
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simplified fabric phase sorptive extraction method for the determination of amphetamine drugs in water samples using liquid chromatography-mass spectrometry
Xiaomeng Ruan,
Limei Xing *,
Ju Peng,
Shiying Li,
Yiqun Song and
Qianqian Sun
*,
Ju Peng,
Shiying Li,
Yiqun Song and
Qianqian Sun
Criminal Investigation Police University of China, China. E-mail: xinglimei1@sina.com; Tel: +86 13898164819
First published on 17th March 2020
Abstract
Fabric phase sorptive extraction (FPSE) can directly extract the target analytes and simultaneously determine many similar substances from complicated sample matrices. Also, it has very high chemical stability. Therefore, we used fabric phase sorptive extraction to analyze three amphetamine drugs (amphetamine (AM), methamphetamine (MAM), and 3,4-methylenedioxymethamphetamine (MDMA)) in water. This was coupled with ultrahigh-performance liquid chromatography and tandem mass spectrometry. The effects of different sorbent chemistries such as sorption time, ratios of back-extraction solvents, back-extraction time, and the salt effect on the extraction efficiency were studied; the optimum operation conditions were determined. Medium polarity polar polymer-coated FPSE media were created using short-chain poly (tetrahydrofuran) (PTHF). This is the most efficient extraction media for the analytes of interest. Under the optimized conditions, the linear range of the three amphetamine drugs were 0.1–150.0 (AM, MAM) and 0.5–200 ng mL−1 (MDMA). The correlation coefficients (γ) were 0.9947 (AM), 0.9925 (MAM), and 0.9918 (MDMA). The detection limits (LOD) were 0.025 ng mL−1 for AM, 0.029 ng mL−1 for MAM, and 0.01 ng mL−1 for MDMA. The corresponding limit of quantification values (LOQ) were 0.083 ng mL−1, 0.097 ng mL−1, and 0.031 ng mL−1, respectively. The recoveries were 73.4–91.6%, 82.6–95.4%, and 92.7–95.3%, respectively, and the relative standard deviations (RSD) were 1.65–6.88%, 1.38–6.11%, and 1.58–7.34%, respectively. Moreover, our method can be successfully applied for the analysis of amphetamines in wastewater samples, and at the same time, lays the foundation for the future detection of such substances.
1. Introduction
At present, the abuse of narcotic drugs and psychotropic substances, and drug crimes worldwide are serious threats to human health and social stability, and present other global hazards.1,2 As drug abuse has become increasingly serious, illegal addition and the loss of various psychotropic drugs have become increasingly threatening to social stability. It has been reported that the drug abuse rate in South Korea and China has steadily increased since the 1980s. Amphetamines are common synthetic addiction agents in Iran, Taiwan, and even the United States, and are, thus, a cause of great concern.3 In 2015, about 2.1 million young people around the world used “ecstasy” and 1.3 million people used amphetamine (AM) and methamphetamine (MAM). The 2016 World Drug Report showed that amphetamine drug consumption is on the rise, and it is the second largest illegal drug.4 Therefore, mastering the patterns, extents, types, and other information of drug abuse is of great significance for the timely and effective planning of anti-drug work. The Sewage Analysis Act was first proposed by the American scholar Daughton CG5 in 2001 and was first implemented in 2005 by Zuccato et al. in Italy. The method, based on human drug abuse, is discharged through the body's metabolic pathways and enters the urban sewage treatment system. By monitoring the concentration of drugs and metabolites in sewage combined with the population of the service area and the flow rate of the sewage treatment plant, drug abuse in the service area is evaluated. In 2016, Been Fred et al. evaluated the differences in drug abuse in different regions by collecting samples from 19 sewage treatment plants in Germany and Switzerland.6 In 2016, Thai PK and other methods used sewage analysis to detect the use of 4-methylmethacinone and 2-methylamino-1-(3,4-methylenedioxyphenyl)-1-propanone in Queensland, Australia.7 Therefore, it is of practical significance to detect amphetamine drugs in sewage.A large number of literature reports have been published on various instrumental methods for measuring such compounds in biological samples and water samples for successfully analyzing drug samples; the methods include gas chromatography-mass spectrometry (GC-MS),8–11 capillary electrophoresis (CE),12–14 and liquid chromatography-mass spectrometry (HPLC-MS).15–17 GC-MS is often used because of its sensitivity. However, samples often require derivatization and higher costs limit their use. Therefore, LC-MS is now widely used for analyzing various trace small molecules in biological matrices and water samples. Compared to GC, LC-MS does not require derivatization and can be used for analyzing non-volatile or semi-volatile compounds.
Although the indexes and functions of modern analytical instruments are improving day by day, such aspects are not suitable for direct use in detection instruments for complex samples in the original state; trace target analysis materials must be extracted and separated via sample pretreatment methods because of the complex composition and low content of target analyte materials. It is only after enrichment and purification that trace target analysis can be carried out using instrument detection and analysis. Therefore, sample pre-processing is very important for analysis and testing in instrumental analysis. Sample pre-processing is related to the integrity of the evidence chain of an entire case and plays an important role in the final determination of criminal facts. Therefore, sample pretreatment is the research content to which every worker in the field of forensic science attaches great importance for a tribunal.
Sample preparation is often required before instrumental analysis. Solid phase extraction (SPE),12,18 solid phase microextraction (SPME),19,20 and liquid–liquid extraction (LLE)20,21 are traditional technologies and are currently widely used as pretreatment techniques. Two of these techniques are currently used for analyzing amphetamines but all the three technologies have certain limitations. SPE requires small particle size filters to prevent clogging of the adsorbent carrier, and this requires a long time and consumes a large amount of organic solvents in the process of resolution.22 In contrast, SPME has a lower cost but the sorbent loading is low and extraction equilibrium time is relatively long. The adsorbent-coated extraction head in SPME equipment may be bent or damaged during operation, resulting in decreased performance with aging.23 Also, the number of commercially available sorbent coatings is limited in SPME; instability of the coating layer in organic solvents, swelling, fracture, and other factors greatly limit the application of SPME in instrumental analysis.24 LLE also has some shortcomings. For example, most of the organic solvents used are toxic and easily emulsify, which prolongs the extraction cycle and causes sample loss.25
As early as the 1990s, Malik et al. developed a sol–gel coating technique for the microextraction of adsorbents to extract complex microsamples.26 In 2014, Kabir and Furton used this technology to develop a new sample extraction technology called fabric phase sorptive extraction (FPSE).27 FPSE is an adsorption extraction technique that uses a sol–gel coating technique to form an ultra-thin coating on a natural or synthetic fabric and then uniformly coats the fabric substrate. After the sol–gel adsorbent coating is applied, the permeability of the substrate remains unchanged and this ensures the fluidity of the sample solution in the extraction system as the point of adsorbent on the coating surface is limited and the mass transfer coefficient increases with the pores of the medium,28 so the extraction can reach equilibrium quickly. FPSE is a simple sample pretreatment technique that consists of two main steps: (1) adsorption of the target substance: the sample is in contact with the FPSE medium and the analytical substance is retained on the adsorbent; (2) desorption of the target: the FPSE medium (in which the target is adsorbed) is fully immersed in the organic solvent and the target analyte is back-extracted into the organic solvent.
In addition, the medium can use a large number of different adsorbent types, including sol–gel polydimethylsiloxane (PDMS), sol–gel polydimethyldiphenylsiloxane (PDMDPS), sol–gel polydiphenylsiloxane, sol–gel C18, sol–gel C8, sol–gel graphene, sol–gel polytetrahydrofuran (PTHF), sol–gel polyethylene glycol (PEG), sol–gel carbowax 20M, and sol–gel polyethylene glycol–polyethylene glycol–polyethylene glycol (PEG–PEG–PEG),27 thereby adsorbing compounds of different polarities.
