
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical fingerprinting and quantitative monitoring of the doping drugs bambuterol and terbutaline in human urine samples using ATR-FTIR coupled with a PLSR chemometric tool†
Faisal K. Algethamia,
Sherif M. Eid *b,
Khadiga M. Kelanicd,
Mohamed R. Elghobashybc and
Mohamed K. Abd El-Rahmanc
*b,
Khadiga M. Kelanicd,
Mohamed R. Elghobashybc and
Mohamed K. Abd El-Rahmanc
aChemistry Department, Faculty of Science, Imam Mohammed ibn Saud Islamic University, Riyadh, Saudi Arabia
bAnalytical Chemistry Department, Faculty of Pharmacy, October 6 University, 6 October City, Giza, Egypt. E-mail: Sheriefmohammed@o6u.edu.eg; Sherief055@icloud.com; Tel: +201111609539
cAnalytical Chemistry Department, Faculty of Pharmacy, Cairo University, El-Kasr El-Aini Street, ET-11562 Cairo, Egypt
dAnalytical Chemistry Department, Faculty of Pharmacy, Modern University for Technology and Information, Cairo, Egypt
First published on 17th February 2020
Abstract
The use of performance-enhancing drugs is prohibited in sports competitions according to the World Anti-Doping Agency (WADA) regulations. Here, ATR-FTIR spectroscopy coupled with a partial least squares regression (PLSR) chemometric tool was used for the detection of the misuse of such substances. Bambuterol and its metabolite terbutaline have been included in the list of prohibited doping agents. Therefore, we used bambuterol and terbutaline as models for the accurate and simultaneous qualitative and quantitative analysis of bambuterol and terbutaline in human urine samples. The method was straightforward and once the urine samples were collected, they could be directly applied to the surface of the ZnSe prism (ATR unit) to get the results within one minute. A calibration set with a partial factorial design was used to develop the PLSR model that could be used to predict the concentration of unknown samples containing the two drugs. The developed method was carefully validated and successfully applied to the urine sample analysis of human volunteers. The drugs were quantified at nanogram level concentrations. A side-by-side comparison of the proposed method with the routine GC-MS method was performed to demonstrate the challenges and opportunities of each method.
1. Introduction
Forensic analysis is expanding, as confirmed from the hundreds of advanced studies published annually.1,2 The World Anti-Doping Agency (WADA) establishes the regulations concerning doping drugs across all sports all over the world.3 The use or attempted use of prohibited substances by athletes is considered a violation of these rules. Since such issues correspond closely to criminal cases under forensic investigations, urine samples are collected in and out of competitions and exposed to a variety of analytical examinations developed to monitor the presence of a banned substance.4,5The WADA rules classify β2-agonists such as bambuterol HCl (BAM) and terbutaline (TER) as stimulants and anabolic agents with a restricted specific regulation.3 Athletes often use BAM for therapeutic purposes as a treatment for exercise-induced asthma.6 However, there are some adverse effects of the use of β2-agonists, such as anabolic and stimulation effects with the increase in myocardial contraction and heart rate, increase in the accretion of muscle proteins and a reduced rate of fat deposition;7 thus, this drug and its metabolite TER8 have been included in the list of prohibited doping agents.3 The detection of BAM and TER as doping drugs for routine analysis in urine samples has been accomplished mainly by chromatography techniques such as GC/MS9,10 and LC-MS/MS.11 Although these methods have high potential in the fight against doping drugs, they suffer from many disadvantages, such as the use of very expensive instruments, being non-portable, need for highly qualified operators, high operation cost, moderate throughput, limitations with respect to solvents and samples, long operation times, and too many sample preparation steps.
It has been proven that attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) has ability for the quantitative and/or qualitative determination of biological factors and samples such as lung and prostate cancer biomarkers,12 protein analysis,13 and petroleum component analysis.14 Chemometrics is a well-established technique in forensic analysis,15,16 and partial least squares regression (PLSR) does not require the accurate knowledge of all the components present in urine samples. It can make concentration predictions even in the presence of interfering compounds.17 Also, the direct application of urine samples to the surface of the ATR prism as a sampling technique is sufficient for the quantitative determination of the early-stage malaria parasite,18 enzyme kinetics parameters,19 biomarkers of glomerulonephritis,20 tumor cells,21 and malignant ovarian tissues.22 A recent study compared the performances of micro-ATR-FTIR and GC-MS as identifying tools for micro-plastics and fibers isolated from different water sources.23
Therefore, this paper describes the application of ATR-FTIR in combination with a PLSR chemometric tool for the determination of the doping drugs BAM and TER. Low detection limits were obtained due to the high selectivity, sensitivity, portability, high throughput analysis, and straightforward nondestructive direct determination capabilities of PLSR/ATR-FTIR.24–26 Only a few microliters (μL) of urine samples were sufficient for the quantitative determination of the analytes. The developed anti-doping method may be considered as a preliminary step, opening the way for further investigations on other doping agents.
