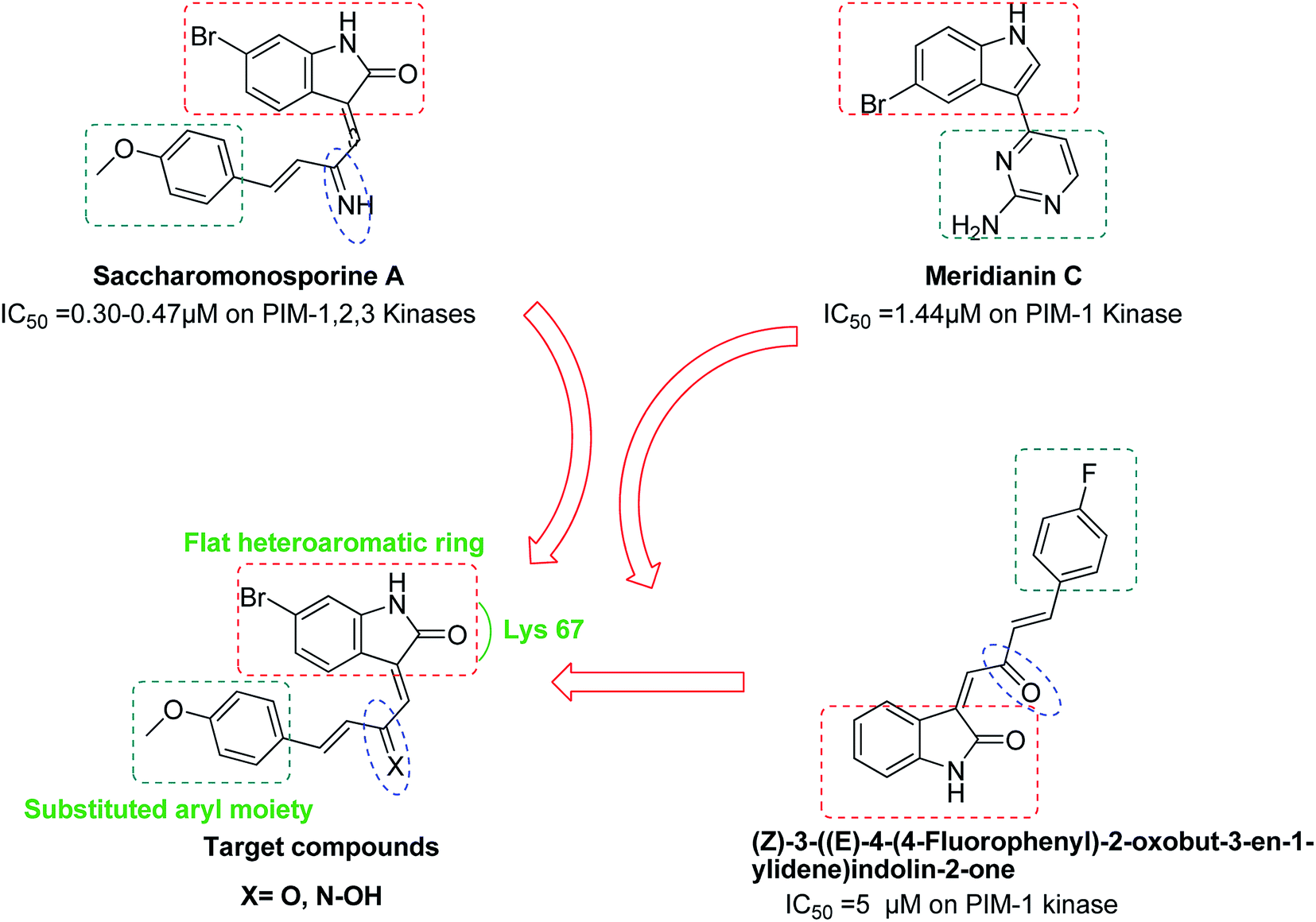
The brominated oxo-indole alkaloid saccharomonosporine A (Fig. 1) was recently isolated from the co-culture of two marine-derived actinomycetes and displayed a potent antiproliferative activity towards a number of leukemia cell lines. Preliminary in vitro and in silico screening highlighted PIM-1 kinase as a possible target for this cytotoxic natural product.29
 | ||
| Fig. 1 Design of the target compounds. | ||
Literature survey showed that the marine alkaloid meridianin C, 4-(5-bromo-1H-indol-3-yl)pyrimidin-2-amine, has an inhibitory activity with IC50 value of 1.44 μM against PIM-1 kinase enzyme.30 Lee et al. reported 3,5-bis(aminopyrimidinyl)indole derivatives at which aminopyrimidine was attached to 5-position of indole to provide meridianin C via two interactions with the side chains of both Lys 67 and Glu 89 of PIM-1.31
In 2015, a series of (Z)-3-functionalized oxo-indoles analogues to saccharomonosporine A were synthesized and they showed moderate selective inhibitory activity against PIM-1 kinase (IC50 5–10 μM)32 (Fig. 1). On the other hand, the natural (E)-3-functionalized oxo-indole, saccharomonosporine A was found to be more potent PIM-1 kinase inhibitor (IC50 0.3 μM), indicating that the (E)-3-functionalized oxo-indole scaffold of saccharomonosporine A is a promising lead for further development. These findings along with the quantity limitation of the naturally produced alkaloid encouraged us to synthesize a number of bioisosters (4–6) for saccharomonosporine A (Fig. 1). The design of this series based on maintaining the common pharmacophoric features shared by PIM kinase inhibitors which involve a flat heteroaromatic ring system to better occupy the ATP binding region of the kinase, a hydrogen bond acceptor represented by carbonyl moiety of indole ring that usually form hydrogen bonds with Lys 67 residues, and a substituted terminal aryl moiety to enhance the potency through possible additional interaction with the enzyme. All synthesized compounds were tested against all PIM kinases and three cancer cell lines namely lung adenocarcinoma H1650, human colon adenocarcinoma HT-29, the human promyelocytic leukemia HL-60, and an ADME/Tox study was also carried out for them.