In the sample preparation process, the FPSE medium of the adsorbed target can be directly introduced into the container with the stripping agent and then, the stripping agent is introduced into GC/LC-MS or another analytical instrument using a syringe. Thus, FPSE is a simpler process as the number of sample processing preparation steps further reduces the potential risks of analyte loss and experimental error. FPSE has a large contact surface area and can quickly adsorb the target analytes. Because of the low viscosity of the organic solvent that is used, it is usually possible to extract the target analyte in 2–10 min and less organic solvent is required. Thus, the target analyte can be back-extracted from the FPSE medium faster and without potential residual risk. In addition, any organic solvent can be used for the reverse-extraction of target analysis and strong chemical bonding between the adsorbent and the fabric matrix allow the fabric phase to be exposed to any selected organic solvent and to harsh chemical conditions (high acid–base).29
The advantages of FPSE are as follows: (1) the operation is simple, the device is cheap, adsorption is fast, and the solvent content is low. (2) the fabric phase can be directly introduced into the sample and the original sample can be absorbed by the target substance. (3) Any organic solvent can be chosen for elution. (4) There are fewer sample processing steps, which thereby reduces the potential sources of errors and the cost of sample preparation. (5) Hundreds of new sol–gel coatings can be used as adsorbents. (6) The chemical bond between the adsorbent and the fabric matrix provides high chemical stability of the fabric phase.22
This technique is used to test amphetamine drugs in water samples, including AM, MAM, and 3,4-methylenedioxymethamphetamine (MDMA). Table 1 shows the chemical structures and basic properties of these three drugs. Drugs such as amphetamine have been included in the scope of illicit drugs but because of illegal and prevalent drug abuse, illegal drug trafficking, new drug abuse, and various drug criminal activities involving amphetamines, the concentration of amphetamines in the environment has reached a very serious level, even in sewage and wastewater. A certain number of drugs can be detected30 and thus, the accurate, simple, and rapid detection of amphetamines using scientific means is of great significance for combating drug abuse and maintaining social stability. In addition, a variety of microextraction methods for determining amphetamines drugs have been published in the literature and most of these are based on adsorbent-based microextraction techniques such as SPE, SPME, and LLE (as mentioned above). Therefore, compared to conventional sample preparation techniques, two new FPSE media were selected for this experiment: sol–gel polytetrahydrofuran (sol–gel PTHF) and sol–gel polyethyleneglycol (sol–gel PEG). These media were evaluated in the extraction process of amphetamines in water samples.
| Compound | Abbreviation | Chemical structure | pKa | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KOW KOW |
|---|---|---|---|---|
| Amphetamine | AM | 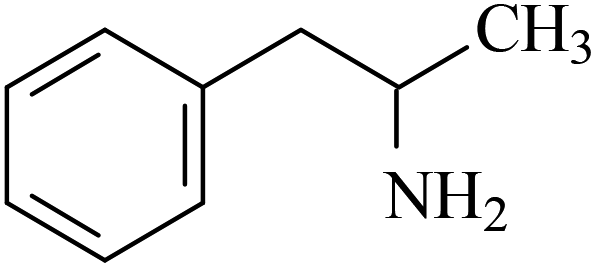 |
9.8 | 1.79 |
| Methamphetamine | MAM | 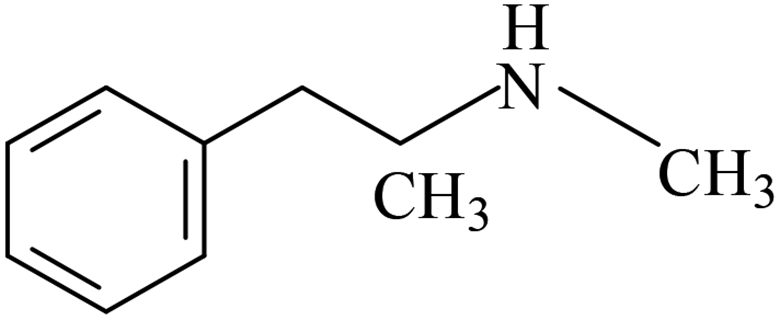 |
9.5 | 2.27 |
| 3,4-Methylenedioxymethamphetamine | MDMA | 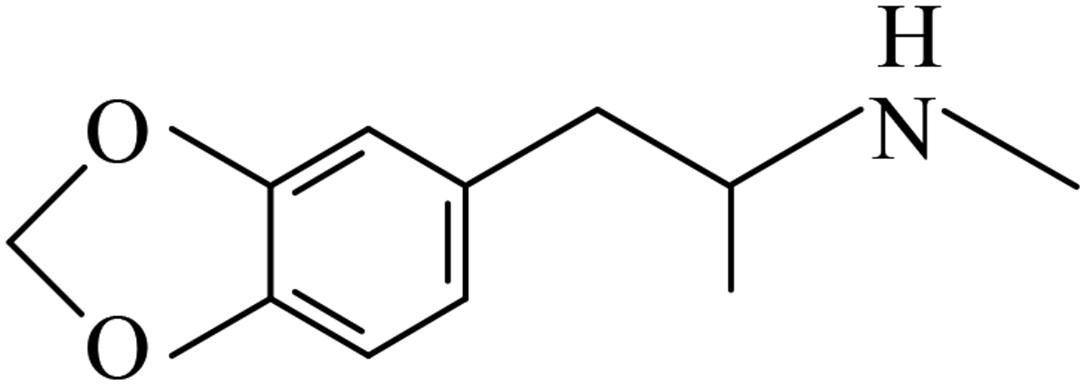 |
9.9 | 2.75 |
2. Experimental
2.1. Chemicals and materials
AM, MAM, and MDMA were purchased from the China Food and Drug Testing Institute. Make a storage solution of 1 mg ml−1 with methanol and store the sample in a refrigerator at 4 °C.Methyltrimethoxysilane (purity 98%), polyethylene (average molecular weight 200), and polytetrahydrofuran (average molecular weight 850) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Trifluoroacetic acid was purchased from Tianjin Kemi European Chemical Reagent and cotton fabric was purchased from Guangzhou Airou Non-woven Fabrics Co., Ltd.
Methanol, acetonitrile, and acetone were chromatographically pure and were purchased from Shandong Yuwang Industrial Co., Ltd. Dichloromethane was chromatographically pure and was purchased from Tianjin Fuyu Fine Chemical Co., Ltd. Hydrochloric acid, NaOH, and NaCl were analytical reagent grade and were purchased from Beijing Chemical Plant. The experimentally used water was deionized water and the blank water sample was distilled water.
2.2. Instrumentation
The instruments used were a Finnigan Surveyor liquid chromatograph with an electrospray ion source, an LXQ linear ion trap mass spectrometer (Thermo Fisher, USA), a KQ-200VDE Dual-Frequency Numerical Control Ultrasonic Cleaner (Kunshan Ultrasonic Instrument Co., Ltd.), an HJ-3 Digital Display Thermostatic Magnetic Stirrer (Gongyi Yuhua Instrument Co., Ltd.), and a PH-10/100 pen-type acidity meter (Shanghai Lichen Bangxi Instrument Technology Co., Ltd.). Also, 100 and 1000 μL pipettes (Gilson, United States) and a 10 μL liquid chromatography micro sampler (Shanghai, China, Pigeon) were used.2.3. Preparation of standards
The stock standard solution (1 mg mL−1) was prepared in methanol. The stock solution was used within 3 months of preparation, was stored at low temperature (−4 °C), and was protected from light. Working standard solutions were prepared by further dilutions of the stock solution to obtain varying concentrations. The dilutions were made using distilled water over the entire linear range and were freshly prepared at the time of use. The concentrations of the working standards ranged from 0.5 to 200 ng mL−1. Aliquots of 10 μL were injected into the LC-MS column and quantitative analysis was based on peak area ratio measurements.2.4. Chromatography and mass spectrometry
The samples were separated and detected using high-performance liquid chromatography (equipped with a diode array detector (DAD)). A Thermo Gold ODS (150 mm × 2.1 mm × 5 μm) was used separately and the column heater remained at 35 °C. The mobile phase was a solution of methanol:0.1% acetic acid (80:20, v/v) and the flow rate was 0.200 mL min−1.
For the mass spectroscopic measurements, an electrospray ionization source (ESI), positive ion mode detection, full ion scanning mode (50–500 m/z), and a capillary temperature of 350 °C were used. The atomized gas, heating auxiliary gas, and purge gas flow rates were 33.00, 8.00, and 2.00 L min−1, respectively. The source voltage was 5.00 kV.