2. Experimental procedures
2.1. Materials
BAM was supplied from AstraZeneca, Egypt. TER was obtained from (Sigma-Aldrich, Germany). Bambec® tablets (AstraZeneca Ind Co., Egypt) labeled to contain 20 mg of BAM per tablet (Batch No. 120065) were purchased from the local community pharmacies, Cairo, Egypt.2.2. Standard and working solutions
Powdered BAM and TER equivalent to 0.01 g were separately placed into two 10 ml volumetric flasks (VFs). The volumes were completed to the mark using human urine. Then, they were stirred until completely dissolved to get 1000 μg ml−1 solution. Only one milliliter was transferred into a 100 ml VF and the volume was completed to the mark using human urine to get a working solution of 10 μg ml−1.2.3. Instrument
An IRaffinity-1 FTIR Spectrometer, (Shimadzu, Japan) fitted with a high energy ceramic light source (single beam), He–Ne laser for alignments, germanium-coated potassium bromide beam splitter, and DLATGS detector equipped with a temperature controlling unit was used in the analysis. A horizontal ATR accessory model 8200HA (PIKE Technology, USA) fitted with a zinc selenide (ZnSe) crystal, which is a rectangular-shaped yellow prism (dimensions: 80 mm long, 10 mm wide, 4 mm thick), was used.2.4. Software
The FTIR instrument was controlled by IRsolution software, version 1.6 (Shimadzu, Japan). The ATR-FTIR absorption spectrum of each sample was obtained with software parameters of SqrTriangle apodization function and 45 scans using a high spectral resolution of 4 cm−1 in the mid-infrared region from 4000 to 400 cm−1; thus, each spectrum gave 2024 data points at 1.93 intervals. The Unscrambler software version X 10.4 (CAMO Software, Norway) was used for PLSR chemometric calculations.2.5. Experimental design for the PLSR model
The experimental design is a significant factor in chemometric measurements. The calibration set concentrations have to be carefully designed to increase the chances of extracting useful information from the prediction samples. To get a representative calibration set, a multilevel partial factorial design27 of two factors (BAM and TER) and five concentration levels was constructed. It required about 25 experiments to study the mixtures, as shown in Table 1. This orthogonal design was obtained after numbering the levels (i.e., the concentrations) from +2 (highest) to −2 (lowest) to get a cyclic generator (−2, −1, 0, 1, 2) to ensure that there is no correlation between the under-investigation analyte concentrations so that the correlation coefficient will be zero.| Coded levels | MIX no. | BAM (ng ml−1) | TER (ng ml−1) | |
|---|---|---|---|---|
| BAM | TER | |||
| 0 | 0 | 1 | 5000 | 5000 |
| 0 | −2 | 2 | 5000 | 50 |
| −2 | −2 | 3 | 50 | 50 |
| −2 | 2 | 4 | 50 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| 2 | −1 | 5 | 10000 |
2500 |
| −1 | 2 | 6 | 2500 | 10000 |
| 2 | 0 | 7 | 10000 |
5000 |
| 0 | −1 | 8 | 5000 | 2500 |
| −1 | −1 | 9 | 2500 | 2500 |
| −1 | 1 | 10 | 2500 | 7500 |
| 1 | 2 | 11 | 7500 | 10000 |
| 2 | 1 | 12 | 10000 |
7500 |
| 1 | 0 | 13 | 7500 | 5000 |
| 0 | 2 | 14 | 5000 | 10000 |
| 2 | 2 | 15 | 10000 |
10000 |
| 2 | −2 | 16 | 10000 |
50 |
| −2 | 1 | 17 | 50 | 7500 |
| 1 | −2 | 18 | 7500 | 50 |
| −2 | 0 | 19 | 50 | 5000 |
| 0 | 1 | 20 | 5000 | 7500 |
| 1 | 1 | 21 | 7500 | 7500 |
| 1 | −1 | 22 | 7500 | 2500 |
| −1 | −2 | 23 | 2500 | 50 |
| −2 | −1 | 24 | 50 | 2500 |
| −1 | 0 | 25 | 2500 | 5000 |
A representative set of 25 mixtures was prepared by taking the proper volumes from the prepared working solutions into the volumetric flasks. Then, the volumes were completed to the mark with human urine. The prepared mixtures were separated into two sets: a calibration set of 15 synthetic mixtures and a validation set of 10 mixtures (Table 1).