2. Results and discussion
2.1. Chemistry
The synthetic routs of the proposed compounds are outlined in Scheme 1. Our initial goal was to prepare(Z)-6-bromo-3-((2E,3E)-2-(hydroxyimino)-4-(4-methoxyphenyl)but-3-enylidene)indolin-2-one (6). This was synthesized through the addition of benzylidene acetones with isatins in presence of catalytic amount of L-arginine at which the final hydroxyl product 4 was separated in a good yield. Compound 4 showed 1H-NMR spectra indicated the products exists as the (E) stereoisomer (J = 16 Hz). Then, targeted compound 5 was obtained via the dehydration reaction of derivative 4 in the presence of CCl4 and PPh3 in moderate yield. The mass spectrum of the latter compound showed molecular ion peaks verified its molecular weight, and generated ionic fragments that confirmed its structure. In addition, the IR data exhibited absorption bands at 3395 (NH) and 1712, 1662 (2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). According to previous reports,33,34 H-4 (δH 8.31 ppm) and H-8 (δH 7.45 ppm) chemical shifts suggest the E configuration at C-3/C-8 double bond. Furthermore, both E and Z conformers' energies were minimized and the distance between H-4, H-10, H-13 and H-14 were determined that were found to be less than 5 Å in E form and more in Z form, from this points the presence of 1H–1H NOESY correlations (cross-peaks) between H-4 and H-10, H-13 and H-14 together with their absence between H-4 and H-8 (Fig. 2, S1 and S2†) confirm the suggestion of E-form. Correlation plots between experimental and calculated chemical shifts (Fig. 3 and S3†) show a higher correlation in the case of E isomer, with R2 of 0.9607 (R2 = 0.901, for Z isomer) (Fig. S4†). Additionally, the calculated relative energy of compound 5 (40.1537 kcal mol−1) was lower if it was in the E configuration. From previous observations and theoretical calculations, the E configuration at C-3/C-8 double bond is unambiguously determined. Moreover, the final oxime derivative 6 was afforded by reacting of compound 5 with NH2OH·HCl in a high yield. This was confirmed by 1H-NMR spectra that indicate the appearance of singlet proton for oxime hydroxyl group at δ 10.41 ppm.
O). According to previous reports,33,34 H-4 (δH 8.31 ppm) and H-8 (δH 7.45 ppm) chemical shifts suggest the E configuration at C-3/C-8 double bond. Furthermore, both E and Z conformers' energies were minimized and the distance between H-4, H-10, H-13 and H-14 were determined that were found to be less than 5 Å in E form and more in Z form, from this points the presence of 1H–1H NOESY correlations (cross-peaks) between H-4 and H-10, H-13 and H-14 together with their absence between H-4 and H-8 (Fig. 2, S1 and S2†) confirm the suggestion of E-form. Correlation plots between experimental and calculated chemical shifts (Fig. 3 and S3†) show a higher correlation in the case of E isomer, with R2 of 0.9607 (R2 = 0.901, for Z isomer) (Fig. S4†). Additionally, the calculated relative energy of compound 5 (40.1537 kcal mol−1) was lower if it was in the E configuration. From previous observations and theoretical calculations, the E configuration at C-3/C-8 double bond is unambiguously determined. Moreover, the final oxime derivative 6 was afforded by reacting of compound 5 with NH2OH·HCl in a high yield. This was confirmed by 1H-NMR spectra that indicate the appearance of singlet proton for oxime hydroxyl group at δ 10.41 ppm.