2.5. Fabric phase sorptive extraction sample preparation process
The substrate selection for the sol–gel coating depends primarily on: (a) the hydrophilicity or hydrophobicity of the target analyte and (b) the strategy for analyte stripping after preconcentration onto the FPSE media.31 This experiment used solvent back extraction, which is known to be hydrophilic, so the fabric substrate selected was cellulose.The fabric was pretreated before it was coated with the sorbent to remove residues of external dirt, particulates, and fabric finishing chemicals, and to activate the surface hydroxyl groups. First, the fabric was soaked in a large amount of deionized water for 15 min and the clean fabric was then placed in a 1 M NaOH solution for 1 h under sonication to activate the silanol groups on the fabric surface. The base treated fabric was then washed several times with a large amount of deionized water. It was then treated with 0.1 M HCl solution for 1 h under sonication to neutralize the hydroxides on the surface. The treated fabric was then rinsed with a copious amount of deionized water and finally placed in a clean beaker with tweezers and dried overnight in a stream of inert gas at a proper temperature. The dried fabrics are were kept in a clean, sealed bottle until the next day.
A coating layer was prepared using a sol–gel method. Briefly, 10 mL of polytetrahydrofuran, 10 mL of sol–gel precursor methyltrimethoxysilane (MTMS), and 20 mL of dichloromethane:acetone (v:v = 50:50) were added sequentially to a beaker. Then, 4 mL of trifluoroacetic acid (containing 5% water) was added as the sol–gel catalyst. The mixture was stirred well and then vortexed for 3 min, centrifuged for 5 min, and ultrasonicated for 2 min. The treated sol–gel solution was transferred to a brown glass bottle.
The pretreated fabrics were immersed in a sol–gel solution and the solution was left for a certain period of time to form the sol–gel coating on the fabrics. The coating was removed and rinsed with dichloromethane:acetone (50:50, v/v) under ultrasonication for 30 min to remove the unreacted contents. When the coating cycle was complete, the solution was discharged from the bottle and the remaining contents of the bottle were then piped into another bottle, which was sealed. A small hole was made during the sealing process to allow nitrogen to circulate and the bottle was placed in an oven. The oven was kept at 40 °C for 12 h. The oven was then closed for 12 h and kept at room temperature. Then, it was opened at 60 °C for 12 h. The temperature was then adjusted to 50 °C and held for 12 h at a constant temperature. The coated fabric was extracted using dichloromethane and acetone (50:50, v/v) sequentially. Before extraction using fabric phase adsorption, the fabric was rinsed with ultrasonic treatment for 30 min to remove non-reactive substances and residual sol–gel solutions from the coated surfaces. The fabric was then dried at 50 °C and stored in an inert atmosphere for 1 h. Finally, the coated fabric was cut into pieces that were 2.0 cm × 2.0 cm and the pieces were stored in a closed glass container to prevent contamination. Compared with a blank fiber (0.0197 g) of the same size, the FPSE weighed 0.0501 g and the sorbent loading per device was 0.0076 g cm−2. The same method was used to prepare the sol–gel polyethylene glycol coating.
2.6. FPSE procedure
A 10 mL sample containing 5 ng mL−1 target analyte was prepared. The pH of the solution was adjusted and then the sample was placed in a vial. The treated sol–gel polytetrahydrofuran-coated FPSE medium was placed in the vial using a tweezer (Fig. 1), and the sample solution was stirred at 1500 rpm and heated at 38 °C to extract the target analyte therein. After 60 min, the FPSE was removed from the vial, immersed in 1 mL of the elution solvent, and left to stand for a while. It was then sonicated for 8 minutes and a clear solution was obtained. The concentrated solution was filtered and 10 μL of the filtered solution was injected into a high-performance liquid chromatograph-mass spectrometer, which was used for detection. | ||
| Fig. 1 Scheme for describing FPSE. | ||
2.7. Method validation
The effectiveness of the method was verified in terms of linearity and precision. The data was assessed using least squares linear regression analysis over the working range. The formula S/N = 3 was used to calculate the limit of detection (LOD) and the formula S/N = 10 was used to calculate the limit of quantitation (LOQ) (in each of these equations, S = is the signal and N = is the noise). The relative standard deviation (RSD%) of repeated measurements was calculated to assess the precision of this method and the concentrations of different standard amphetamines were analyzed to determine the accuracy.3. Results and discussion
3.1. Chemistry of the substrate and sol–gel PTHF coating
Currently, many adsorbents use different porous substrates as the support because the fabrics themselves are porous substrates. Also, there are many sol–gel reactive functional groups on the surface of the fabrics and thus, the sol–gel solution can be firmly coated on the fabrics. Kabir and coworkers developed microextraction sorbents that use fabrics as the matrix.32 In just a few years, hundreds of kinds of sol–gel solutions with different uses have been reported.The preparation of sol–gel polytetrahydrofuran (PTHF) coatings on the fabrics involves the following reactions: hydrolysis of sol–gel precursors catalyzed by MTMS, polymerization of MTMS after hydrolysis (resulting in the formation of a three-dimensional sol–gel network on the fabrics), random incorporation of the sol–gel PTHF to the sol–gel network, and chemical fixation of the grown sol–gel network to the flexible cellulose substrate via condensation. The sol–gel PTHF can be coated on the fabrics via chemical bonding.
During the polycondensation process, the grown sol–gel PTHF network reacts with hydroxyl groups on the surface of the fabrics and this results in a covalently bonded sol–gel poly-tetrahydrofuran coating that is uniformly distributed throughout the fabric. The coating is chemically stable, and has good selectivity for efficient and fast analyte extraction. The chemical representation of the sol–gel PTHF coated cellulose substrate reaction scheme is shown in Fig. 2.
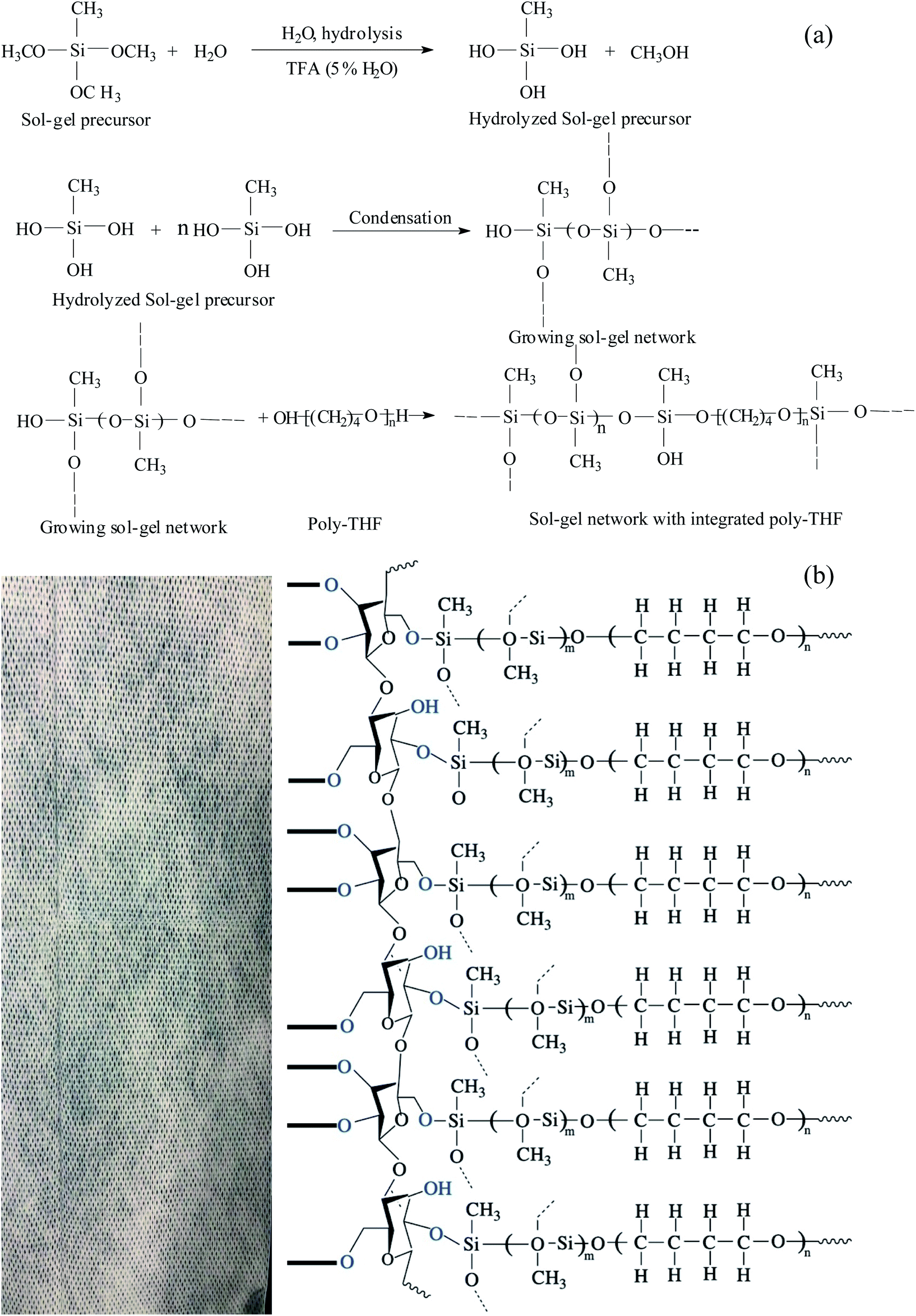 | ||
| Fig. 2 Schematic diagram of (a) sol–gel PTHF and (b) sol–gel PTHF-coated FPSE media. | ||
3.2. Characterization of sol–gel PTHF-coated FPSE media
Scanning electron microscopy (SEM) and Fourier transform-infrared spectroscopy (FT-IR) were used to characterize the sol–gel short-chain polytetrahydrofuran-coated FPSE medium. The surface morphology of the sol–gel tetrahydrofuran network and chemical integration of the fabric substrate were studied using these methods. | ||
| Fig. 3 SEM images of (a) uncoated substrate surface at 320× magnification and of (b) coated substrate surface at 300× magnification. | ||
As seen in these SEM images, each of the fabrics is composed of many microfabrics and is designed from macroscopic pores that are generated during the weaving process. Because the low viscosity sol solution enters the fabric matrix, a sol–gel PTHF-coated ultrathin film is uniformly formed on the surface of the cellulose microfabric. Therefore, as seen from the SEM image of the sol–gel PTHF-coated extraction medium, the coating is not limited to the surface of the cellulose substrate; it also occurs in the three-dimensional matrix. The sol–gel coating does not block the pores and this allows the sample matrix to penetrate through the body, and such a structure helps to complete the feature extraction balance in a short period of time.