2.6. Spectral data acquisition and calibration steps
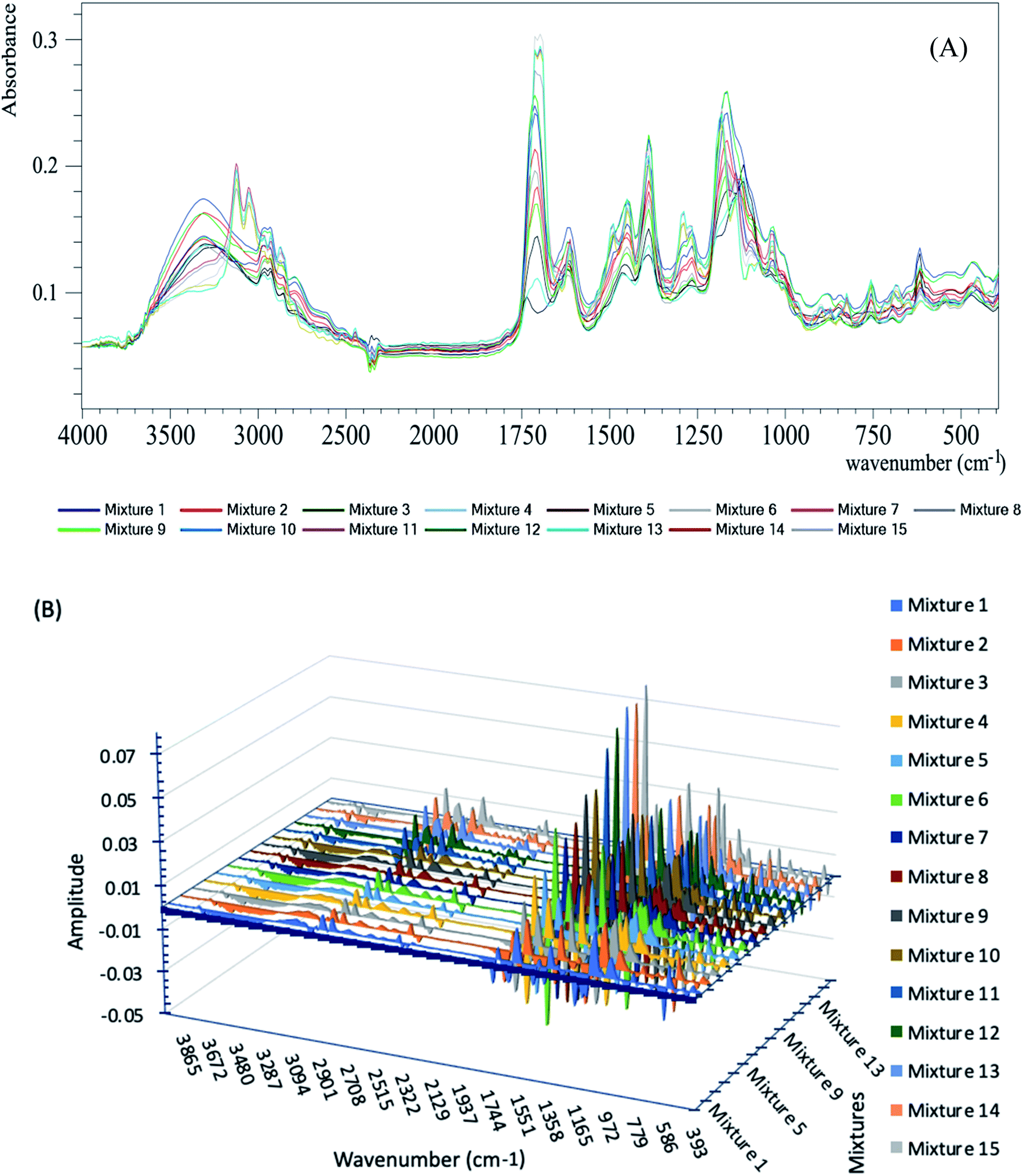 | ||
| Fig. 1 (A) ATR-FTIR absorption spectra of the calibration set mixtures; (B) 3D plot of the 1st derivative of the calibration set mixtures. | ||
Complete sample dryness was ensured by the application of 10 μL of human urine over the ATR prism; then, a spectrum was obtained each minute during the air-drying process. Complete sample dryness occurred after two minutes. The cleanness of the ATR prism was tested regularly between the measurements, where an absorption spectrum was recorded without the sample to make sure that it had no peaks higher than the normal environmental noise level.
The derivative calculations were applied to the recorded spectra from the earlier steps using the following parameters: derivative order: 1, polynomial order: 2 and smoothing points: 5, as shown in Fig. 1B.
These pre-processing steps could be registered on the Unscrambler X software to be performed automatically for the prediction of unknown sample concentrations.
Finally, the PLSR chemometric algorithm29 was applied to the derivative spectra obtained from the last step (shown in Fig. 1B). The recorded absorbance spectra were related to the concentrations by Beer's law.30 The Regression calibration curves were plotted between the known and predicted concentrations.
2.7. Analysis of drugs in urine samples
Healthy volunteers were fully informed verbally and in writing about the objectives and nature of this study. They received a single oral dose of 20 mg Bambec® tablets in the morning after breakfast and no extra food was allowed for at least two hours (h). Urine samples were collected in sterile screw-top containers in consecutive pools with at least 5 drops of each sample during random time intervals. The volunteers took a pre-dose sample and then at 0–2, 2–4, 4–6, 6–8, 8–10, 10–12, and 12–24 hours after the dose. The collected urine volume was measured after every period. The containers were labeled with date and time and then stored at −80 °C. The concentrations of BAM and TER were determined following the described procedure. About 10 μL of the sample was transferred from each container to be placed over the ZnSe prism and rapidly air-dried. The samples were scanned using the same device parameters mentioned above and the collected data stored in a computer. PLSR chemometric calculations were performed on the stored spectra.The study involving human participants was performed in strict accordance with the institutional ethical standards of the Helsinki Declaration of 1964 and its later amendments. It was approved by the local research ethical committee of faculty of pharmacy, Cairo University, Egypt. Healthy volunteers were fully informed verbally about the objectives, nature and possible risks of this study and a written informed consent was obtained from all the volunteers involved in the study.