 | ||
| Scheme 1 Reagents and conditions: (a) CH3COCH3, H2O, 0.5% NaOH, r.t. overnight; (b) 10% L-Arg, MeOH, r.t., overnight; (c) CCl4, PPh3, THF, reflux, overnight, (d) NH2OH·HCl, EtOH, pyridine, reflux, 6 h. | ||
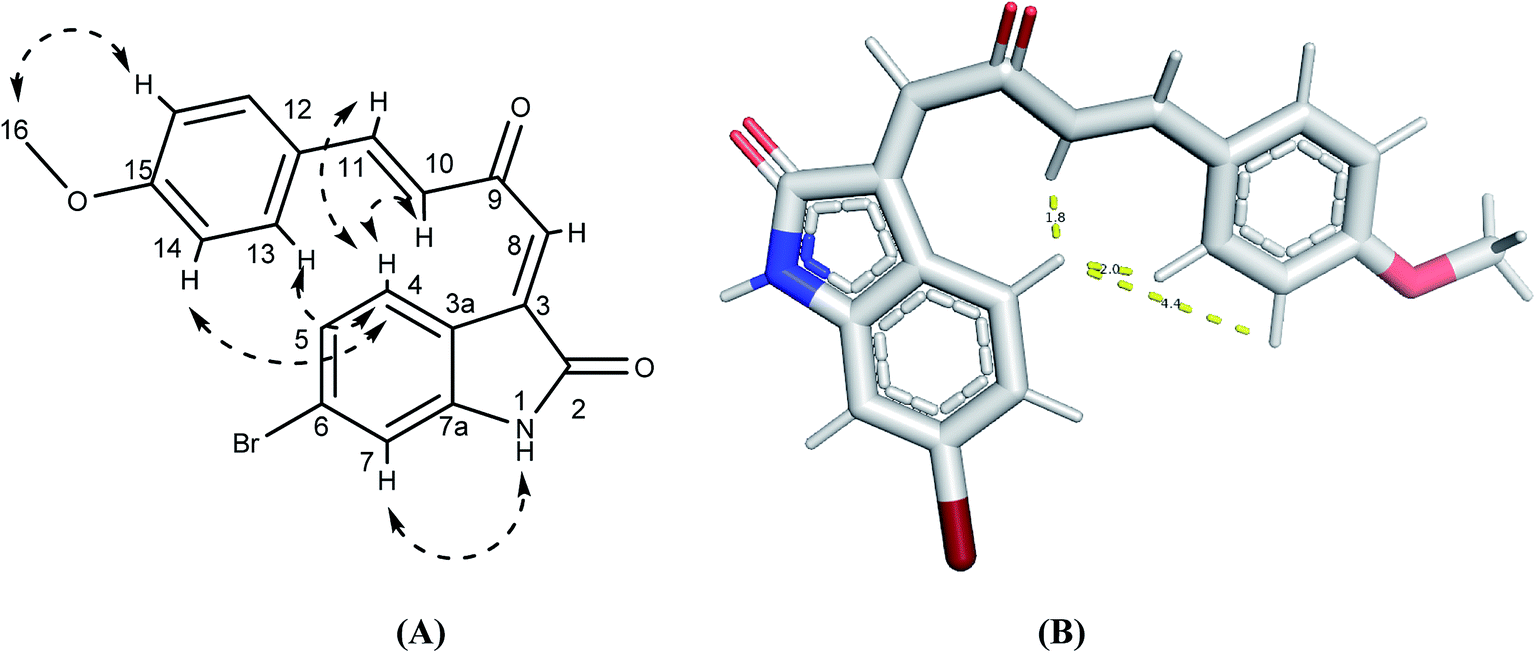 | ||
| Fig. 2 (A and B) 1H–1H NOESY correlations of compound 5. | ||
 | ||
| Fig. 3 Theoretical calculations of 1H NMR chemical shifts of compound 5 in E isomer. | ||
2.2. PIM kinases enzyme inhibition
All the synthesized compounds 4–6 as well as the natural Sach A were tested for their ability to inhibit all PIM kinases (1–3) using quercetin as a reference compound, and the results in terms of IC50 were shown in Table 1 and represented graphically in Fig. 4.| Compound number | IC50 (μM) | ||
|---|---|---|---|
| PIM-1 | PIM-2 | PIM-3 | |
| 4 | 1.62 ± 0.02 | 2.46 ± 0.025 | 1.74 ± 0.022 |
| 5 | 0.37 ± 0.011 | 0.41 ± 0.012 | 0.3 ± 0.026 |
| 6 | 0.47 ± 0.013 | 0.22 ± 0.01 | 0.36 ± 0.012 |
| Sach A | 0.3 ± 0.02 | 0.51 ± 0.011 | 0.47 ± 0.012 |
| Querecitin | 0.86 ± 0.01 | 0.64 ± 0.01 | 0.83 ± 0.013 |
 | ||
| Fig. 4 IC50 in μM of compounds 4–6 and Sach A on all PIM kinases (1–3). | ||
All the tested compounds showed potent inhibitory activity against the three PIM kinase isoforms (IC50 0.22–2.46 μM). Both compounds 5 and 6 showed comparable inhibitory activity to Sach A (IC50 0.22–0.47 μM), with an overall improvement, particularly against both PIM-2 and PIM-3. Compound 4 was the least active one (IC50 1.62–2.46 μM), indicating that a planer structure is an important feature for the highly potent inhibitory activity. The previous report32 on similar C3-functionalized oxo-indole derivatives were significantly less active in comparison to our studied ones, suggesting that an E configuration around the double bond at C-3 is also an essential structural feature to obtain potent inhibitors. Based on these results, compound 5 was selected as a lead compound for further study because of its best potency against PIM kinases.
2.3. Antiproliferative activity
Three cancer cell lines namely lung adenocarcinoma H1650, the human promyelocytic leukemia HL-60 and the human colon adenocarcinoma HT-29 cell lines were chosen to evaluate the antiproliferative activity of the three synthesized compounds (Table 2). Compounds 5 and 6 displayed potent antiproliferative activity against HL-60 and HT-29 cells (IC50 1.4, 1.7 and 3.3, 3.9 μM, respectively) compared to the reported values of cytotoxic activity of Sach A.29 While all the synthesized compounds don't show any activity against lung adenocarcinoma H1650 which came in accordance with the previously reported anti-proliferative activity of Sach A. These results adhere with the PIM kinase enzyme inhibition results.2.4. Docking study
For better understanding of the potency of compound 5 and guiding further SAR studies, we evaluated the interaction between all the synthesized compounds and PIM kinases (PIM-1, PIM-2 and PIM-3). The binding affinity score of the poses in addition to its orientation into the active site is selected in a manner similar to the co-crystallized ligands orientation; and keeping the binding interactions, especially hydrogen bond formation and hydrophobic groups. The estimated binding affinity of the co-crystallized ligand of PIM-1 kinase (3UMW) was −8.011 kcal mol−1 with of complex hydrogen network and also a number of amino acids including, Glu 121, Lys 67, Glu 171 and Asp 128, thereby giving an evidence about the importance of hydrogen bond formation for effective enzyme binding. Modeling studies suggest that E isomer of compound 5 that showed IC50 0.37 μM binds to the active site of PIM-1 kinase with a binding affinity −6.248 kcal mol−1 via strong hydrogen-bonding interaction of its carbonyl group with Lys 67 amino acid (Fig. 5). In addition, two hydrophobic interactions with residues Val 52 were observed. As for the synthesized derivative 4 with a binding affinity −4.984 kcal mol−1 with one hydrophobic interaction with Val 52 that is consistent with the higher IC50 value (IC50 1.62 μM). In case of PIM-2 kinase (PDB code 4X7Q), compound 5 with binding affinity score −5.854 kcal mol−1 forms hydrophobic interactions with Val 46 and Ile 141 with its indole ring in addition to another interaction of methoxy substituted phenyl ring with Leu 38. Regarding PIM-3 kinase binding site residues, Ser 367 made hydrogen bond interaction with NH group of indole moiety of derivative 5 in addition to two hydrophobic interactions of both phenyl rings via Thr 371 and Arg 312 amino acids. Furthermore, the results of docking were consistent with IC50 value of compound 4, which forms one hydrophobic interaction with amino acids residues Pro 204 (Fig. 5).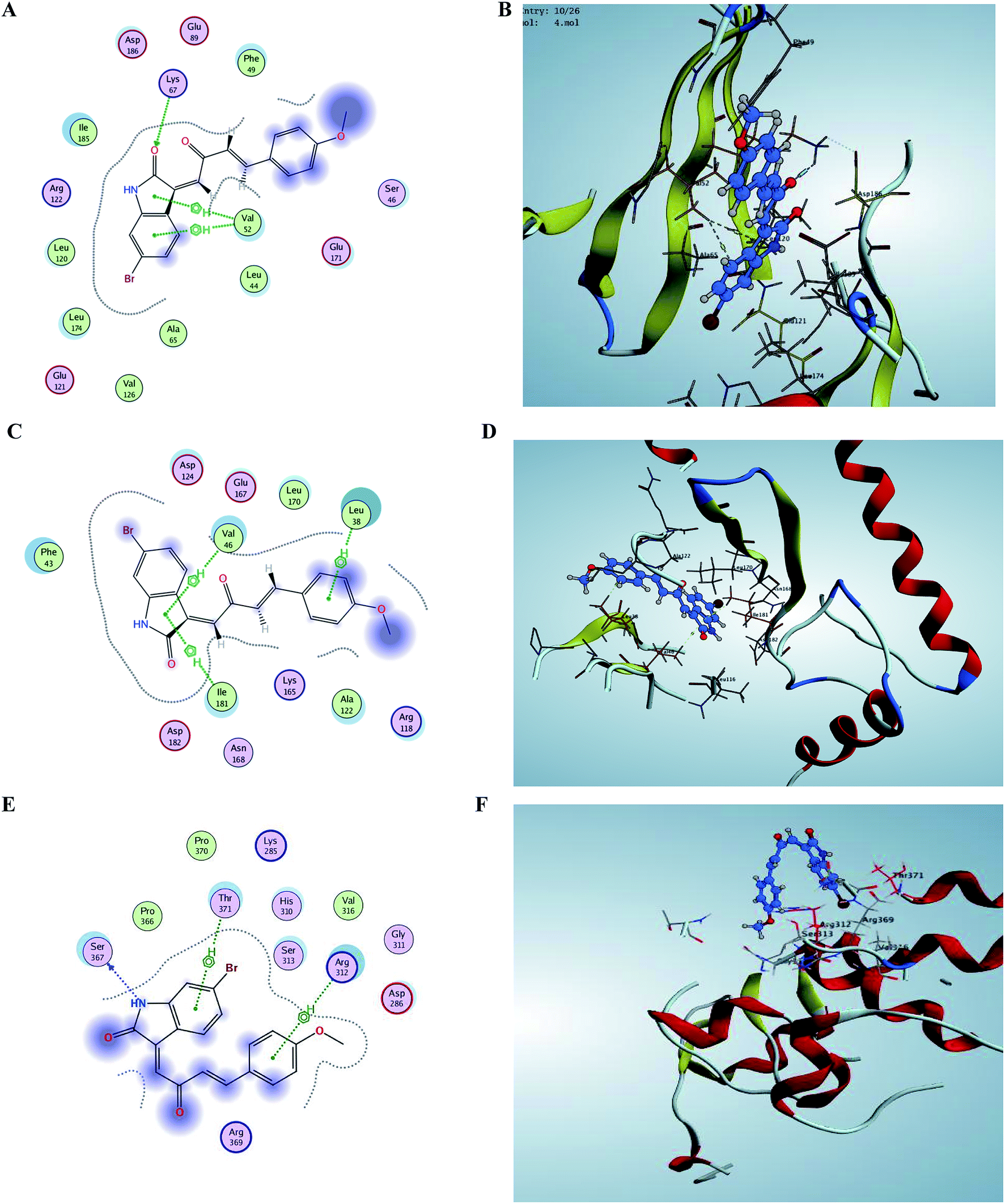 | ||
| Fig. 5 Comparison between binding sites of compound 5 within crystal structure of PIM-1 (PDB code 3 UMW), PIM-2 (PDB code 4X7Q) and modeled PIM-3. (A and B) active site of PIM-1 shows some hydrogen bonding and hydrophobic interaction with important active site residues. (C and D) Small hydrophobic active site of PIM-2 have Leu 38, Val 46, and Ile 141. All of these residues are conserved in PIM-1. (E and F) Binding site of PIM-3 also contains some hydrophobic residues. | ||
2.5. Homology modeling
3D structure of PIM-3 was obtained by homology model, using Swiss model software (Fig. 6). The sequence identity of 100% with the 3D structure of human PIM-1 protein (PDB code 4JX3) was used as template for homology model generation. Validation of the model was carried out by using QMEAN scoring function, and DFIRE. Additionally, in order to guarantee the correct modeling of the target protein, other tools for local model quality are taken into consideration which are graphical plots of ANOLEA mean force potential, GROMOS empirical force field energy and the neural network-based approach. It was found that both the target and the template indicated the close structural identity with their RMSD values 0.77 Å. This encouraged us to further used this model in this study.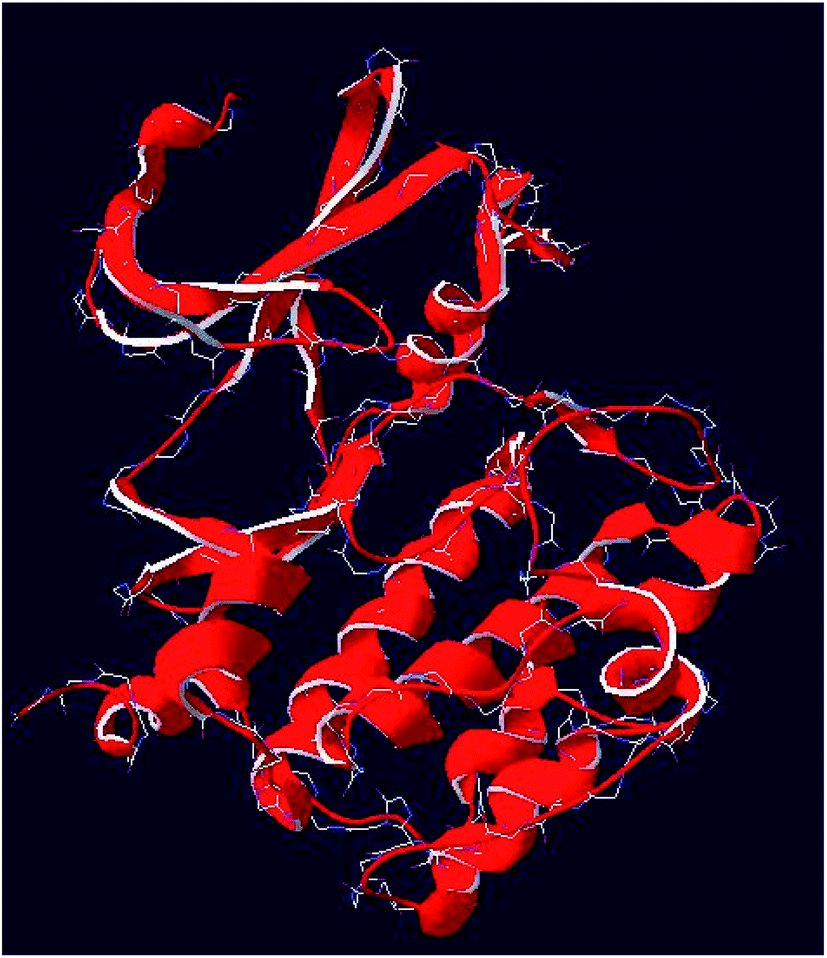 | ||
| Fig. 6 Generated homology model of PIM-3 kinase. | ||
2.6. In silico molecular and ADME properties
In most drug discovery projects sorting out the ADME/T issues is the most challenging part of the project as pharmacokinetic (ADME) and pharmacodynamic (e.g., toxicological) properties (drug-likeness) are of great importance during pre-clinical evaluation to optimize a lead compound into a successful drug candidate, and to minimize attrition rates during clinical trials.A drug-like molecule (DLM) possesses the physicochemical properties that might enable it to become a drug should a disease-modifying receptor be identified. Generally speaking, an orally available molecule that satisfies Lipinski's rule (Lipinski-compliant) and shows a balance between lipophilicity and hydrophilicity would qualify it to be a drug-like molecule.35
An orally active compound should obey both Lipinski's and Veber's rules, as shown from Table 3, compound 5 followed all Lipinski's and Veber's rules. Yet both compounds 4 & 6 obeys all Lipinski's and Veber's rules with one exception have higher ASA_P value than the acceptable level. However, the overall drug-likeness properties are excellent which highlight their potential to pass the drug development process.
| Compound | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (o/w) (<5) P (o/w) (<5) |
MW (<500) | a_acca (≤10) | a_donb (≤5) | B_rotNc (≤10) | ASA_Pd (<140 Å) |
|---|---|---|---|---|---|---|
| a Number of hydrogen-bond acceptors (a_acc).b Number of hydrogen-bond donors (a_don).c Number of rotatable bonds (B_rotN).d Polar surface area (ASA_P). | ||||||
| 4 | 3.248 | 402.244 | 4 | 2 | 5 | 163.022 |
| 5 | 4.229 | 384.229 | 3 | 1 | 4 | 140.683 |
| 6 | 4.475 | 399.244 | 4 | 2 | 4 | 167.762 |
| Sach A | 4.33 | 383.039 | 3 | 2 | 4 | 158.075 |
Moreover, all the target compounds were predicted for pharmacokinetic properties, namely, blood–brain barrier (BBB) penetration, gastrointestinal absorption, solubility, inhibition of CYP2D6, and bioavailability. All the target compounds in addition to saccharomonosporin A showed good BBB penetration, high GIT absorption, moderately water solubility, no inhibition of CYP2D6 except for compound 4, and bioavailability of 0.55 (Table 4).
| Compound | BBBa | GIT absorptionb | Solubilityc | CYP2D6d | Bioavailability scoree |
|---|---|---|---|---|---|
| a Predicts ability of the compound to penetrate the blood brain barrier (BBB) according to the yolk of the boiled egg.b Predicts gastrointestinal absorption according to the white of the boiled egg.c Predicts the solubility of each compound in water. Levels <−10, <−6, <−4, <−2, <0 correspond to insoluble, poorly soluble, moderately soluble, soluble, very soluble, respectively.d Predicts the cytochrome P450, 2D6 inhibition.e Predicts the bioavailability score. | |||||
| 4 | Yes | High | −4.24 | Yes | 0.55 |
| 5 | Yes | High | −4.33 | No | 0.55 |
| 6 | Yes | High | −4.92 | No | 0.55 |
| Sach A | Yes | High | −4.57 | No | 0.55 |
Toxicity screening results of preADMET for the synthesized compounds and Sach A showed mutagenicity against AMES with medium risk of hERG_inhibition. The target derivatives showed negative TA100_NA stain and for the other strains used namely TA1535_10RLl and TA1535_NA. Moreover, the oxime 6 showed no potential rodent carcinogenicity except against rat which the rest of the compounds lacked. Also, only compound 4 has a slight increase in the toxicity value against algeae_at, daphnia_at, medaka_at & minnow_at, Table 5. The results of in silico screening highlights that derivative 4 was the most toxic analogue while both derivatives 5 & 6 have more positive results when compared with the natural analogue Sach A.
| Molecule | 4 | 5 | 6 | Sach A |
|---|---|---|---|---|
| Algae_at | 0.0255 | 0.0215 | 0.015 | 0.016 |
| Ames_test | Mutagen | Mutagen | Mutagen | Mutagen |
| Carcino_Mouse | Positive | Positive | Negative | Positive |
| Carcino_Rat | Negative | Positive | Positive | Positive |
| Daphnia_at | 0.0485 | 0.024 | 0.021 | 0.022 |
| hERG_inhibition | Medium risk | Medium risk | Medium risk | Medium risk |
| Medaka_at | 0.005 | 0.001 | 0.001 | 0.001 |
| Minnow_at | 0.019 | 0.003 | 0.003 | 0.003 |
| TA100_10RLl | Positive | Negative | Positive | Negative |
| TA100_NA | Negative | Negative | Negative | Negative |
| TA1535_10RLl | Negative | Negative | Negative | Negative |
| TA1535_NA | Negative | Negative | Negative | Negative |
3. Experimental
3.1. Chemicals and instrumental analysis
Starting materials and reagents were purchased from Sigma-Aldrich or Acros Organics. Melting points were recorded on Gallen Kamp apparatus and were uncorrected. FT-IR spectra were recorded on a Shimadzu IR 435 spectrophotometer. 1H-NMR spectra were recorded in δ scale given in ppm on a Varian 400 MHz, spectrophotometer. Coupling patterns are described as follows: s, singlet; d, doublet, dd, doubled doublet; t, triplet; m, multiplet. J describes a coupling constant. The coupling constants were rounded off to one decimal place. MS spectra mass were recorded on Hewlett Packard 5988 spectrometer (70 eV). Elemental analysis was performed at the Microanalytical Center, Al-Azhar University.6-Bromoindoline-2,3-dione (3), 6-bromoisatin. By adjusting the temperature at 35 °C, chloral hydrate (5 g, 0.03 mol) and Na2SO4 (35 g) were dissolved in water (70 mL) and warmed in a 500 mL beaker. This was followed by the addition of a warm solution of the 3-bromo aniline (0.276 mol) in water (20 mL), and a concentrated HCl (30 mL) in which a white precipitate of the amine sulfate was formed. After that, hydroxylamine hydrochloride (6.1 g) dissolved in water (27.5 mL) was warmed at the same temperature and added to the previous mixture. The mixture was stirred and heated on a hot plate at 80–90 °C for 2 h and then allowed to cool for 1 h, by which time the temperature had fallen to 50 °C and filtered. 100 mL H2O was added to the pale cream product with stirring so that the whole mixture washed to filter. Drying overnight at 40 °C gave the isonitrosoacetanilide intermediate (Scheme 1).
The dry isonitrosoacetanilide was added portion-wise over 30 min to sulfuric acid (100 mL) in a 500 mL beaker after warming on a hot plate to 60 °C with strong stirring such that the temperature did not exceed 65 °C. The temperature was raised to 80 °C and the mixture was heated for 15 min, and then cooled to 70 °C. The solution was poured on to crushed ice (500 mL) and left to stand for 1 h before filtering the orange-red precipitate. The product was further washed by water (40 mL) with stirring and filtered to give a mixture of 6-bromoisatin and 4-bromoisatin. The crude product was dissolved in a 10% aqueous solution of NaOH (20 mL) at 60 °C and then acidified with acetic acid (6 mL). After standing 0.5 h and cooling to 35 °C, the 4-bromoisatin precipitate was filtered and washed with water (5 mL). A concentrated HCl solution (6 mL) was used for acidification of the combined filtrate and washings in which the precipitated 6-bromoisatin was filtered off and washed with water (5 mL) after standing for 2 h at 5 °C to yield 49% as reported.
O); MS (Mw: 402.24): m/z 402.99 (M+, 7.10%), 383.09 (6.84%), 356.05 (76.65%), 63.26 (100%); anal. calcd for C19H16BrNO4: C, 56.73; H, 4.01; N, 3.84; found: C, 57.01; H, 4.23; N, 3.69.O); MS (Mw: 383.02): 385.08 (M + 2, 100%), 383.08 (M+, 92.65%), 356.07 (48.56%); anal. calcd for C19H14BrNO3: C, 59.39; H, 3.67; N, 3.65; found: C, 59.56; H, 3.73; N, 3.91 (S5–S7†).O); MS (Mw: 399.24): m/z 399.26 (M+, 33.26%), 401.67 (M + 2, 48.32%), 310.82 (59.52%), 71.42 (100%); anal. calcd for C19H15BrN2O3: C, 57.16; H, 3.79; N, 7.05; found: C, 57.34; H, 3.68; N, 7.30.3.2. Biological evaluation
Calculation of [P33]-ATP specific activity (SA) (cpm pmol−1)
| Specific activity (SA) = cpm for 5 μL [33P]-ATP/pmoles of ATP (in 5 μL of a 250 μM ATP stock solution, i.e. 1250 pmol). |
Kinase specific activity (SA) (pmol min−1 μg−1 or mmol min−1 mg−1)
| Corrected cpm from reaction/[(SA of 33P-ATP) × (reaction time in min) × (enzyme amount in μg or mg)] × [(reaction volume)/(spot volume)]. |
P), and number of rotatable bonds (B_rotN), polar surface area (ASA_P).4. Conclusion
In this study, analogues for saccharomonosporine A were designed and synthesized via a simple straightforward method as PIM kinase inhibitors. All the synthesized derivatives were evaluated for their PIM kinases inhibitory activity, which revealed the significant inhibition of derivative 5 with IC50 of 0.37, 0.41 & 0.30 μM against PIM-1, 2 & 3, respectively. Moreover, the molecular docking of the active analogue revealed its ability to bind into the active site of PIM-1 via hydrogen bond interaction with Lys 67 amino acid and two hydrophobic interactions with residues Val 52. Furthermore, its binding interaction with PIM-2 kinase via Ile 181, Val 46 & Leu 38 was consistent with its significant PIM-2 inhibitory effect. Homology modeling was carried out to obtain 3D structure of PIM-3, which was further used to study the interaction of the designed molecules in its binding site. Finally, ADMET study showed the drug like properties and pharmacokinetics properties for both analogues 5 & 6 with the least toxic prediction values compared to the hydroxylated derivative 4.Conflicts of interest
There are no conflicts to declare.References
- A. Tawfike, R. Abdo, E. Z. Attia, S. Y. Desoukey, D. Hajjar, A. A. Makki, R. Edrada-Ebel, P. Schupp and U. R. Abdelmohsen, New bioactive metabolites from the elicited marine sponge-derived bacterium Actinokineospora spheciospongiae sp. nov, AMB Express, 2019, 9, 12–21 CrossRef PubMed.
- E. M. Gordon, K. K. Sankhala, N. Chawla and S. P. Chawla, Trabectedin for soft tissue sarcoma: current status and future perspectives, Adv. Ther., 2016, 33, 1055–1071 CrossRef CAS PubMed.
- Y. Hirata and D. Uemura, Halichondrins-antitumor polyether macrolides from a marine sponge, Pure Appl. Chem., 1986, 58, 701–710 CAS.
- W. Ibrahim, J. Refaat, E. Z. Attia, D. Hajjar, M. A. Anany, S. Y. Desoukey, M. Fouad, M. S. Kamel, H. Wajant, T. A. M. Gulder and U. R. Abdelmohsen, New cytotoxic cyclic peptide from the marine sponge-associated Nocardiopsis sp. UR67, Mar. Drugs, 2018, 16, 290–303 CrossRef PubMed.
- A. H. Ibrahim, S. Y. Desoukey, M. Fouad, M. S. Kamel, T. A. M. Gulder and U. R. Abdelmohsen, Natural product potential of the genus Nocardiopsis, Mar. Drugs, 2018, 16, 147–159 CrossRef PubMed.
- C. Y. Yim, T. C. Le, T. G. Lee, I. Yang, H. Choi, J. Lee, K. Y. Kang, J. S. Lee, K. M. Lim and S. T. Yee, et al., Saccharomonopyrones A–C, new a-pyrones from a marine sediment-derived bacterium Saccharomonospora sp. CNQ-490, Mar. Drugs, 2017, 15, 239–246 CrossRef PubMed.
- C. Olano, C. M_endez and J. A. Salas, Antitumor compounds from actinomycetes: from gene clusters to new derivatives by combinatorial biosynthesis, Nat. Prod. Rep., 2009, 26, 628–660 RSC.
- C. Olano, C. M_endez and J. A. Salas, Antitumor compounds from marine actinomycetes, Mar. Drugs, 2009, 7, 210–248 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, Natural products as sources of new drugs over the last 25 years, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS PubMed.
- O. F. Davies-Bolorunduro, I. A. Adeleye, M. O. Akinleye and P. G. Wang, Anticancer potential of metabolic compounds from marine actinomycetes isolated from Lagos lagoon sediment, J. Pharm. Anal., 2019, 9, 201–208 CrossRef PubMed.
- N. Yi-Lei, W. Yun-Dan, W. Chuan-Xi, L. Ru, X. Yang and F. Dong-Sheng, et al., Compounds from marine-derived Verrucosispora sp. FIM06054 and their potential antitumour activities, Nat. Prod. Res., 2014, 28, 2134–2139 CrossRef PubMed.
- M. C. Nawijn, A. Alendar and A. Berns, For better or for worse: the role of Pim oncogenes in tumorigenesis, Nat. Rev. Cancer, 2011, 11, 23–34 CrossRef CAS PubMed.
- D. Drygin, M. Haddach, F. Pierre and D. M. Ryckman, Potential use of selective and nonselective pim kinase inhibitors for cancer therapy, J. Med. Chem., 2012, 55, 8199–8208 CrossRef CAS PubMed.
- Y. Alvarado, F. J. Giles and R. T. Swords, The PIM kinases in hematological cancers, Expert Rev. Hematol., 2012, 5, 81–96 CrossRef CAS PubMed.
- H. Mikkers, M. Nawijn, J. Allen, C. Brouwers, E. Verhoeven and J. Jonkers, et al., Mice deficient for all PIM kinases display reduced body size and impaired responses to hematopoietic growth factors, Mol. Cell. Biol., 2004, 24, 6104–6115 CrossRef CAS PubMed.
- A. L. Merke, E. Meggers and M. Ocker, PIM1 kinase as a target for cancer therapy, Expert Opin. Invest. Drugs, 2012, 21, 425–436 CrossRef PubMed.
- P. D. Garcia, J. L. Langowski, Y. Wang, M. Chen, J. Castillo and C. Fanton, et al., Pan-PIM kinase inhibition provides a novel therapy for treating hematologic cancers, Clin. Cancer Res., 2014, 20, 1834–1845 CrossRef CAS PubMed.
- S. Kapoor, K. Natarajan, P. R. Baldwin, K. A. Doshi, R. G. Lapidus and T. J. Mathias, et al., Concurrent inhibition of pim and FLT3 kinases enhances apoptosis of FLT3-ITD acute myeloid leukemia cells through increased Mcl-1 proteasomal degradation, Clin. Cancer Res., 2018, 24, 234–247 CrossRef CAS PubMed.
- B. Claude, P. James, T. Elina, G. Alexandre, C. Mark, B. Matthieu, E. Paul, M. Sachin, J. John, F. Luke, P. Vinod, H. Andrew, L. Mikhail, W. Anlai, S. Frank and A. H. Shih-Min, Discovery of N-substituted 7-azaindoles as Pan-PIM kinases inhibitors – lead optimization – part III, Bioorg. Med. Chem. Lett., 2019, 29, 491–495 CrossRef PubMed.
- B. H. Jiang, G. Jiang and J. Z. Zheng, et al., Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1, Cell Growth, 2001, 12, 363–369 CAS.
- U. Weirauch, N. Beckmann, M. Thomas, A. Grünweller, K. Huber and F. Bracher, et al., Functional role and therapeutic potential of the pim-1 kinase in colon carcinoma, Neoplasia, 2013, 15, 783–794 CrossRef CAS PubMed.
- Z. Xinning, S. Mengqiu, K. K. Joydeb, L. Mee-Hyun and L. Zhen-Zhen, PIM Kinase as an Executional Target in Cancer, J. Cancer Prev., 2018, 23, 109–116 CrossRef PubMed.
- Y. B. Fan, M. Huang, Y. Cao, P. Gong, W. B. Liu and S. Y. Jin, et al., Usnic acid is a novel Pim-1 inhibitor with the abilities of inhibiting growth and inducing apoptosis in human myeloid leukemia cells, RSC Adv., 2016, 6, 24091–24096 RSC.
- V. Munugalavadla, L. Berry and Y. H. Chen, A selective PIM kinase inhibitor is highly active in multiple myeloma: mechanism of action and signal transduction studies, Blood, 2010, 116, 1662–1663 CrossRef.
- L. S. Chen, S. Redkar, D. Bearss, W. G. Wierda and V. Gandhi, Pim kinase inhibitor, SGI-1776, induces apoptosis in chronic lymphocytic leukemia cells, Blood, 2009, 114, 4150–4157 CrossRef CAS PubMed.
- K. T. Kim, M. Levis and D. Small, Constitutively activated FLT3 phosphorylates BAD partially through pim-1, Br. J. Haematol., 2006, 134, 500–509 CrossRef CAS PubMed.
- K. T. Kim, K. Baird, J. Y. Ahn, P. Meltzer, M. Lilly, M. Levis and D. Small, Pim-1 is up-regulated by constitutively activated FLT3 and plays a role in FLT3-mediated cell survival, Blood, 2005, 105, 1759–1767 CrossRef CAS PubMed.
- M. Haddach, J. Michaux, M. K. Schwaebe, F. Pierre, S. E. O'Brien, C. Borsan and A. Siddiqui-Jain, Discovery of CX-6258. A potent, selective, and orally efficacious pan-Pim kinases inhibitor, ACS Med. Chem. Lett., 2011, 3, 135–139 CrossRef PubMed.
- S. S. El-Hawary, A. M. Sayed, R. Mohammed, M. A. Khanfar, M. E. Rateb, T. A. Mohammed, D. Hajjar, H. M. Hassan, T. A. M. Gulder and U. R. Abdelmohsen, New pim-1 kinase inhibitor from the co-culture of two sponge-associated actinomycetes, Front. Chem., 2018, 6, 538 CrossRef CAS PubMed.
- M. Gompel, M. Leost, E. B. De Kier Joffe, L. Puricelli, L. H. Franco, J. Palermo and L. Meijer, Meridianins, a new family of protein kinase inhibitors isolated from the ascidian Aplidium meridianum, Bioorg. Med. Chem. Lett., 2004, 14, 1703–1707 CrossRef CAS PubMed.
- J. Lee, K. N. More, S. Yang and V. S. Hong, 3,5-Bis(aminopyrimidinyl)indole Derivatives: Synthesis and Evaluation of Pim Kinase Inhibitory Activities, Bull. Korean Chem. Soc., 2014, 35, 2123–2129 CrossRef CAS.
- H. B. Sun, X. Y. Wang, G. B. Li, L. D. Zhang, J. Liu and L. F. Zhao, Design, synthesis and biological evaluation of novel C3-functionalized oxindoles as potential Pim-1 kinase inhibitors, RSC Adv., 2015, 5, 29456–29466 RSC.
- G. Faita, M. Mella, P. Righetti and G. Tacconi, An easy lewis acid-mediated isomerization from (E)-to (Z)-oxoindolin-3-ylidene ketones, Tetrahedron, 1994, 50, 10955–10962 CrossRef CAS.
- S. J. Edeson, J. Jiang, S. Swanson, P. A. Procopiou, H. Adams, A. J. Meijer and J. P. Harrity, Studies on the stereochemical assignment of 3-acylidene 2-oxindoles, Org. Biomol. Chem., 2014, 12, 3201–3210 RSC.
- N. Hirofumi, S. Nae, P. Lorien, T. Yukio, A. Masanao, T. Keiko, Y. Shigeyuki, T. Akiko, K. Hirotatsu, O. Takayoshi and N. Tetsuo, Rational Evolution of a Novel Type of Potent and Selective Proviral Integration Site in Moloney Murine Leukemia Virus Kinase 1 (PIM1) Inhibitor from a Screening-Hit Compound, J. Med. Chem., 2012, 55, 5151–5164 CrossRef PubMed.
- D. B. Kitchen, H. Decornez, J. R. Furr and J. Bajorath, Docking and Scoring in Virtual Screening for Drug Discovery: Methods and Applications, Nat. Rev. Drug Discovery, 2004, 3, 935–949 CrossRef CAS PubMed.
- Y. N. Kumar, P. S. Kumar, G. Sowjenya, V. K. Rao, S. Yeswanth, U. V. Prasad, J. A. Pradeepkiran, P. Sarma and M. Bhaskar, Comparison and Correlation of Binding Mode of ATP in the Kinase Domains of Hexokinase Family, Bioinformation, 2012, 8, 543 CrossRef PubMed.
- A. Grover, Drug Design: Principles and Applications, Springer Singapore, 2017, p. 113 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10216g |
| This journal is © The Royal Society of Chemistry 2020 |