In this experiment, we used FT-IR to analyze the structural changes in the FPSE medium before and after coating. For these tests, the sample tested was placed directly on the OMNIC sampler germanium crystal and the spectrum was scanned in the range of 4000–500 cm−1.
Fig. 4 depicts the FT-IR spectra of an uncoated cellulose substrate (top) and a sol–gel polytetrahydrofuran-coated FPSE medium (bottom). The FT-IR spectra of uncoated cellulose fabrics show characteristic absorptions between 3343 cm−1, 2900 cm−1, and 1428 cm−1. The characteristic absorptions of –OH association, –CH2 symmetry extension, and –CH2 deformation vibrations are observed. The characteristic absorptions of C–O stretching and C–H bending vibrations were also observed at 1107 cm−1, 1056 cm−1, and 1033 cm−1. Because of the functional groups of polytetrafluoroethylene combined with the –OH groups on the fabric, there are, therefore, no characteristic bands for the sol–gel polytetrafluoroethylene coating at about 3000 cm−1, 2942 cm−1, and 2864 cm−1 (corresponding to –CH2 antisymmetric scaling), at 2805 cm−1 and 2334 cm−1 (both corresponding to –CH2 symmetric scaling), or –CH2 at 1491 cm−1, 1373 cm−1, and 1270 cm−1 (corresponding to deformation vibrations). These observations strongly suggest the presence of sol–gel PTHF coatings.33 The sol–gel-polytetrahydrofuran-coated FPSE medium has clean bands in the FT-IR spectrum at 1107 cm−1 and 1029 cm−1, and these indicate SiO–C–C bonding, which may be derived from the sol–gel PTHF network and cellulose in the successful combination of substrates.34
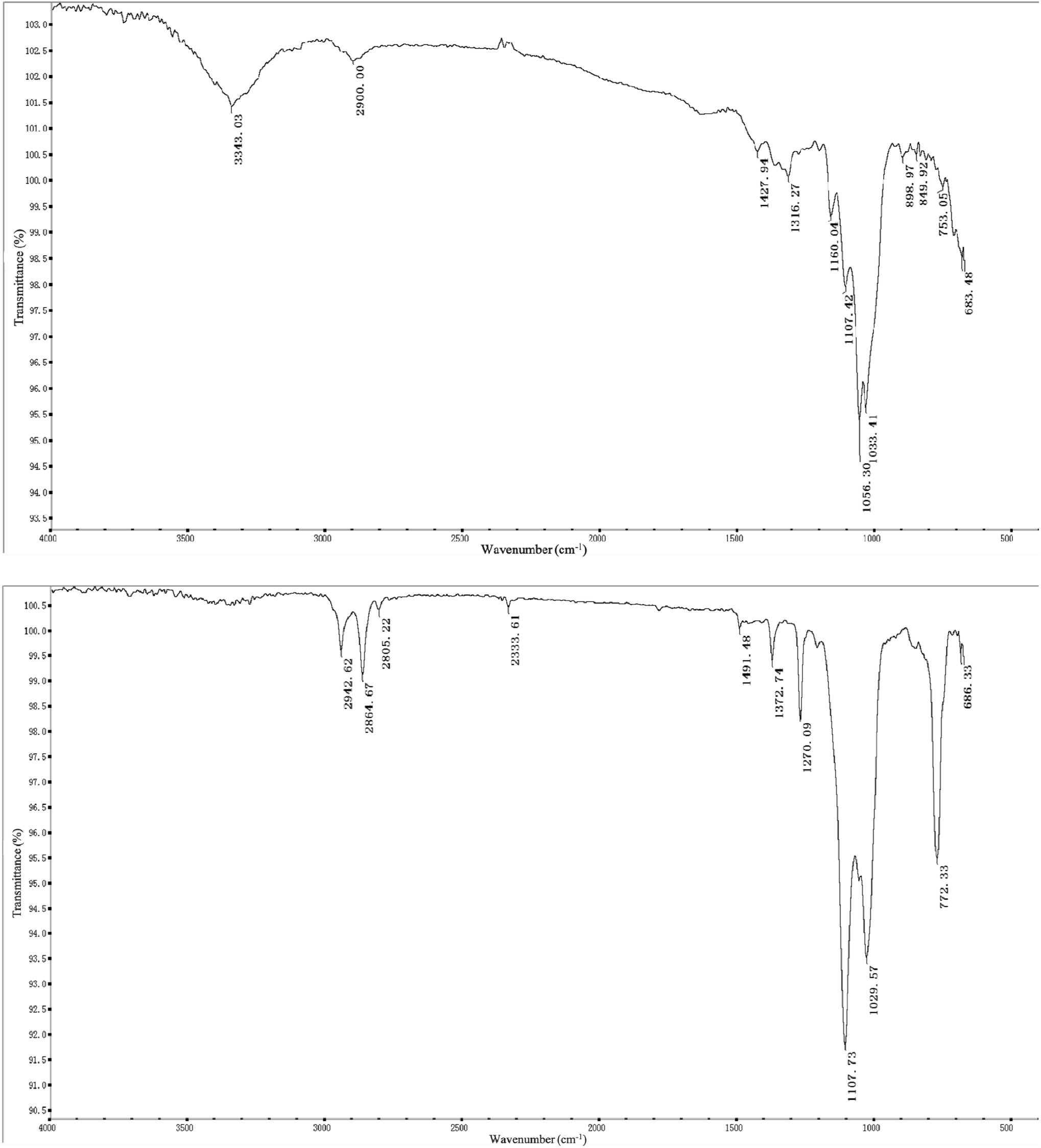 | ||
| Fig. 4 FT-IR spectra of uncoated cellulose substrate (top) and sol–gel poly-THF coated FPSE medium (bottom). | ||
3.3. Optimization of FPSE conditions
Developing methods for the determination of amphetamines using FPSE-LC-MS requires the optimization of parameters that affect the efficiency of the method. These parameters include the extraction time, back-extraction solvent, back-extraction time, and the salt effect. The maximum value of peak area was used to evaluate the extraction efficiency of amphetamines in the water samples. Each factor was assessed three times and all of the experiments were performed at 1500 rpm with stirring.35Because of the polarity of amphetamine drugs, we chose sol–gel PTHF and sol–gel PEG as the coating layers. We determined from our experiment that sol–gel PTHF provides a high recovery rate for the analyte. Therefore, the sol–gel PTHF-coated fabric phase was selected as the optimized experimental operation.