3. Results and discussion
Based on the fact that an infrared spectrum is considered as a fingerprint of each compound, structurally related compounds can be differentiated using their spectra. To the best of our knowledge, this is the first method for the determination of BAM and TER as doping drugs in urine samples using the ATR-FTIR technique coupled with PLSR chemometrics. FTIR spectroscopy is a well-established technique with a wide application range. The direct application of biological samples over the ATR prism is a widely used sampling technique with high sensitivity and selectivity, as reported in literature.18,20–22 The PLSR algorithm was chosen as it does not require prior accurate knowledge of all the components present in the samples under investigation.The analysis of BAM in the presence of its metabolite TER is a challenge as they are structurally related compounds. Fig. 2 shows the structures of BAM and TER. BAM (RS-5-(2-tert-butylamino-1-hydroxyethyl)-m-phenylene bis dimethyl carbamate) is metabolized in the liver by the cleavage of the ester bond and replacement of the branches of N,N-dialkyl carbamate by OH groups to give TER.31 This hydrolysis causes the main differences in the structures.8
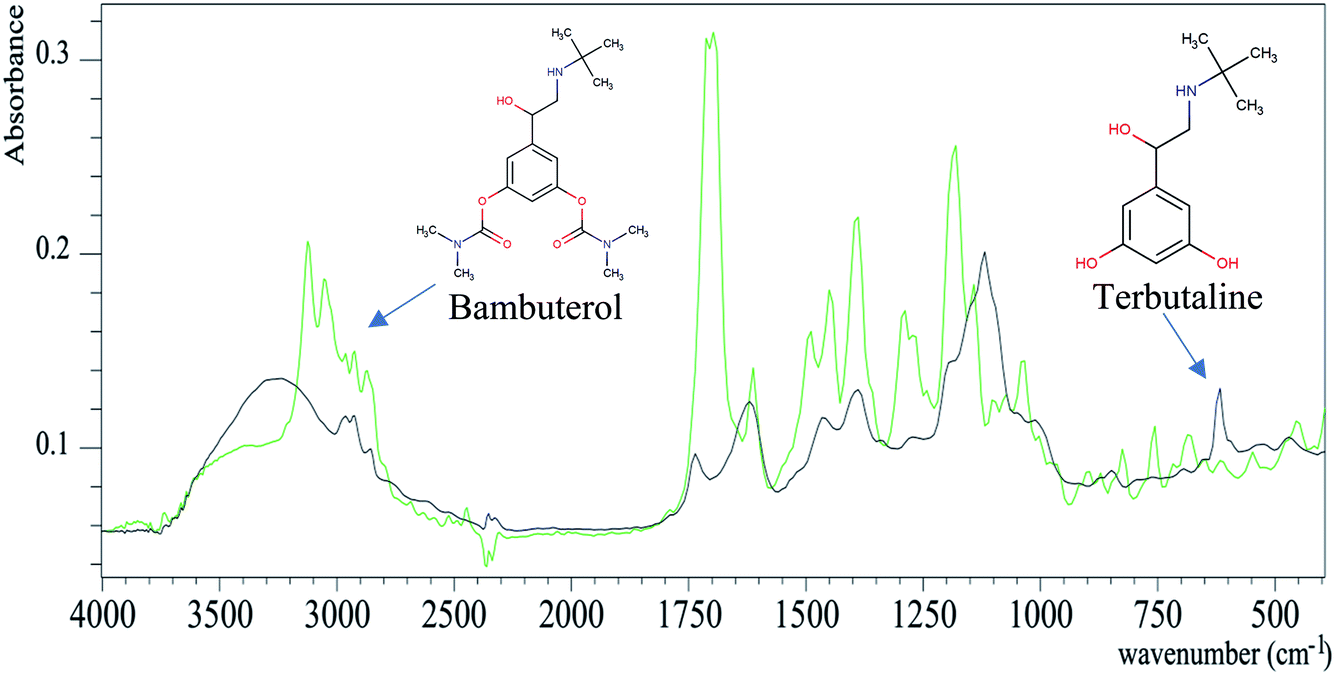 | ||
| Fig. 2 ATR-FTIR absorption spectra of bambuterol (green) and terbutaline (black). | ||
Fig. 2 shows the overlay plot of the two spectra of BAM and TER and their characteristic peaks can be distinguished. For more illustration, the spectra were analyzed by the assignment function of the ACD/SpecManager software to show the peak differences between BAM and TER, as summarized in Table 1S.† The FTIR spectra of BAM and TER can be considered as their own fingerprints. The BAM spectrum has a characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group that shows a peak at 1720 cm−1, which disappears in the TER spectrum. The proposed method was convenient for the simultaneous qualitative and quantitative determination of BAM and TER.
O group that shows a peak at 1720 cm−1, which disappears in the TER spectrum. The proposed method was convenient for the simultaneous qualitative and quantitative determination of BAM and TER.