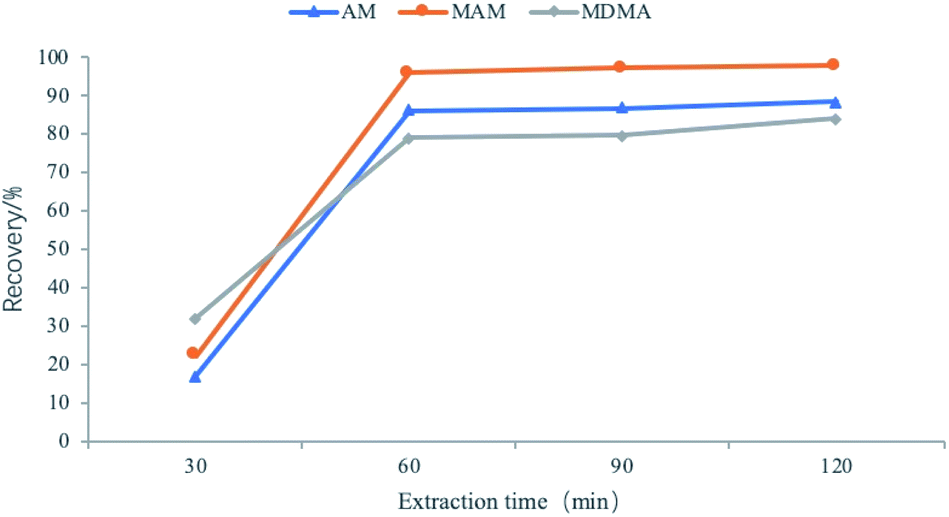 | ||
| Fig. 5 Effects of extraction time. | ||
:methanol = 3:7. The volume of the extracted solvent was fixed at 1.0 mL for all further experiments.
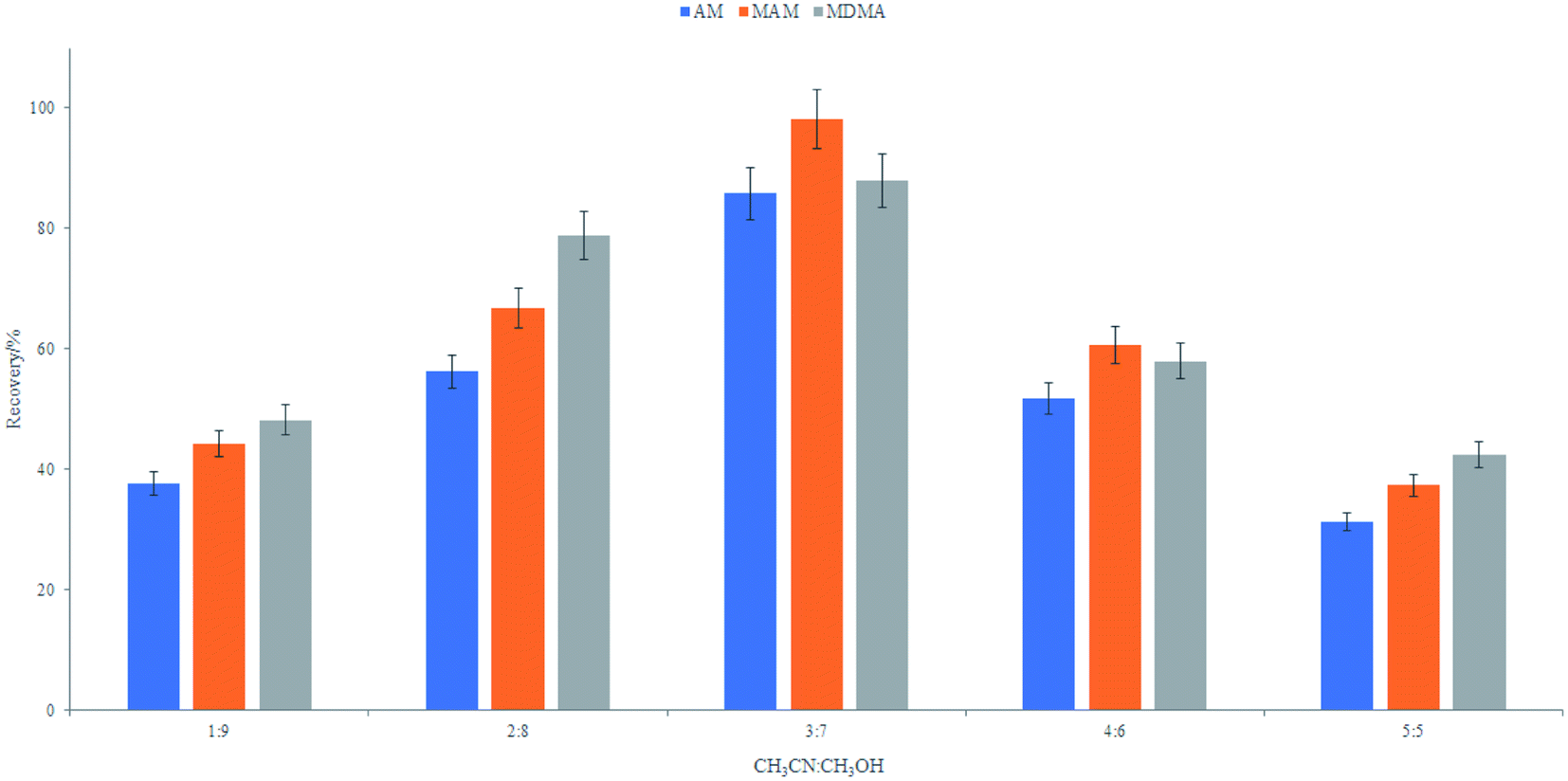 | ||
| Fig. 6 Effects of elution solvent ratio. | ||
:methanol (in the ratio of 3:7) for 4 to 12 min. The largest number of amphetamine-type drugs can be laundered in 8 min (Fig. 7). Therefore, a back-extraction time of 8 min was optimal and was used for further experimentation.