3.1. PLSR model calibration and validation
In order to get a representative calibration set, the calibration samples have to be distributed over the concentration space of both analytes. Thus, a representative wide range calibration set of uncorrelated mixtures was constructed. It was efficiently applied for the analysis of BAM with a linear range of 50–10000 ng ml−1, covering the reported excretion range of BAM in urine samples.11 The method showed high accuracy over the calibration range with the mean recovery percent values of 98.92 and 99.97% for BAM and TER, respectively.
The performance of multicomponent analysis is based on the spectral model used and the wavelength selected. PLSR with a non-linear iterative partial least squares algorithm (NIPALS) was calculated using the Unscrambler software on the Savitzky–Golay first derivative algorithm for the selected spectrum range (400–4000 cm−1), due to which all the available measured wavelengths are often used without neglecting any region that may contain important data.
The key step in building the PLSR model is the selection of optimum number of factors to achieve correct concentration prediction in PLSR calibrations to prevent data over-fitting. PLSR models with 1 to 7 factors were calculated for each wavelength region and validated using “leave-one-out cross-validation”.32,33 Briefly, the model is repeatedly recalculated leaving out a single sample individually at a time and then used to predict the concentration of the left out observation. PLSR calibration using 14-mixture spectra was performed and the regression equation was calculated. The concentration of the left-out sample was calculated using the obtained regression equation. The same procedure was employed for the 15-mixture analysis; each sample was left out once and the predicted concentration was calculated for each sample. The predicted concentrations were plotted against the known concentrations of each mixture. The obtained recoveries (between 95 and 105%) indicate the validity of the method for its application to unknown samples. The latent factors that explain the spectral matrix are sorted in decreasing order according to their contributions to the spectral features. Using the optimal pre-processing method, latent factors were selected with a cross-validation method, as mentioned above. This method depended on the selection of the model including the smallest number of factors that give the minimal root mean square error of calibration (RMSEC) and the minimum root mean square error of validation (RMSEV), by plotting the difference of RMSEV against the number of factors for the analysis of each mixture. The PLSR method proved that (2) factors were found to be optimum for BAM and TER determination. The PLSR calibration model was constructed to achieve correct quantification and prediction with minimum errors.34,35
The PLSR model's predictive ability was evaluated using two procedures: the 1st procedure was achieved by plotting the reference concentrations against the corresponding predicted concentrations, as shown in Fig. 3; then, the internal validation method was performed (estimation of BAM concentrations in its own calibration set). A satisfactory correlation coefficient (r2) (0.9988) and good recovery (101.09%) were obtained, indicating high predictive abilities of the model. The 2nd procedure for the determination of the predictive ability of the method was conducted by calculating the standard error of prediction (SEP).36
 | ||
| Fig. 3 Plots of reference concentrations against the predicted concentrations of the calibration sets of (A) BAM and (B) TER. | ||
Table 2 summarizes the characteristic parameters of PLSR chemometric urine analysis. The obtained very low BIAS (0.00048) means a very small acceptable error of prediction. Correlation is the linear relationship between the predicted and reference values in the plot, which is very close to 1, indicating that the predicted and reference concentrations are highly correlated. RMSEC measures the dispersion of the calibration samples around the regression line. BIAS is the mean of all the numbers that lie above or below the regression line and the values close to zero show a random distribution of points around the regression line. The obtained RMSEC and standard error of calibration (SEC) are very close, indicating insignificant BIAS.
| Parameter | Compound | |
|---|---|---|
| BAM | TER | |
| Elements | 15 | 15 |
| Range (ng ml−1) | 50–10000 |
50–10000 |
| Slope | 0.9842 | 0.9892 |
| Offset | 76.98 | 53.97 |
| Correlation | 0.9921 | 0.9945 |
| R2 | 0.9842 | 0.9892 |
| RMSEC | 456.01 | 365.67 |
| SEP | 432.87 | 510.50 |
| Bias | 13 × 10−4 | 13.3 × 10−4 |
External validation was achieved by a validation set of 10 mixtures and it was not implemented in model building. The concentrations of BAM and TER in the validation set solutions were selected randomly but were in the range of the calibration set. The method has high resolving power with good mean recoveries (97.3 and 99.1%) for BAM and TER, respectively.
Regression coefficient plots were obtained to summarize the relationship between all predictors and responses by analyzing the 15-mixture 1st derivative spectra to find the areas (highlighted with black marks) that the software depended on during the building of the model for the detection of either BAM or TER, as shown in Fig. 4. This plot proves and validates the ability of the developed method to differentiate between BAM and TER within the matrix of urine components.
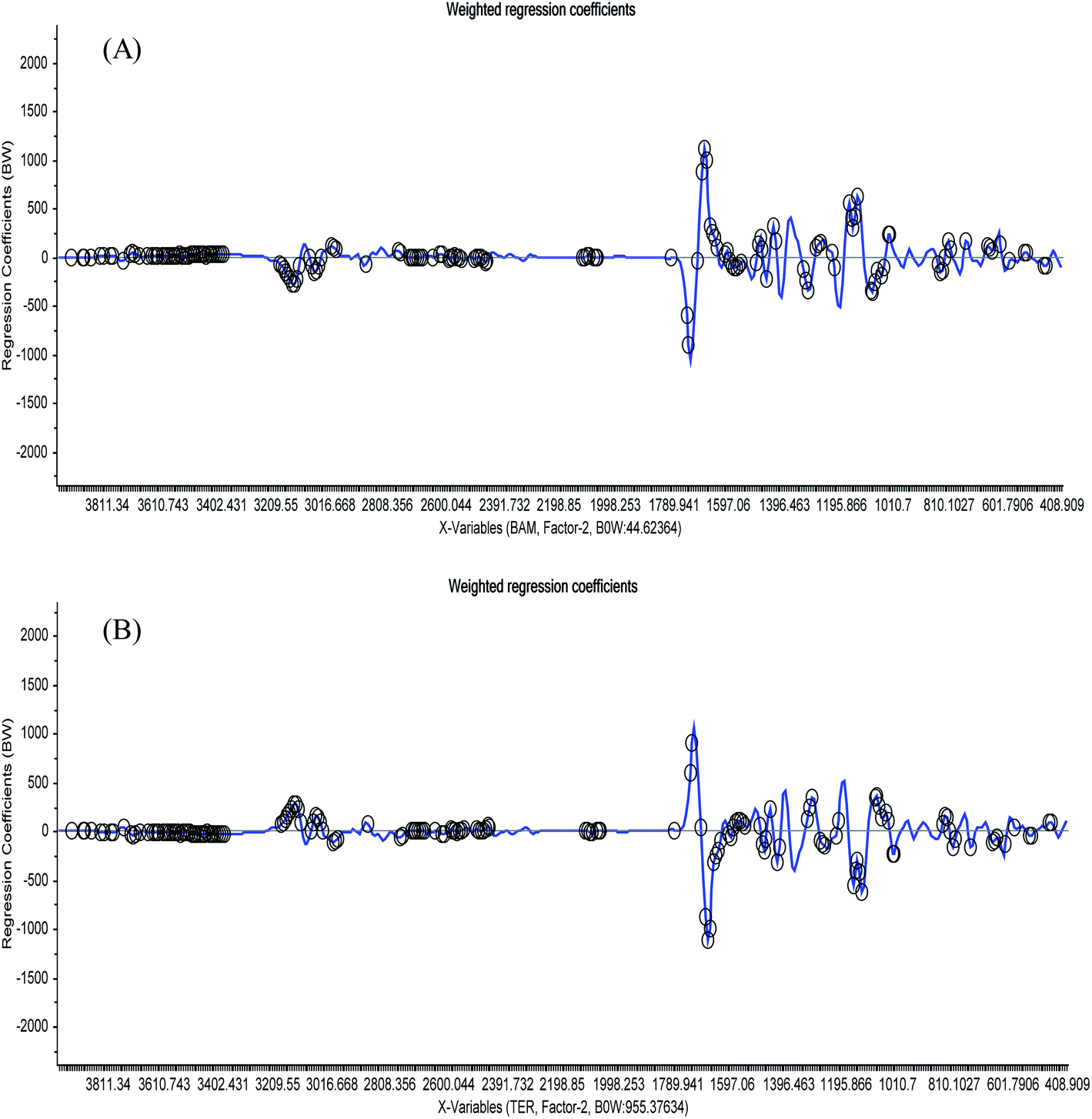 | ||
| Fig. 4 Plots of the regression coefficient weights against the wavelength variables of the 1st derivative of (A) BAM and (B) TER. The important variables used by the software during building the PLSR model are highlighted by black circles except the spectral areas above 3250 cm−1. | ||
3.2. Quantification of BAM and TER in human urine samples
The proposed PLSR chemometric algorithm was applied for the quantification of BAM and TER in the collected urine samples. They can be detected and quantified in the urine of healthy volunteers up to about 24 h after receiving a single oral dose of Bambec® tablets.Urine samples collected from the same volunteer before and after the oral administration of the Bambec® tablets were scanned. The urine sample that did not contain any drug was used as a blank and scanned as a background spectrum. Then, the samples collected after the oral administration of Bambec® tablets were scanned against this background to subtract any substances that may be found in urine and may interfere with the analysis results. Thus, the obtained peaks were due to the presence of BAM and TER in urine with different concentrations, as confirmed from Fig. 1S.†
Fig. 5 shows the obtained spectra of human volunteers' urine at different times (1, 2 and 6 h). It can be easily noticed that the peak intensity moderately increases with time, which may be due to the increased excretion of the drugs in urine with time. The obtained spectra were highly related to the spectra of original BAM and TER shown in Fig. 2.
 | ||
| Fig. 5 Typical ATR-FTIR spectra of the urine samples obtained from volunteers at various time intervals (1, 2 and 6 h) after a single oral dose of 20 mg bambuterol tablet. | ||
The mean cumulative excretion profiles of BAM and TER in healthy volunteers are shown in Fig. 6. It shows that the highest drug concentration in urine is detected within the first six hours; then, the concentration decreases gradually with the analysis time. Most of the drugs (BAM and TER) were eliminated within 10 hours of oral administration. The maximum elimination rates of BAM and TER were 180 and 530 ng ml−1 h−1, respectively, and this was observed within 0–5 h after their oral administration.