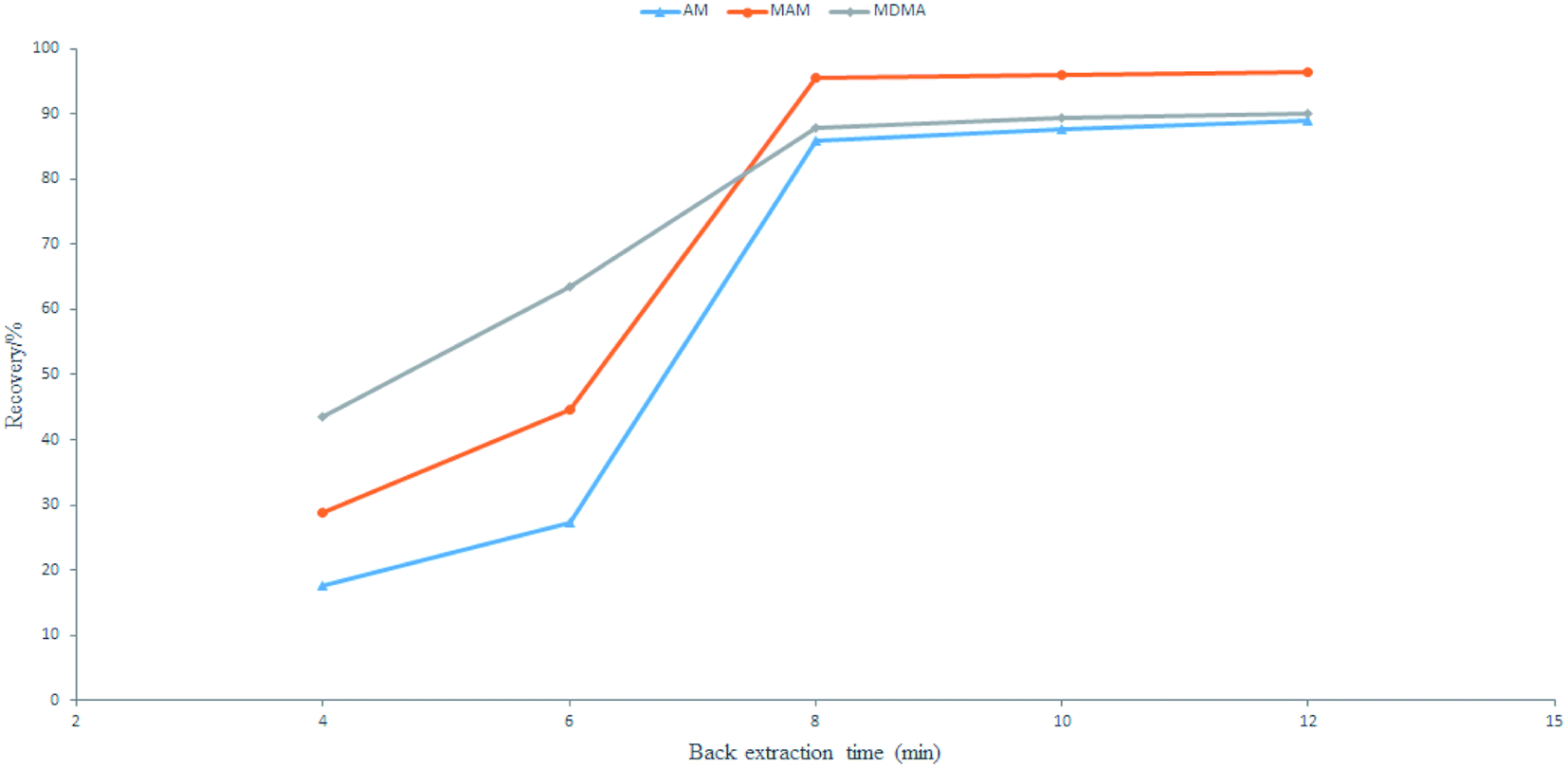 | ||
| Fig. 7 Effects of back extraction time. | ||
KOW < 3, ionic strength can have a great impact on extraction.35–38 Therefore, salt effect can facilitate the movement of a target analyte in the PTHF direction. Different amounts of NaCl were added to the aqueous solution to analyze the effects of the analyte on the adsorption of the FPSE media. The concentrations of NaCl in the aqueous solution varied from 0% to 3% (w/v). However, the recovery value changed sharply with the adsorption characteristics when NaCl was added. Other authors have reported in the related literature that ionic strength does not improve efficiency.19,38 Some scholars have explained that this is because NaCl helps to move analytes to the surface of water and this makes their interaction with the fabric PTHF weaker.19 Also, higher NaCl content may increase electrostatic interaction between the analyte and salt ions, which can also reduce the extraction efficiency. Because NaCl can have a negative impact on the extraction efficiency, we chose not to add NaCl in our experiment.In summary, the employed conditions for the FPSE procedure were as follows: 60 min of extraction time, no added salt, pH 11, back-extraction time of 8 min, and 1 mL of acetonitrile:methanol at 3:7 as the back-extraction solvent. The enrichment factor achieved using these conditions was more than 10.
3.4. Matrix effects
Since the matrix often significantly interferes with the analysis of analytes and affects the accuracy of the results, it is necessary to determine the matrix effect of amphetamine drugs in urban river water. Matrix effects are considered to be suitable in the range from 80% to 120% because they indicate that the matrix effects in this experiment are small. Standard solutions of the three amphetamine drugs were measured and the peak area of each analyte was recorded as A. Water samples from different sources were processed by FPSE, then the standard analytes were added, and the peak area for each analyte was recorded as B. Matrix effect (ME%) = A/B × 100%.39 The results show that the matrix effect values of AM, MAM, and MDMA are between 0.8872 and 1.2041 in the selected municipal wastewater; thus, matrix effects are not significant.3.5. Method validation
A blank water sample was tested as described above. Secondary ion chromatograms of 5 ng mL−1 AM, MAM, and MDMA solutions were compared to examine the effects of matrix components on the test. The parent ion of MAM is m/z 150 and the secondary ion is m/z 119. The parent ion of AM is m/z 136 and the secondary ion is m/z 119. The parent ion of MDMA is m/z 194 and the secondary ion of m/z 163. After treatment with FPSE, each of the drugs (AM, MAM, and MDMA) have good chromatographic behavior, the peak shape is symmetrical (Fig. 8), and the matrix impurities in the water sample do not interfere with the determinations made using the optimized conditions.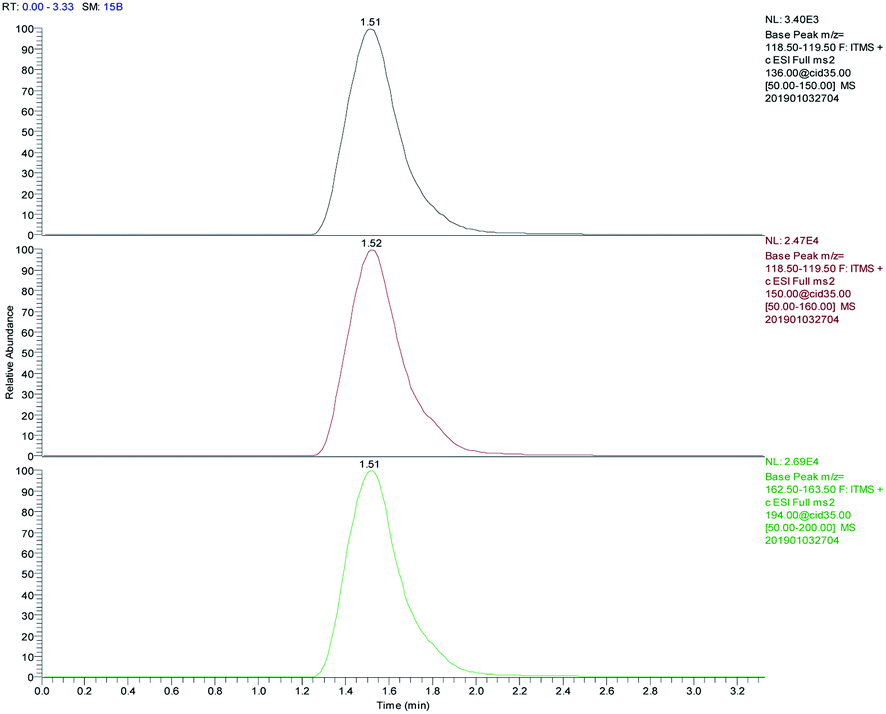 | ||
| Fig. 8 The chromatographic behavior of AM, MAM, MDMA after FPSE (from top to bottom). | ||
The samples were spiked with standard solutions of different concentrations of three amphetamine drugs, and the relative recoveries and precisions of the same measured object in parallel at 5 different time points in a day were calculated. The results are shown in Table 2 for the adsorption and extraction efficiency and the stability of the test method. The recoveries of AM, MAM, and MDMA in the water samples were all higher than 70% when FPSE was used with the optimized experimental conditions. The relative standard deviation is less than 10% and this indicates that the method is reliable for the quantitative analysis of three drugs.
| Analyte | Concentration (ng mL−1) | Recovery (%, n = 5) | RSD (%, n = 5) |
|---|---|---|---|
| Amphetamine | 5 | 73.4 | 6.88 |
| 50 | 92.3 | 5.30 | |
| 150 | 91.6 | 1.65 | |
| Methamphetamine | 5 | 82.6 | 6.11 |
| 50 | 89.5 | 4.97 | |
| 150 | 95.4 | 1.38 | |
| 3,4-Methylenedioxymethamphetamine | 5 | 92.7 | 7.34 |
| 50 | 94.7 | 6.32 | |
| 150 | 95.3 | 1.58 |
The optimized FPSE-LC-MS conditions that were established above were used to prepare calibration curves for the analytes spiked with the concentrations of 0.5, 1.0, 2.0, 5.0, 10.0, 15.0, 20.0, 40.0, 50.0, 100.0, 150.0, and 200.0 ng mL−1. Using the experimental method described above, the values of peak area were plotted versus drug concentration and linear regression was performed to determine the regression equation. From the fitting results (Table 3), the linear range for the quantitative analysis of AM and MAM in water is 0.1–150.0 ng mL−1 and that for the quantitative analysis of MDMA is 0.5–200.0 ng mL−1. The linear equation correlation coefficients (γ) are higher than 0.99. This shows that the working curves for the three amphetamine drugs in water that were established using this method have a good linear relationship within the concentration range studied.
| Analyte | Regression equation | Linear range (ng mL−1) | R2 | LOD (ng mL−1) | LOQ (ng mL−1) |
|---|---|---|---|---|---|
| AM | Y = 283X + 29.123 | 0.1–150.0 | 0.9986 | 0.025 | 0.083 |
| MAM | Y = 1244.6X + 204.36 | 0.1–150.0 | 0.9925 | 0.029 | 0.097 |
| MDMA | Y = 1763.2X + 291.16 | 0.5–150.0 | 0.9918 | 0.010 | 0.031 |
The LODs of the three drugs were calculated using a S/N ratio of 3. Similarly, the LOQs were estimated as the S/N ratio of 10 (S/N = 10, n = 5). Also, as seen in Table 3, the LODs for AM, MAM, and MDMA in water were 0.025, 0.029, and 0.01 ng mL−1, respectively. The LOQs were 0.083, 0.097, and 0.031 ng mL−1, respectively.