 | ||
| Fig. 6 Cumulative excretion of BAM and TER in urine samples of healthy volunteers after a single oral dose of BAMBIC® tablet. | ||
3.3. Challenges and opportunities of the proposed method in comparison with GC-MS
In order to compare the proposed ATR-FTIR multivariate method with the reported GC-MS method37 for the quantification of β2-agonists in human urine, a statistical comparison was applied. The developed ATR-FTIR-PLSR algorithm was tested for the quantification of BAM and TER. Then, the obtained results were compared with the reported data, as summarized in Table 3. The computed t and f values were compared to t and f tabulated values. The computed values were smaller than the tabulated values, indicating that there was no remarkable difference between the results obtained through different methodologies and the proposed method was suitable for the quantification of BAM in biological samples.| Parameter | PLSR chemometrics urine analysis | GC-MS reported method ref. 9 | ||
|---|---|---|---|---|
| BAM | TER | BAM | TER | |
| a The values in parenthesis are the corresponding theoretical values of t and f at the 95% confidence level. | ||||
| M.R.% | 98.92 | 99.97 | 98 | 100.2 |
| S.D. | 0.982 | 1.67 | 0.4 | 0.3 |
| N | 5 | 5 | 5 | 5 |
| Standard error | 0.439 | 0.747 | ||
| Student's t testa | 1.94 (2.31) | 0.303 (2.31) | ||
| F testa | 0.166 (6.39) | 0.032 (6.39) | ||
Obviously, the ATR-FTIR method was straightforward; once the sample was collected (10 μL of urine), it could be directly placed over the ZnSe prism and scanned using the FTIR device to detect if the doping agent is present or not. Also, it has many advantages, which are as follows: nondestructive techniques, a wide range of samples with different masses can be analyzed, provides a spectrum of the analyte, which is considered as a fingerprint of each compound, only one minute is needed to get the results, and no chemical extraction or sample pre-treatment is required.
The disadvantages of the GC-MS method are as follows: it is a destructive technique, i.e., measurements cannot be repeated using the same sample, inorganic compounds cannot be detected, nonvolatile substances have to be chemically modified to be volatile, several sample pre-treatments have to be performed before measurements, not portable, chemical extraction steps of the analytes from the urine samples are required, analysis time is nearly 15 minutes for each sample, needs highly qualified operators, and the instruments are very expensive.
However, it gives complete data about the chemical composition of the sample and contamination that may be present in the sample.
The application of ATR-FTIR as a universal technique for the determination of doping drugs still needs much more investigation and application to all the doping drugs under all possible conditions. Our study can be considered as a preliminary study that opens the way for much more ATR-FTIR measurements for doping control.
4. Conclusion
The experiments illustrate that ATR-FTIR coupled with a chemometric tool is qualitatively and quantitatively useful for the determination of the doping drug BAM and its metabolite TER in human urine samples. The drugs can be detected in the matrix of human urine without any sample pre-treatment or several preparation steps. Only 10 μL sample was sufficient for the analysis, which is less than one drop of volume and nearly the same as the volume used for regular HPLC injections. It was also a viable approach for the quantification of very small concentrations that reach nanogram levels with high selectivity and accuracy.It has many advantages in comparison with the traditional techniques by eliminating the errors during sample pre-treatment that can consume time and may lead to bad recoveries.
Conflicts of interest
The authors declare that there is no conflict of interest.References
- H. Lin, Y. Zhang, Q. Wang, B. Li, P. Huang and Z. J. Wang, Sci. Rep., 2017, 7, 13254 CrossRef PubMed.
- S. Mc Shine, K. Suhling, A. Beavil, B. Daniel and N. Frascione, Anal. Methods, 2017, 9, 2007–2013 RSC.
- M. Thevis, T. Kuuranne, K. Walpurgis, H. Geyer and W. Schänzer, Drug Test. Anal., 2016, 8, 7–29 CrossRef CAS PubMed.
- N. Jan, F. Marclay, N. Schmutz, M. Smith, A. Lacoste, V. Castella and P. Mangin, Forensic Sci. Int., 2011, 213, 109–113 CrossRef CAS PubMed.
- K. C. Doty and I. K. Lednev, ACS Cent. Sci., 2018, 4, 862–867 CrossRef CAS PubMed.
- P. S. Bajwa, J. Sharma, A. Bhargava, S. Sharma, A. R. Sharma and B. Sharma, J. Biomed. Pharm. Res., 2017, 6, 106–117 CAS.
- D. R. Mottram and N. Chester, Drugs in Sport, Taylor & Francis, 2014 Search PubMed.
- M. K. A. El-Rahman, S. M. Eid, M. R. Elghobashy and K. M. Kelani, Sens. Actuators, B, 2019, 285, 216–223 CrossRef.
- W. Van Gansbeke, M. Polet, F. Hooghe, C. Devos and P. Van Eenoo, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 1001, 221–240 CrossRef CAS PubMed.
- P. Van Eenoo, W. Van Gansbeke, N. De Brabanter, K. Deventer and F. T. Delbeke, J. Chromatogr. A, 2011, 1218, 3306–3316 CrossRef CAS PubMed.
- T. Zhou, T. Zhao, Q. Cheng, S. Liu, L. Xu and W. Tan, Biomed. Chromatogr., 2014, 28, 994–1002 CrossRef CAS PubMed.
- P. D. Lewis, K. E. Lewis, R. Ghosal, S. Bayliss, A. J. Lloyd, J. Wills, R. Godfrey, P. Kloer and L. A. Mur, BMC Cancer, 2010, 10, 640 CrossRef CAS PubMed.
- F. Bonnier, S. Rubin, L. Debelle, L. Ventéo, M. Pluot, B. Baehrel, M. Manfait and G. Sockalingum, J. Biophotonics, 2008, 1, 204–214 CrossRef CAS PubMed.
- P. Filgueiras, C. Sad, A. Loureiro, M. Santos, E. Castro, J. Dias and R. Poppi, Fuel, 2014, 116, 123–130 CrossRef CAS.
- R. Kumar and V. Sharma, TrAC, Trends Anal. Chem., 2018, 105, 191–201 CrossRef CAS.
- V. Sharma and R. Kumar, TrAC, Trends Anal. Chem., 2018, 107, 181–195 CrossRef CAS.
- S. Eid, S. Soliman, M. Elghobashy and O. Abdalla, Vib. Spectrosc., 2020, 106, 102995 CrossRef.
- A. Khoshmanesh, M. Dixon, S. Kenny, L. Tilley, D. McNaughton and B. Wood, Anal. Chem., 2014, 86, 4379–4386 CrossRef CAS PubMed.
- S. Eid, M. El-Rahman, M. Elghobashy and K. Kelani, Anal. Chim. Acta, 2018, 1005, 70–80 CrossRef CAS PubMed.
- M.-C. Yu, P. Rich, L. Foreman, J. Smith, M.-S. Yu, A. Tanna, V. Dibbur, R. Unwin and F. W. Tam, Sci. Rep., 2017, 7, 4601 CrossRef PubMed.
- H. Ghimire, M. Venkataramani, Z. Bian, Y. Liu and A. Perera, Sci. Rep., 2017, 7, 16993 CrossRef PubMed.
- G. Theophilou, K. M. Lima, P. L. Martin-Hirsch, H. F. Stringfellow and F. L. Martin, Analyst, 2016, 141, 585–594 RSC.
- A. Käppler, M. Fischer, B. M. Scholz-Böttcher, S. Oberbeckmann, M. Labrenz, D. Fischer, K.-J. Eichhorn and B. Voit, Anal. Bioanal. Chem., 2018, 410, 5313–5327 CrossRef PubMed.
- N. W. Turner, M. Cauchi, E. V. Piletska, C. Preston and S. A. Piletsky, Biosens. Bioelectron., 2009, 24, 3322–3328 CrossRef CAS PubMed.
- M. Dendisova, A. Jeništová, A. Parchaňská-Kokaislová, P. Matějka, V. Prokopec and M. Švecová, Anal. Chim. Acta, 2018, 1031, 1–4 CrossRef CAS PubMed.
- M. Boulet-Audet, S. G. Kazarian and B. Byrne, Sci. Rep., 2016, 6, 30526 CrossRef CAS PubMed.
- R. G. Brereton, Chemometrics: data analysis for the laboratory and chemical plant, John Wiley & Sons, 2003 Search PubMed.
- P. A. Gorry, Anal. Chem., 1990, 62, 570–573 CrossRef CAS.
- S. Wold, M. Sjöström and L. Eriksson, Chemom. Intell. Lab. Syst., 2001, 58, 109–130 CrossRef CAS.
- T. J. Bruno and P. D. N. Svoronos, CRC Handbook of Fundamental Spectroscopic Correlation Charts, CRC Press, 2005 Search PubMed.
- W. E. Hassan, S. M. Eid, K. M. Kelani and A. A. Shalaby, Drug Invent. Today, 2013, 5, 2–7 CrossRef CAS.
- R. Kohavi, Int. Joint Conf. Artif. Intell., 1995, 14, 1137–1145 Search PubMed.
- C. J. Willmott, Phys. Geogr., 1981, 2, 184–194 CrossRef.
- D. M. Haaland and E. V. Thomas, Anal. Chem., 1988, 60, 1193–1202 CrossRef CAS.
- N. Zhao, Z.-s. Wu, Q. Zhang, X.-y. Shi, Q. Ma and Y.-j. Qiao, Sci. Rep., 2015, 5, 11647 CrossRef PubMed.
- N. K. M. Faber and R. Bro, Chemom. Intell. Lab. Syst., 2002, 61, 133–149 CrossRef CAS.
- M. J. Kang, Y. H. Hwang, W. Lee and D. H. Kim, Rapid Commun. Mass Spectrom., 2007, 21, 252–264 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10033d |
| This journal is © The Royal Society of Chemistry 2020 |