3.6. Method comparison
In terms of the LOD and LOQ values, the newly developed and validated FPSE-LC-MS method was used to compare the detection of amphetamine drugs with other reported methods. The comparative data are listed in Table 4. In these existing studies, component extraction was performed by conventional extraction techniques, such as SPE and SPME. The LOD and LOQ of amphetamine drugs in this study are basically higher than those reported in these literatures. Only one paper has lower data than the procedure of this study but SPME has some disadvantages (see Introduction). Therefore, the FPSE-LC-MS program selected in this study does not require complicated equipment configuration, and the obtained LOD and LOQ values are low.| Analytes | Matrix | Sample preparation methoda | Analytical instrument | LOD | LOQ | Reference |
|---|---|---|---|---|---|---|
| a SPE: solid phase extraction; SPME: solid phase microextraction; LPE: liquid phase extraction; MIP: molecular imprinting. | ||||||
| AM and its derivates | Urine | SPE | CE-MS | 0.31–4.29 ng mL−1 | 1.00–13.98 ng mL−1 | 13 |
| AM | Water urine | SPME | GC-MS | 0.4–2 ng L−1, 0.09–0.2 ng L−1 | 10.4–6.8 ng L−1, 0.3–0.6 ng L−1 | 19 |
| MAM | ||||||
| MDMA | ||||||
| MDA | ||||||
| MDEA | ||||||
| AM | Saliva | LPE | GC-MS | 1–5 μg L−1 | 10 μg L−1 | 21 |
| MAM | ||||||
| MDMA | ||||||
| MDA | ||||||
| AM | Oral fluid | MIP | UHPLC-MS | 0.03–1.3 μg L−1 | — | 40 |
| AM | Water | FPSE | LC-MS | 0.01–0.025 ng mL−1 | 0.031–0.083 ng mL−1 | This work |
| MAM | ||||||
| MDMA | ||||||
4. Application to sewage samples
To further evaluate the method's applicability, real water samples were also used. The FPSE-LC-MS method was applied to 100 mL of non-spiked wastewater samples to measure three of the studied analytes. The water samples were taken from municipal sewage outlets of Xinkaihe River in Huanggu District and from Weigong Nullah in Tiexi District (City in northern China). The measured concentration for AM in the Xinkai River water sample was 123.2 ng L−1, that for MAM was 169.6 ng L−1, and that for MDMA was 72.5 ng L−1. Although AM and MAM were not detected in the Weigong open channel water sample, where the concentration for MDMA was 63.4 ng L−1.5. Conclusions
In the last few decades, sample preparation was the most time consuming and challenging step in the entire analytical protocol. This was particularly true when analytes were present in trace amounts and at ultratrace concentrations in various environmental matrices. Several analytical methods for the analysis of different drugs have been reported in the literature, and efforts are being made to develop more efficient and selective methods for sample preparation. Conventional pretreatment methods are gradually being replaced by microextraction techniques to reduce the volume of sample and solvent required. The novel fabric phase sorptive extraction procedure has the advantages of high efficiency, low cost, and small solvent consumption, which is in agreement with the criteria of green chemistry.In this paper, a method based on fabric phase sorptive extraction coupled to LC-MS has been developed for the analysis of analyzing amphetamines. This procedure allows for the direct determination of target substances using an FPSE medium. There is no complicated pretreatment step and this provides a simple method for analyzing such drugs. Lower detection limits were obtained for AM, MAM, and MDMA with this method, and the analysis time was shorter, which makes the method more economical and greener. The verification parameters of this method include the range, linearity, precision, and accuracy; good results were obtained for these parameters. The real water samples were tested and traces of amphetamine in water were detected. The effectiveness of the method was proved by the analysis of actual water samples. Therefore, FPSE-LC-MS can be used as effective analytical method for detection for detecting the presence of drugs in water samples. This method can objectively assess the amount of drug abuse in a certain area and is an important means of monitoring poisonous conditions. Also, this study shows that the method is rapid, accurate, reproducible, and sensitive. Also, it meets the actual combat requirements of the physical security of public and chemical inspection work.
On the basis of this research, it can provide the basis for the detection of other sample types (such as urine, blood, hair, saliva, and other biological samples) using FPSE-LC-MS, thereby establishing a more efficient and applicable sample pretreatment method. Additionally, GC, GC-MS, HPLC-MS, and other analytical instruments were used to examine the drug-detecting effect of the biological samples that were pretreated with FPSE; thus, the application range of FPSE was expanded. In future, research will have the challenge to develop new coatings and materials to determine any analytes in complex sample matrices.
Conflicts of interest
We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.References
- A. M. Barr, W. J. Panenka, W. MacEwan, A. E. Thornton, D. J. Lang, W. G. Loner and T. Lecomte, The need for speed: an update on methamphetamine addiction, J. Psychiatr. Neurosci., 2006, 31, 301–313 Search PubMed.
- A. C. Parrott, MDMA in humans: factors which affect the neuropsychobiological profiles of recreational ecstasy users, the integrative role of bioenergetic stress, J. Psychopharmacol., 2006, 20, 147–163 CrossRef CAS PubMed.
- N. j. Kwon and E. Han, A commentary on the effects of methamphetamine and the status of methamphetamine abuse among youths in South Korea, Japan, and China, Forensic Sci. Int., 2018, 286, 81–85 CrossRef CAS PubMed.
- F. Accioni, D. García-Gómez, E. Girela and S. Rubioa, SUPRAS extraction approach for matrix-independent determination of amphetamine-type stimulants by LC-MS/MS, Talanta, 2018, 182, 574–582 CrossRef CAS PubMed.
- C. G. Daughton, Illicit drugs in municipal sewage: proposed new non-intrusive tool to heighten public awareness of societal use of illicit-abused drugs and their potential for ecological consequences, Pharmaceuticals and Care Products in the Environment, American Chemical Society, 2001, pp. 348–364 Search PubMed.
- F. Been, L. Bijlsma and L. Benaglia, et al., Assessing geographical differences in illicit drug consumption: a comparison of results from epidemiological and wastewater data in Germany and Switzerland, Drug Alcohol Depend., 2016, 161, 189–199 CrossRef PubMed.
- P. K. Thai, F. Y. Lai and M. Edirisinghe, et al., Monitoring temporal changes in use of two cathinones in a large urban catchment in Queensland, Australia, Sci. Total Environ., 2016, 545, 250–255 CrossRef PubMed.
- M. Gilbert, S. Odoardi, M. Cassar and S. S. Rossi, Rapid and simple procedure for the determination of cathinones, amphetamine-like stimulants and other new psychoactive substances in blood and urine by GC-MS, J. Pharm. Biomed. Anal., 2018, 149, 494–501 CrossRef PubMed.
- A. A. S. Marais and J. B. Laurens, Rapid GC-MS confirmation of amphetamines in urine by extractive acylation, Forensic Sci. Int., 2009, 183, 78–86 CrossRef CAS PubMed.
- S. Y. Kim, J. Y. Kim, W. Kwon, M. K. In, Y. E. Kim and K.-J. Paeng, Method development for simultaneous determination of amphetamine type stimulants and cannabinoids in urine using GC-MS, Microchem. J., 2013, 110, 326–333 CrossRef CAS.
- M. Kacper Woźniak, M. Wiergowski, J. Aszyk, P. Kubica, J. Namieśnik and M. Biziuk, Application of gas chromatography–tandem mass spectrometry forthe determination of amphetamine-type stimulants in blood and urine, J. Pharm. Biomed. Anal., 2018, 148, 58–64 CrossRef PubMed.
- G. Boatto, M. Nieddu, A. Carta, A. Pau, M. Palomba, B. Asproni and R. Cerri, Determination of amphetamine-derived designer drugs in human urine by SPE extraction and capillary electrophoresis with mass spectrometry detection, J. Chromatogr. B, 2005, 814, 93–98 CrossRef CAS PubMed.
- G. Boatto, M. Virginia Faedda, A. Pau, B. Asproni, S. Menconi and R. Cerri, Determination of amphetamines in human whole blood by capillary electrophoresis with photodiode array detection, J. Pharm. Biomed. Anal., 2002, 29, 1073–1080 CrossRef CAS PubMed.
- X. Cui, C. Ni, C. Liang, F. Gong, R. Wang, G. Chen and Y. Zhang, Screening and quantification of forty-six drugs for abuse and toxic compounds in human whole blood by capillary electrophoresis: application of forensic cases, Microchem. J., 2019, 144, 403–410 CrossRef CAS.
- F. Vaiano, F. P. Busardò, D. Palumbo, C. Kyriakou, A. Fioravanti, V. Catalani, F. Mari and E. Bertol, A novel screening method for 64 new psychoactive substances and 5 amphetamines in blood by LC–MS/MS and application to real cases, J. Pharm. Biomed. Anal., 2016, 129, 441–449 CrossRef CAS PubMed.
- M. Concheiro, S. M. d. S. S. Simoes, O. Quintela, A. d. Castro, M. J. R. Dias, A. Cruz and M. Lopez-Rivadulla, Fast LC–MS/MS method for the determination of amphetamine, methamphetamine, MDA, MDMA, MDEA, MBDB and PMA in urine, Forensic Sci. Int., 2007, 171, 44–51 CrossRef CAS PubMed.
- M. Cheze, M. Deveaux, C. Martin, M. Lhermitte and P. Gilbert, Simultaneous analysis of six amphetamines and analogues in hair, blood and urine by LC-ESI-MS/MS: application to the determination of MDMA after low Ecstasy intake, Forensic Sci. Int., 2007, 170, 100–104 CrossRef CAS PubMed.
- I. Senta, I. Krizman, M. Ahel and S. Terzic, Multiresidual analysis of emerging amphetamine-like psychoactivesubstances in wastewater and river water, J. Chromatogr. A, 2015, 1425, 204–212 CrossRef CAS PubMed.
- I. Racamonde, R. Rodil, J. B. Quintana and R. Cela, In-sample derivatization-solid-phase microextraction of amphetamines and ecstasy related stimulants from water and urine, Anal. Chim. Acta, 2013, 770, 75–84 CrossRef CAS PubMed.
- J. Lee, Y. Park, W. Yang, H. Chung, W. Choi, H. Inoue, K. Kuwayama and J. Park, Cross-examination of liquid–liquid extraction (LLE) and solid-phase microextraction (SPME) methods for impurity profiling of methamphetamine, Forensic Sci. Int., 2012, 215, 175–178 CrossRef CAS PubMed.
- P. Meng and Y. Wang, Small volume liquid extraction of amphetamines in saliva, Forensic Sci. Int., 2010, 197, 80–84 CrossRef CAS PubMed.
- R. Kumar, H. Gaurav, A. Kumar Malika, A. Kabirb and G. Kenneth, Furton. Efficient analysis of selected estrogens using fabric phase sorptive extraction and high-performance liquid chromatography-fluorescence detection, J. Chromatogr. A, 2014, 1359, 16–25 CrossRef CAS PubMed.
- A. Kabir, K. G. Furton and A. Malik, Innovations in sol-gel microextraction phases for solvent-free sample preparation in analytical chemistry, Trends Anal. Chem., 2013, 45, 197–218 CrossRef CAS.
- C. Nerín, J. Salafranca, M. Aznar and R. Batlle, Critical review on recent developments in solventless techniques for extraction of analytes, Anal. Bioanal. Chem., 2009, 393, 809–833 CrossRef PubMed.
- M. R. Fuh, T. Y. Wu and T. Y. Lin, Determination of amphetamine and methamphetamine in urine by solid phase extraction and ion-pair liquid chromatography–electrospray-tandem mass spectrometry, Talanta, 2006, 68, 987–991 CrossRef CAS PubMed.
- S. L. Chong, D. X. Wang, J. D. Hayes, B. W. Wilhite and A. Malik, Sol–gel coating technology for the preparation of solid-phase microextraction fabrics of enhanced thermal stability, Anal. Chem., 1997, 69, 3889–3898 CrossRef CAS PubMed.
- A. Kabir and K. G. Furton, Fabric phase sorptive extractors (FPSE), US Patent Application: 14/216121, 2014.
- P. S. Wei and S. Y. Hsiao, Effects of mass transfer coefficient on pore shape in solid, Int. J. Heat Mass Tran., 2016, 103, 931–939 CrossRef.
- I. Racamonde, R. Rodil, J. B. Quintana, B. J. Sieira, A. Kabir, K. G. Furton and R. Cela, Fabric Phase Sorptive Extraction: A New Sorptive Microextraction Technique for the Determination of Non-steroidal Anti-inflammatory Drugs from Environmental Water Samples, Anal. Chim. Acta, 2015, 865, 22–30 CrossRef CAS PubMed.
- N. Daglioglu, E. Y. Guzelb and S. Kilercioglu, Assessment of illicit drugs in wastewater and estimation of drugs of abuse in Adana Province, Turkey, Forensic Sci. Int., 2019, 294, 132–139 CrossRef CAS PubMed.
- S. S. Lakade, F. Borrull, K. G. Furton, A. Kabir, N. ú. Fontanals and R. M. Marcé, Comparative Study of Different Fabric Phase Sorptive Extraction Sorbents to Determine Emerging Contaminants from Environmental Water Using Liquid Chromatography-Tandem Mass Spectrometry, Talanta, 2015, 144, 1342–1351 CrossRef CAS PubMed.
- A. Kabir, K. G. Furton and A. Malik, Innovations in sol-gel microextraction phases for solvent-free sample preparation in analytical chemistry, Trends Anal. Chem., 2013, 45, 197–218 CrossRef CAS.
- A. Montarsolo, M. Periolatto, M. Zerbola, R. Mossotti and F. Ferrero, Hydrophobic sol-gel finishing for textiles: improvement by plasma pre-treatment, Text. Res. J., 2013, 83, 1190–1200 CrossRef.
- M. Aznar, P. Alfaro, C. Nerin, A. Kabir and K. G. Furton, Fabric phase sorptive extraction: an innovative sample preparation approach applied to the analysis of specific migration from food packaging, Anal. Chim. Acta, 2016, 936, 97–107 CrossRef CAS PubMed.
- A. Kabir, C. HamLet and A. Malik, Parts per quadrillion level ultra-trace determination of polar and nonpolar compounds via solvent-free capillary microextraction on surface-bonded sol–gel polytetrahydrofuran coating and gas chromatography–flame ionization detection, J. Chromatogr. A, 2004, 1047, 1–13 CrossRef CAS PubMed.
- M. Roldán-Pijuán, R. Lucena, S. Cárdenas, M. Valcárcel, A. Kabir and K. G. Furton, Stir fabric phase sorptive extraction for the determination of triazine herbicides in environmental waters by liquid chromatography, J. Chromatogr. A, 2015, 1376, 108–120 CrossRef PubMed.
- C. Almeida and J. M. F. Nogueira, Determination of steroid sex hormones in water and urine matrices by stir bar sorptive extraction and liquid chromatography with diode array detection, J. Pharmaceut. Biomed. Anal., 2006, 41, 1303–1311 CrossRef CAS PubMed.
- S. Montesdeoca-Esponda, Z. Sosa-Ferrera, A. Kabir, K. G. Furton and J. J. Santana-Rodríguez, Fabric phase sorptive extraction followed by UHPLC-MS/MS for the analysis of benzotriazole UV stabilizers in sewage samples, Anal. Bioanal. Chem., 2015, 407, 8137–8150 CrossRef CAS PubMed.
- H. Stahnke, T. Reemtsma and L. Alder, Compensation of matrix effects by postcolumn infusion of a monitor substance in multiresidue analysis with LC-MS/MS, Anal. Chem., 2009, 81, 2185–2192 CrossRef CAS PubMed.
- A. Sorribes-Soriano, A. Francesc, E. Turrillas, S. Armenta, A. s. Pedro and J. M. Herrero-Martínez, Amphetamine-type stimulants analysis in oral fluid based on molecularly imprinting extraction, Anal. Chim. Acta, 2019, 1052, 73–83 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |