
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn investigation of the metabolic activity, isozyme contribution, species differences and potential drug–drug interactions of PI-103, and the identification of efflux transporters for PI-103-O-glucuronide in HeLa1A9 cells†
Li Gaoab,
Zifei Qin *ab,
Beibei Zhangab,
Zhao Yinab,
Xiaojian Zhangab and
Jing Yang*ab
*ab,
Beibei Zhangab,
Zhao Yinab,
Xiaojian Zhangab and
Jing Yang*ab
aDepartment of Pharmacy, The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China. E-mail: qzf1989@163.com
bHenan Key Laboratory of Precision Clinical Pharmacy, Zhengzhou University, Zhengzhou 450052, China. E-mail: jingyang_0101@163.com
First published on 5th March 2020
Abstract
PI-103 is a phosphatidylinositol 3-kinase inhibitor that includes multiple receptor affinity modifications, and it is also a therapeutic drug candidate primarily for human malignant tumors. However, its metabolic fate and potential drug–drug interactions involving human cytochrome P450 (CYP) and UDP-glucuronosyltransferases (UGT) enzymes remain unknown. In this study, our results demonstrated that the intrinsic clearance (CLint) values of oxidated metabolite (M1) in human liver microsomes (HLM) and human intestine microsomes (HIM) were 3.10 and 0.08 μL min−1 mg−1, respectively, while PI-103 underwent efficient glucuronidation with CLint values of 15.59 and 211.04 μL min−1 mg−1 for mono-glucuronide (M2) by HLM and HIM, respectively. Additionally, reaction phenotyping results indicated that CYP1A1 (51.50 μL min−1 mg−1), 1A2 (46.96 μL min−1 mg−1), and UGT1A1 (18.80 μL min−1 mg−1), 1A7 (8.52 μL min−1 mg−1), 1A8 (8.38 μL min−1 mg−1), 1A9 (34.62 μL min−1 mg−1), 1A10 (107.01 μL min−1 mg−1) were the most important contributors for the oxidation and glucuronidation of PI-103. Chemical inhibition assays also suggest that CYP1A2 and UGT1A1, 1A9 play a predominant role in the metabolism of PI-103 in HLM. Significant activity correlations were detected between phenacetin-N-deacetylation and M1 (r = 0.760, p = 0.004) as well as β-estradiol-3-O-glucuronide and M2 (r = 0.589, p = 0.044), and propofol-O-glucuronidation and M2 (r = 0.717, p = 0.009). Furthermore, the metabolism of PI-103 revealed marked species differences, and dogs, rats, mice and mini-pigs were not the appropriate animal models. Gene silencing of breast cancer resistance protein (BCRP) or multidrug resistance-associated protein (MRPs) transporter results indicated that M2 was mainly excreted by BCRP, MRP1 and MRP4 transporters. Moreover, PI-103 displayed broad-spectrum inhibition towards human CYPs and UGTs isozymes with IC50 values ranging from 0.33 to 6.89 μM. Among them, PI-103 showed potent non-competitive inhibitory effects against CYP1A2, 2C19, 2E1 with IC50 and Ki values of less than 1 μM. In addition, PI-103 exhibited moderate non-competitive inhibition against UGT1A7, 2B7, and moderate mixed-type inhibition towards CYP2B6, 2C9 and UGT1A3. Their IC50 and Ki values were 1.16–6.89 and 0.56–5.64 μM, respectively. In contrast, PI-103 could activate the activity of UGT1A4 in a mechanistic two-site model with a Ki value of 13.76 μM. Taken together, PI-103 was subjected to significant hepatic and intestinal metabolism. CYP1A1, 1A2 and UGT1A1, 1A7, 1A8, 1A9, 1A10 were the main contributing isozymes, whereas BCRP, MRP1 and MRP4 contributed most to the efflux excretion of M2. Meanwhile, PI-103 had a potent and broad-spectrum inhibitory effect against human CYPs and UGTs isozymes. These findings could improve understanding of the metabolic fates and efflux transport of PI-103. The inhibited human CYP and UGT activities could trigger harmful DDIs when PI-103 is co-administered with clinical drugs primarily cleared by these CYPs or UGTs isoforms. Additional in vivo studies are required to evaluate the clinical significance of the data presented herein.
Introduction
Phosphatidylinositol 3-kinase (PI3K) is a family of intracellular signal transducer enzymes which are capable of phosphorylating the hydroxyl groups of the inositol ring of phosphatidylinositol.1 Class I PI3Ks including a catalytic (p110α, p110β, p110δ, or p110γ) and a regulatory (p85α, p55α, p50α, p85β, p55γ) subunit, play critical roles in cell survival, proliferation, and differentiation in normal cells.2 p110α and p110β are ubiquitously expressed in all cells, while p110δ is expressed primarily in leukocytes and p110γ expression is largely restricted to hematopoietic cells.1 Briefly, the PI3K pathway is usually activated by cell-surface receptors such as G-protein-coupled receptors and tyrosine kinase receptors.3 However, aberrant activation of this pathway is frequently involved in human tumorigenesis.4 Owing to the importance of the PI3K signaling pathway in the development of human cancers, this pathway has received increased attention for specific therapeutic applications.PI-103, as a potent PI3K inhibitor, inhibited human cancer cell lines with IC50 values of 8, 88, 48 and 150 nM for p110α, p110β, p110δ and p110γ, respectively.5 Furthermore, PI-103 could also inhibit the target of DNA-PK (IC50 = 2 nM).5 In addition, PI-103 inhibits the leukemic proliferation, clonogenicity of leukemic progenitors, and induce mitochondrial apoptosis, especially in the compartment containing leukemic stem cells.6 Furthermore, the rational combination of PI-103 and other chemotherapeutic drugs or radiotherapy show particular promise for inhibiting the growth of cancer cells and tumor in animals.7,8 Importantly, PI-103 produces little toxicity in mice,9 and has little impact on normal hematopoietic progenitors.6 Despite the increasing understanding of the pharmacological properties of PI-103, we know nothing about the metabolic fate, efflux transport and potential drug–drug interactions (DDI) involving human drug-metabolizing enzymes (DMEs), mainly cytochrome P450s (CYPs) and UDP-glucuronosyltransferases (UGTs).
Traditionally, the metabolic studies in vitro are an essential component of the clinical development of a drug candidate because they relate preclinical studies to patient treatment. Additionally, human CYPs and UGTs are primarily responsible for the elimination and detoxification of xenobiotics (e.g., clinical drugs, carcinogens, pollutants) and maintaining the balance of endogenous substances (e.g., bilirubin, estrogens, bile acids).10–12 Inhibition of human CYP or UGT functions can not only trigger potentially adverse clinical DDIs, but could also result in metabolic disorders for endogenous molecules.13–15 Considering the potential co-administration of PI-103 with other chemotherapeutic drugs, it is crucial to evaluate the potential risks of DDI in clinics.
For these goals, this study was first conducted to characterize the metabolic fates of PI-103 in HLM and HIM by ultra-high-performance liquid chromatography coupled to quadrupole time-of-flight tandem mass spectrometry (UHPLC/Q-TOF-MS) and ultra-high-performance liquid chromatography coupled with triple quadrupole tandem mass spectrometry (UHPLC/TQD-MS). Second, the in vitro metabolism involving human CYPs and UGTs was investigated. Third, a previously established UGT1A1-overexpressing HeLa cell model was applied to investigate the active function of efflux transporters, mainly including breast cancer resistance protein (BCRP) and multidrug resistance-associated proteins (MRPs) for the glucuronide. Finally, several widely recognized probe substrates of human CYPs and UGTs were selected to evaluate the potential DDI. Taken together, this study presents the preclinical metabolic fates and potential DDI of PI-103. These collective results may facilitate the design of appropriate clinical regimens for evaluating this drug candidate in phase I clinical trials.
Experimental
Chemicals and reagents
PI-103 (purity > 98%) was purchased from Guangzhou Fans Biotechnology Co., Ltd (Guangzhou, China). Probe substrates and their corresponding metabolites for human CYPs (e.g., ethoxyresorufin, resorufin, phenacetin, paracetamol, coumarin, 7-hydroxycoumarin, bupropion, hydroxybupropion, paclitaxel, 6α-hydroxy-paclitaxel, tolbutamide, 4-hydroxytolbutamide, S-mephenytoin, 4-hydroxymephenytoin, chlorzoxazone, 6-hydroxychlorzoxazone, nifedipine, oxidized nifedipine) were all obtained Sigma-Aldrich (St. Louis, MO, USA). Specific substrates for human UGTs, including β-estradiol, trifluoperazine (TFP), propofol, 4-methylumbelliferone (4-MU), were all also purchased from Sigma-Aldrich (St. Louis, MO, USA). The corresponding glucuronides (e.g., 4-MU-glucuronide, β-estradiol-3-glucuronide, TFP-N-glucuronide, propofol-O-glucuronide) were all obtained from Toronto Research Chemicals (Ontario, Canada).Alamethicin, D-saccharic-1,4-lactone, magnesium chloride (MgCl2), nicotinamide adenine dinucleotide phosphate (NADPH), and uridine 5′-diphospho-glucuronosyltransferase (UDPGA) were all obtained from Sigma-Aldrich (St. Louis, MO, USA). Pooled human liver microsomes (n = 20) and pooled human intestine microsomes (n = 50), individual human liver microsomes (iHLM), dogs liver microsomes (DLM), mice liver microsomes (MLM), rats liver microsomes (RLM), mini-pig liver microsomes (MpLM), and expressed cytochrome P450 proteins (CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4 and 3A5), recombinant UDP-glucuronosyltransferase enzymes (UGT1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B10, 2B15 and 2B17) were all purchased form Corning Biosciences (Corning, NY, USA). All other chemicals and reagents were of analytical grade or the highest grade commercially available.
Incubation conditions
For the phase I metabolism studies, a series of PI-103 standard solutions (0.1–80 μM) were incubated with drug metabolizing enzymes, MgCl2 (5 mM), and NADPH (1 mM) in Tris–HCl buffer (50 mM, pH = 7.4) in incubation system (100 μL) as described recently. The reaction was terminated by ice-cold acetonitrile (100 μL) after incubation at 37 °C for 60 min. In addition, after centrifuging at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800g for 10 min, the supernatant (8 μL) was injected into UHPLC system (Waters, Manchester, UK).
800g for 10 min, the supernatant (8 μL) was injected into UHPLC system (Waters, Manchester, UK).
For the glucuronidation assays, a total volume of 100 μL incubation solutions contained Tris–HCl buffer (50 mM, pH = 7.4), MgCl2 (4.0 mM), alamethicin (22 μg mL−1), saccharolactone (4.4 mM), drug metabolizing enzymes, serial of PI-103 standard solutions (0.1–80 μM) and UDPGA solutions (3.5 mM). After 30 min, ice-cold acetonitrile (100 μL) was added into the incubation mixture to terminate the reaction. In addition, the supernatant (8 μL) was subjected to UHPLC analyses after centrifuging at 13800g for 10 min.
Preliminary experiment was performed to ensure that the metabolic rates of PI-103 were determined under linear conditions with respect to the incubation time (60 min for phase I metabolism and 30 min for glucuronidation) and protein concentration (0.1 mg mL−1 for phase I metabolism and 0.05 mg mL−1 for glucuronidation). Moreover, incubation without NADPH and UDPGA served as negative control to confirm the formed metabolites were NADPH-dependent and UDPGA-dependent, respectively. Similarly, the incubation system for pooled HLM, pooled HIM, individual HLM (n = 12), animal liver microsomes, and each expressed CYP and UGT enzyme was the same as above. All experiments were conducted in triplicate.
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Zhengzhou University and experiments were approved by the Animal Ethics Committee of Zhengzhou University.
Enzyme kinetics evaluation
Two kinetic models including the Michaelis–Menten equation and the substrate inhibition equation, were fitted to the data of metabolic activities versus substrate concentrations, and displayed in eqn (1) and (2), respectively. Meanwhile, appropriate models were selected by visual inspection of the Eadie–Hofstee plot.16 The relative kinetic parameters were as follow. V is the formation rate of BV-related metabolites. Vmax is the maximal velocity. Km is the Michaelis constant and [S] is the substrate concentration. Ksi is the substrate inhibition constant. The intrinsic clearance (CLint) values were derived using Vmax/Km for the Michaelis–Menten and substrate inhibition models.16 Model fitting and parameter estimation were performed using Graphpad Prism V5 software (SanDiego, CA).
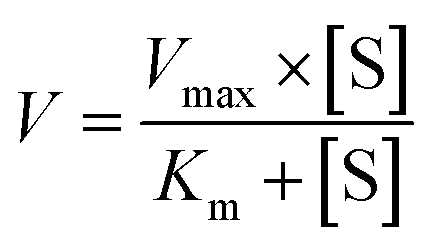 | (1) |
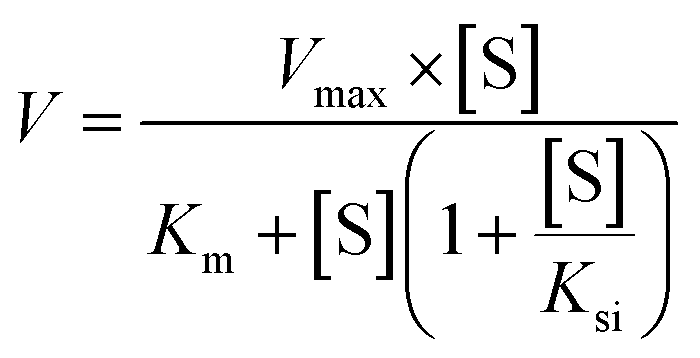 | (2) |
Chemical inhibition assays
Metabolic activities of PI-103 in pooled HLM was measured in the absence or presence of an inhibitor for human CYPs as described previously.17,18 For phase I metabolism, α-naphthoflavone (0.1 μM), montelukast (1.5 μM), sulfaphenazole (3 μM), omeprazole (2 μM), quinidine (0.5 μM), 4-methylpyrazole (3 μM) and ketoconazole (0.1 μM) were used as the inhibitors for CYP1A2, 2C8, 2C9, 2C19, 2D6, 2E1 and 3A, respectively. Similarly, nilotinib (10 μM), protopanaxatriol (500 μM) and glycyrrhetinic acid (20 μM) were all selected as the inhibitors of UGT1A1, whereas CDCA (20 μM), androsterone (10 μM) and amitriptyline (10 μM) were used as the inhibitors for UGT1A3, 1A9 and 2B7, respectively, to investigate their potential inhibitory effects on the glucuronidation in pooled HLM. The incubations conditions were same as these of in vitro metabolism assays.Activity correlation analyses
The metabolic activities of PI-103 and several probe substrates for CYPs and UGTs isozymes were determined by individual HLM (n = 12) according to the assay protocol as published previously.19 PI-103, phenacetin and nifedipine, which the concentrations were 2.5, 100 and 50 μM, respectively, were all incubated with NADPH-supplemented individual HLM (n = 12). Likewise, PI-103, β-estradiol and propofol were incubated with UDPGA-supplemented individual HLM (n = 12) with concentrations of 2.5, 50 and 40 μM, respectively.Furthermore, correlation analyses were performed between PI-103-mono-oxidation (M1) and phenacetin-N-deacetylation, nifedipine-oxidation, respectively. Similarly, correlation analyses were also performed between PI-103-O-glucuronidation (M2) and β-estradiol-3-O-glucuronidation, and propofol-O-glucuronidation, respectively. These correlation (Pearson) analyses were performed using GraphPad Prism V5 software.
Species difference analyses
To explore the appropriate animal model, a series of PI-103 solutions (0.1–80 μM) was incubated with four animal liver microsomes (DLM, MLM, RLM and MpLM) to determine the metabolic rates for phase I metabolism and glucuronidation, respectively. Kinetic parameters were derived from the appropriate model fitting. In addition, the CLint values for the metabolic activities of PI-103 by HLM and each animal liver microsomes were used as the evaluation parameters to estimate species diversity as published previously.20Glucuronide excretion experiments
The human UGT1A9 gene (NM_021027.2) was synthesized and subcloned into the pcDNA3.1(±) vector. The vector carrying the UGT1A9 gene was transiently transfected into HeLa cells using a modified calcium precipitation method as previously described.21 The HeLa cells stably transfected with UGT1A9 were called engineered HeLa1A9 cells. After transfection for 48 h, HeLa1A9 cells were used for PI-103-O-glucuronide excretion experiments. In brief, HeLa1A9 cells were incubated with 2 mL of Hank's buffered salt solution (HBSS) containing PI-103 at 5 μM as a negative control. At each time point (30, 60, 90, and 120 min), 200 μL of incubation solution was removed and immediately replenished with the same volume of HBSS solution containing PI-103 at a concentration of 5 μM. The cells were collected 120 min later and processed as described previously.21 The collected samples were treated with an equal volume of ice-cold acetonitrile and centrifuged at 13800g for 10 min. The samples were subjected to UHPLC analyses to calculate the excreted glucuronide concentration, the excretion rate of glucuronide, the intracellular glucuronide concentration and the fraction metabolized (fmet) value. Similarly, a previously established shRNA plasmid of BCRP, MRP1, MRP3, or MRP4 was transfected into the HeLa1A9 cells following an identical procedure to determine the roles of BCRP, MRP1, MRP3 or MRP4 in the efflux excretion of PI-103-O-glucuronide, respectively.
The excretion rate of PI-103-O-glucuronide was calculated according to eqn (3). The apparent efflux clearance for PI-103-O-glucuronide was derived by ER/Ci, where Ci is the intracellular concentration of PI-103-O-glucuronide. The fmet value reflected the extent of PI-103-O-glucuronide in the HeLa1A9 cells and was calculated as eqn (4). Where V is the volume of the incubation medium; C is the cumulative concentration of PI-103-O-glucuronide; t is the incubation time. Here, dC/dt describes the changes in the PI-103-O-glucuronide levels with time.
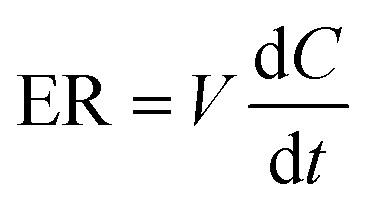 | (3) |
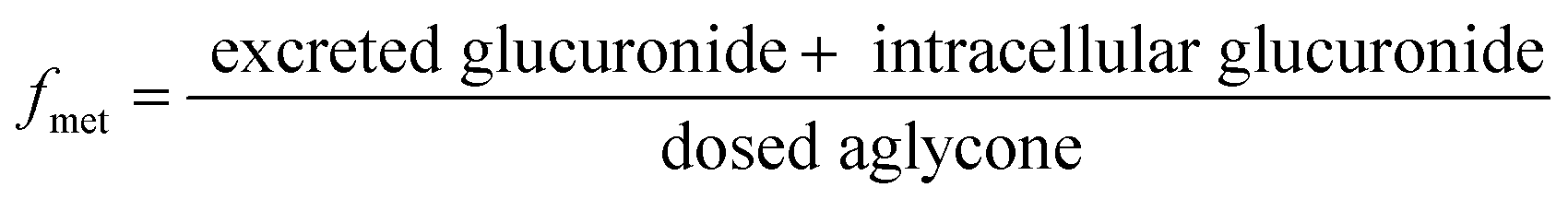 | (4) |
Inhibitory effects of PI-103 against human CYPs and UGTs
In this study, ethoxyresorufin-deethylation, phenacetin-N-deacetylation, coumarin-7-hydroxylation, bupropion-hydroxylation, paclitaxel-6α-hydroxylation, tolbutamide-4-hydroxylation, mephenytoin-4-hydroxylation, chlorzoxazone-6-hydroxylation and nifedipine-oxidation were utilized as the selective marker reactions for CYP1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2E1, 3A4 and 3A5, respectively. The concentrations for these substrates approximately equaled their respective Km values as follows: 1 μM for ethoxyresorufin, 100 μM for phenacetin, 100 μM for coumarin, 100 μM for bupropion, 60 μM for paclitaxel, 200 μM for tolbutamide, 100 μM for mephenytoin, 200 μM for chlorzoxazone and 40 μM for nifedipine, respectively. The specific substrates were incubated with individual CYP isozymes at different protein concentrations (0.05 mg mL−1 for CYP1A1, 1A2, 3A4 and 3A5; 0.1 mg mL−1 for CYP2A6, 2B6, 2C8, 2C9, 2C19 and 2E1) in the absence (control) and presence of PI-103. Aliquots of the supernatant were then collected for further UHPLC-TQD-MS analyses.Likewise, β-estradiol, TFP and propofol were typically used as the specific probe substrates for UGT1A1, 1A4 and 1A9, respectively, while 4-MU was used as the non-selective substrate for UGT1A3, 1A6, 1A7, 1A8, 1A10, 2B4, 2B7, 2B10, 2B15 and 2B17. Corresponding to the reported values of Km for each UGT isozyme, the concentrations of these probe substrates were as follows: 60 μM β-estradiol for UGT1A1, 1200 μM 4-MU for UGT1A3, 40 μM TFP for UGT1A4, 110 μM 4-MU for UGT1A6, 30 μM 4-MU for UGT1A7, 750 μM 4-MU for UGT1A8, 40 μM propofol for UGT1A9, 110 μM 4-MU for UGT1A10, 1000 μM 4-MU for UGT2B4, 350 μM 4-MU for UGT2B7, 1000 μM 4-MU for UGT2B10, 350 μM 4-MU for UGT2B15 and 350 μM SAHA for UGT2B17. The protein concentrations were 0.125, 0.05, 0.1, 0.025, 0.05, 0.025, 0.05, 0.05, 0.25, 0.05, 0.05, 0.2 and 0.5 mg mL−1 for UGT1A1, 1A3, 1A4, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B10, 2B15 and 2B17, respectively. Similarly, each supernatant was injected into the UHPLC-TQD-MS for analyses.
Preliminary experiments were performed to ensure that the experimental conditions were conducted within the linear ranges of the incubation times and protein concentrations. All incubations in this study were performed in triplicate. For those CYP and UGT isozymes which were strongly inhibited by PI-103, the half-inhibition concentration (IC50) values were determined using various PI-103 concentrations by non-linear regression analyses in the incubation conditions described above. The inhibition of human CYPs or UGTs was classified to four categories as follows, potent (IC50 values < 1 μM), moderate (IC50 values between 1 and 10 μM), weak (IC50 values over 10 μM), or no inhibition (IC50 values over 100 μM).
Inhibition or activation kinetic analyses of PI-103 on human CYPs and UGTs
The inhibition constant (Ki) values were determined by various concentrations of the CYP or UGT isozyme substrates in the presence or absence of PI-103 with the program GraphPad Prism V5 software. Three kinetic models were applied to calculate the Ki values by nonlinear regression using eqn (5), (6) and (7) for competitive inhibition, noncompetitive inhibition, and mixed-type inhibition, respectively. The inhibition kinetic type was evaluated by determining the intersection point in the Dixon and Lineweaver–Burk plots. The parameters were as follows. V is the velocity of the reaction; [S] and [I] are the concentrations of the substrate and inhibitor, respectively; Ki is the constant describing the affinity between the inhibitor and the enzyme; Km is the substrate concentration at half of the maximum velocity (Vmax) of the reaction. The αKi describes the affinity of the inhibitor to the complex of enzyme and substrate; when α is very large (α > 1), the binding of inhibitor would prevent the binding of the substrate, and the mixed inhibition model becomes identical to competitive inhibition. Goodness of fit was assessed by the Akaike information criterion (AIC) and the Schwartz information criterion (SC) values. The model with the smallest AIC and SC values was regarded as the best model.
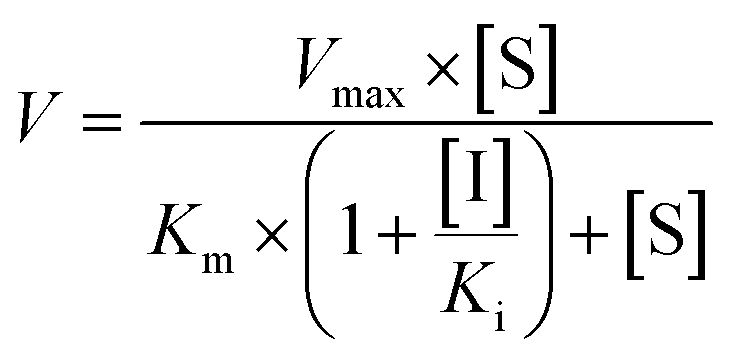 | (5) |
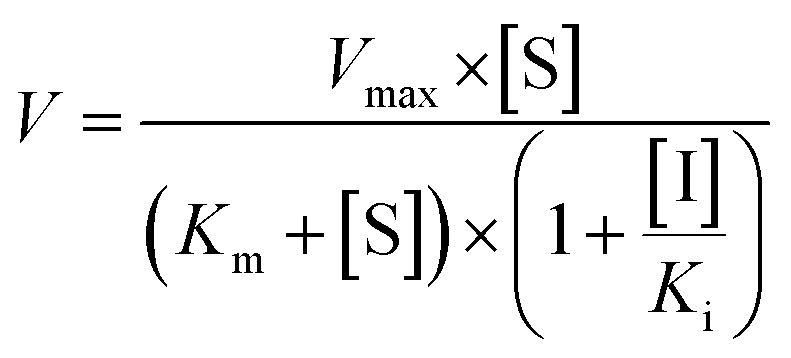 | (6) |
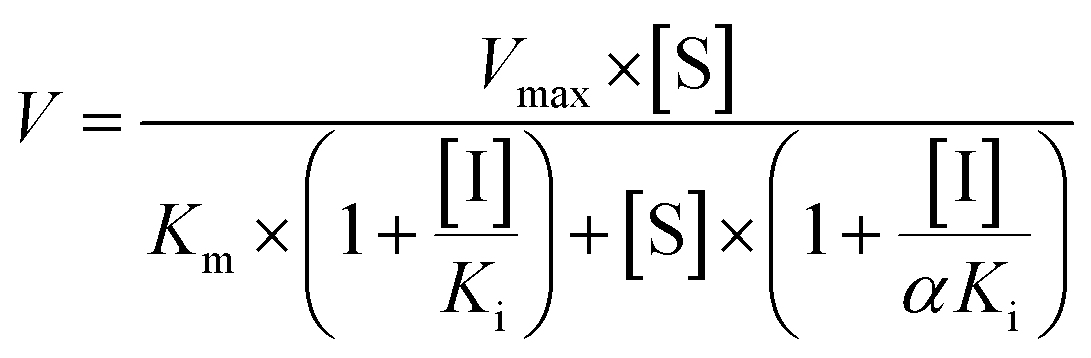 | (7) |
In addition, a mechanistic two-site model was traditionally used to describe the data when activation was observed.22 We assumed that the enzyme had two binding sites, a reaction and an allosteric sites. The substrate only bound the reaction site, while the modifier could bond to both two sites. The Ki value was the binding affinity constant of the modifier (inducer).
Analytical conditions
Due to lack of reference standards, the quantification of formed metabolites was referred to the standard curve of PI-103 based on the assume that PI-103 and its metabolites have similar UV absorbance maxima.16 Therefore, a practical UHPLC analyses were performed on an Acquity™ UHPLC I-Class system equipped with PDA detector (Waters, Manchester, UK). Chromatographic separation was performed on BEH C18 column (2.1 mm × 50 mm, 1.7 μm, Waters, Ireland) with water (A) and acetonitrile (B) (both including 0.1% formic acid, v/v) as the mobile phase. The column temperature was maintained at 35 °C. The gradient elution program was 10% from 0 to 0.5 min, 10–50% B from 0.5 to 2.0 min, 50–100% B from 2.0 to 3.0 min, maintaining 100% B from 3.0 to 3.2 min, 100–10% B from 3.2 to 3.5 min and keeping 10% B from 3.5 to 4.0 min at a flow rate of 0.3 mL min−1. The detection wavelength was set at 270 nm.To better analyze the metabolites, the UHPLC system was coupled to a hybrid quadrupole orthogonal time-of-flight tandem mass spectrometer (SYNAPT G2 HDMS, Waters, Manchester, UK) equipped with electrospray ionization (ESI). The operating parameters were as follows: capillary voltage, 3 kV (ESI+); sample cone voltage, 35 V; extraction cone voltage, 4 V; source temperature, 100 °C; de-solvation temperature, 300 °C; cone gas flow, 50 L h−1 and de-solvation gas flow, 800 L h−1. The full scan mass range was 50–1500 Da. The method employed lock spray with leucine enkephalin (m/z 556.2771 in positive ion mode) to ensure mass accuracy.
For quantification of the metabolites of specific substrates, UHPLC system was coupled to a triple quadrupole mass spectrometer (Waters Xevo TQD, Waters, Manchester, UK) equipped with ESI mode. The mass spectrometers were adjusted as follows: capillary voltage, 3.5 kV (ESI+) or 1.5 kV (ESI−); cone voltage, 50 V (ESI+) or 50 V (ESI−); source temperature, 350 °C; desolvation gas flow, 650 L h−1. The chromatographic separation and mobile phase were same as those above. Their detailed UHPLC and multiple reaction monitoring mode (MRM) conditions were listed in Table S1.† All experimental data were collected in centroid mode and processed using Masslynx 4.1 software.
Statistical analyses
Data are expressed as the mean ± SD (standard deviation). Mean differences between treatment and control groups were analyzed by two-tailed Student's t test. The level of significance was set at p < 0.05 (*), p < 0.01 (**) or p < 0.001 (***).Results
Structural identification of PI-103 related metabolites
PI-103, the parent compound, was defined as M0 with the [M + H]+ ion at m/z 349.130. It eluted at 3.39 min with the extracted ion chromatogram (EIC) shown in Fig. 1A. In addition, M0 gave the fragment ions at m/z 305.104, 278.093 and 262.062 in the MS/MS spectra (Fig. 1B). After incubation, two additional peaks were produced and eluted at 2.89 min (M1) and 2.60 min (M2) in phase I and the glucuronidation system, respectively (Fig. 1A). The mother and daughter ions of M1 were at 365.125, 321.099, 294.088 and 278.057 (Fig. 1B), which were all 16 Da higher than those of M0. Likewise, the [M + H]+ ion of M2 was at m/z 525.162 (Fig. 1B), of which the fragment ions were similar to those of M0. Hence, M1 was were identified as the mono-oxidized metabolite of PI-103, while M2 was considered the glucuronide of PI-103. Their structures are shown in Fig. 1C.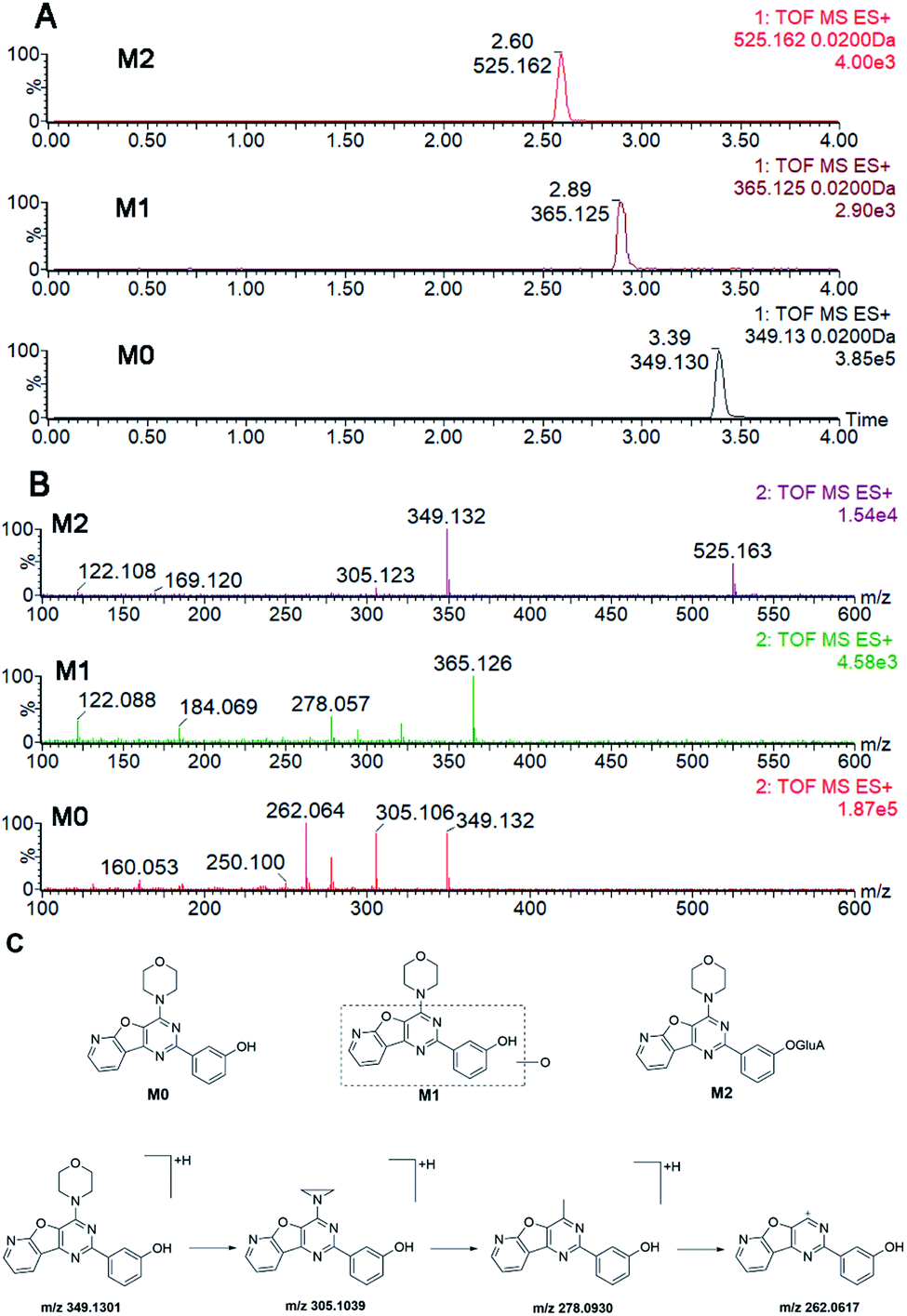 | ||
| Fig. 1 Structure analyses of PI-103 and its metabolites, M1 and M2. (A) The extracted ion chromatograms of PI-103, M1 and M2; (B) the MS/MS spectra of PI-103, M1 and M2; (C) the structure of PI-103, M1 and M2. | ||
Phase I metabolism of PI-103 in HLM, HIM and recombinant CYP enzymes
As shown in Fig. S1A and B,† kinetic profiling revealed that the formation of M1 in pooled HLM and HIM both followed the classical Michaelis–Menten equation. The maximal metabolic fates of M1 in HLM and HIM were 6.75 and 0.46 pmol per min per mg protein, respectively (Table 1). Meanwhile, the intrinsic clearance (CLint) values of M1 were 3.10 and 0.08 μL min−1 mg−1 in HLM and HIM, respectively, following Km values of 2.17 and 5.60 μM, respectively (Table 1). These results indicated that PI-103 was more likely subjected to be transformed in the liver than in the intestine.| Enzymes | Metabolites | Vmax (pmol min−1 mg−1) | Km (μM) | Ksi (μM) | CLint (μL min−1 mg−1) | Model |
|---|---|---|---|---|---|---|
| a HLM, human liver microsomes; HIM, human intestine microsomes; DLM, dog liver microsomes; MLM, mice liver microsomes; RLM, rat liver microsomes; MpLM, mini-pig liver microsomes; SI, substrate inhibition model; MM, Michaelis–Menten model. N.A.: not available. | ||||||
| HLM | M1 | 6.75 ± 0.25 | 2.17 ± 0.35 | N.A. | 3.10 ± 0.51 | MM |
| M2 | 41.19 ± 1.74 | 2.64 ± 0.48 | N.A. | 15.59 ± 2.88 | MM | |
| HIM | M1 | 0.46 ± 0.03 | 5.60 ± 1.06 | N.A. | 0.08 ± 0.01 | MM |
| M2 | 371.0 ± 12.34 | 1.76 ± 0.27 | N.A. | 211.04 ± 32.83 | MM | |
| CYP1A1 | M1 | 36.21 ± 0.90 | 0.70 ± 0.09 | N.A. | 51.50 ± 6.79 | MM |
| CYP1A2 | M1 | 40.62 ± 0.65 | 0.87 ± 0.07 | N.A. | 46.96 ± 3.88 | MM |
| CYP1B1 | M1 | 2.19 ± 0.14 | 1.29 ± 0.39 | N.A. | 1.71 ± 0.54 | MM |
| CYP3A4 | M1 | 1.46 ± 0.06 | 1.72 ± 0.35 | N.A. | 0.85 ± 0.18 | MM |
| CYP3A5 | M1 | 2.58 ± 0.07 | 1.82 ± 0.22 | N.A. | 1.42 ± 0.17 | MM |
| UGT1A1 | M2 | 19.03 ± 0.21 | 1.01 ± 0.06 | N.A. | 18.80 ± 1.05 | MM |
| UGT1A3 | M2 | 3.20 ± 0.07 | 1.67 ± 0.16 | N.A. | 1.91 ± 0.19 | MM |
| UGT1A7 | M2 | 6.63 ± 0.38 | 0.78 ± 0.15 | 270.9 ± 122.7 | 8.52 ± 1.75 | SI |
| UGT1A8 | M2 | 16.71 ± 0.56 | 1.99 ± 0.30 | N.A. | 8.38 ± 1.30 | MM |
| UGT1A9 | M2 | 122.9 ± 11.38 | 3.55 ± 0.74 | 86.85 ± 24.31 | 34.62 ± 7.92 | SI |
| UGT1A10 | M2 | 145.20 ± 1.76 | 1.40 ± 0.08 | N.A. | 107.01 ± 6.08 | MM |
| DLM | M1 | 6.18 ± 0.09 | 0.65 ± 0.05 | N.A. | 9.45 ± 0.75 | MM |
| M2 | 432.2 ± 14.97 | 2.99 ± 0.43 | N.A. | 144.69 ± 21.41 | MM | |
| MLM | M1 | 13.56 ± 0.65 | 1.52 ± 0.22 | 202.6 ± 56.03 | 8.91 ± 1.34 | SI |
| M2 | 273.6 ± 8.32 | 2.63 ± 0.34 | N.A. | 105.37 ± 13.87 | MM | |
| RLM | M1 | 9.56 ± 0.30 | 0.92 ± 0.14 | N.A. | 10.42 ± 1.64 | MM |
| M2 | 95.31 ± 2.98 | 0.71 ± 0.12 | N.A. | 133.39 ± 22.00 | MM | |
| MpLM | M1 | 20.46 ± 0.47 | 1.21 ± 0.14 | N.A. | 16.87 ± 1.92 | MM |
| M2 | 373.1 ± 10.79 | 2.57 ± 0.32 | N.A. | 145.23 ± 18.44 | MM | |
Furthermore, twelve CYP isozymes were analyzed for their catalysis activities of PI-103. The results suggested that CYP1A1, 1A2, 1B1, 2B6, 2D6, 2E1, 2C8, 2C9, 2C19, 3A4 and 3A5 all participated in the phase I metabolism of PI-103 at 1 and 10 μM (Fig. S2A†). However, due to a concentration under the limit of quantification, we were unable to determine the kinetic parameters in the absence of a full kinetic profile in CYP2B6, 2D6, 2E1, 2C8, 2C9 and 2C19. The kinetic profiles of M1 in CYP1A1 (Fig. S3A†), 1A2 (Fig. S3B†), 1B1 (Fig. S3C†), 3A4 (Fig. S3D†) and 3A5 (Fig. S3E†) were all well modeled by the Michaelis–Menten equation, which followed the same kinetic as the formation of M1 in HLM (Fig. S1A†) and HIM (Fig. S1B†). In addition, the CLint values of M1 (Fig. 2A) by CYP1A1, 1A2, 1B1, 3A4, and 3A5 were 51.50, 46.96, 1.71, 0.85, 1.42 μL min−1 mg−1, respectively (Table 1), which indicated that CYP1A1 exhibited the highest activity toward the formation of M1. Furthermore, the Km values ranged from 0.70 to 1.82 μM (Table 1), which suggested that these CYP isozymes exhibited a strong affinity towards PI-103.
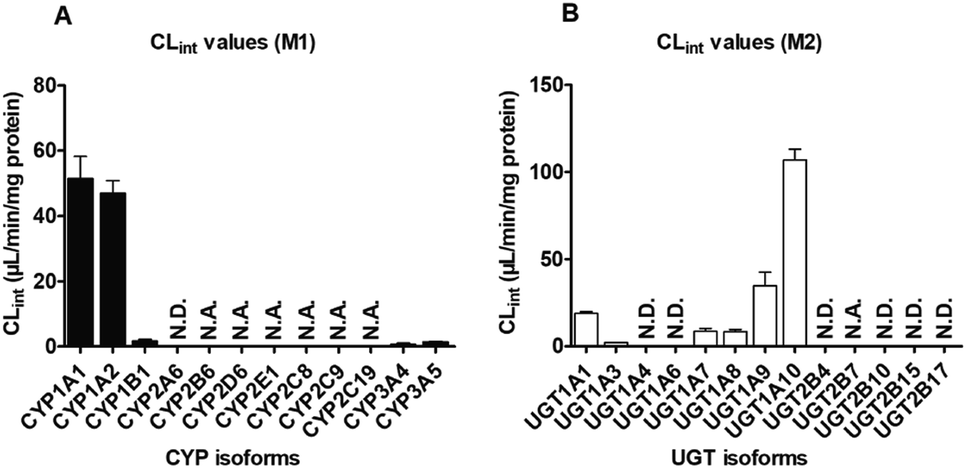 | ||
| Fig. 2 The intrinsic clearance (CLint) values of PI-103 by expressed isozymes. (A) Twelve CYP isoforms; (B) thirteen UGT isozymes. All experiments were performed in triplicate. Data are expressed as the mean ± SD. N.A.: unable to determine the kinetic parameters in the absence of a full kinetic profile. N.D.: not detected. | ||
Glucuronidation of PI-103 in HLM, HIM and expressed UGT enzymes
Like phase I metabolism, the formation of M2 in pooled HLM (Fig. S1C†) and HIM (Fig. S1D†) both followed the Michaelis–Menten equation with small Km values of 2.64 and 1.76 μM, respectively (Table 1), suggesting that PI-103 was a high affinity substrate for HLM and HIM. Furthermore, the Vmax values for M2 in HLM and HIM were 41.19 and 371.0 pmol min−1 mg−1, respectively, with the CLint values of 15.59 and 211.04 μL min−1 mg−1, respectively (Table 1). Notably, the glucuronidation of PI-103 was more efficient in the intestine than in the liver.Of thirteen tested UGT enzymes, UGT1A1, 1A3, 1A7, 1A8, 1A9, 1A10 and 2B7 were responsible for the glucuronidation of PI-103 at 1 and 10 μM (Fig. S2B†). Likewise, it was difficult to obtain a full kinetic profile of M2 in UGT2B7 due to the low concentration of the glucuronide. Of note, the glucuronidation of PI-103 mediated by UGT1A7 (Fig. S4C†) and 1A9 (Fig. S4E†) both followed the substrate inhibition equation, which did not always follow the same kinetics as in HLM (Fig. S1C†) and HIM (Fig. S1D†). Additionally, the kinetic profiles of M2 by UGT1A1 (Fig. S4A†), 1A3 (Fig. S4B†), 1A8 (Fig. S4D†) and 1A10 (Fig. S4F†) all followed Michaelis–Menten kinetics. The CLint value (Fig. 2B) for UGT1A10 was 107.01 μL min−1 mg−1 (Table 1). By contrast, the glucuronidation of PI-103 (Fig. 2B) by UGT1A1 (18.80 μL min−1 mg−1), 1A3 (1.91 μL min−1 mg−1), 1A7 (8.52 μL min−1 mg−1), 1A8 (8.38 μL min−1 mg−1) and 1A9 (34.62 μL min−1 mg−1) was significantly less efficient (Table 1). The Km values by these UGT isozymes were between 0.78 and 3.55 μM, which also illustrated that PI-103 could easily undergo glucuronidation.
Chemical inhibition assays
As shown in Fig. S5A,† only α-naphthoflavone (0.1 μM) significantly inhibited in HLM, with a remaining activity of 53.37% of the control values, while other chemical inhibitors did not alter the phase I metabolism of PI-103 in HLM. These findings suggested that CYP1A2 played an important role in the oxidation of PI-103 in HLM.In addition, nilotinib (10 μM) and glycyrrhetinic acid (20 μM) both produced inhibition in HLM, decreasing the activity to 82.37% and 78.63% of the control values, respectively (Fig. S5B†). Furthermore, the glucuronidation of PI-103 by HLM was decreased to 69.36% of control values in the presence of androsterone (10 μM) (Fig. S5B†). In contrast, no changes were detected when treated with CDCA (20 μM) and amitriptyline (10 μM) (Fig. S5B†). These results illustrated UGT1A1 and UGT1A9 were the important isozymes for the glucuronidation of PI-103.
Activity correlation analyses
We found that the generation of M1 was strongly correlated with phenacetin-N-deacetylation with correlation factors (r = 0.760, p = 0.004) (Fig. S6A†). In contrast, no significant correlation was observed between the formation of M1 and nifedipine-oxidation (r = 0.412, p = 0.184) (Fig. S6B†). These findings suggested that CYP1A2 contributed more to the glucuronidation of PI-103 than CYP3A4.Similarly, there was a correlation between the glucuronidation of PI-103 and specific marker reactions of UGT isozyme substrates. The production of M2 was significantly correlated with β-estradiol-3-O-glucuronidation (r = 0.589, p = 0.044) (Fig. S6C†) and propofol-O-glucuronidation (r = 0.717, p = 0.009) (Fig. S6D†), respectively. These results indicated that the contribution of UGT1A1 and 1A9 to the glucuronidation of PI-103 in liver was appreciable.
Species differences
On the whole, M1 was also only metabolite of PI-103 after incubation with animal liver microsomes. Except for MLM (substrate inhibition kinetics, Fig. S7B†), kinetic profiles all followed the Michaelis–Menten equation by DLM (Fig. S7A†), RLM (Fig. S7C†) and MpLM (Fig. S7D†). The apparent Km values were 0.65, 1.52, 0.92 and 1.21 μM for the formation of M1 by DLM, MLM, RLM and MpLM, respectively (Table 1). The catalytic efficiencies (reflected by CLint values, Fig. 3A) for M1 by human and animal liver microsomes followed the order of MpLM (16.87 μL min−1 mg−1) > RLM (10.42 μL min−1 mg−1) > DLM (9.45 μL min−1 mg−1) > MLM (8.91 μL min−1 mg−1) > HLM (3.10 μL min−1 mg−1).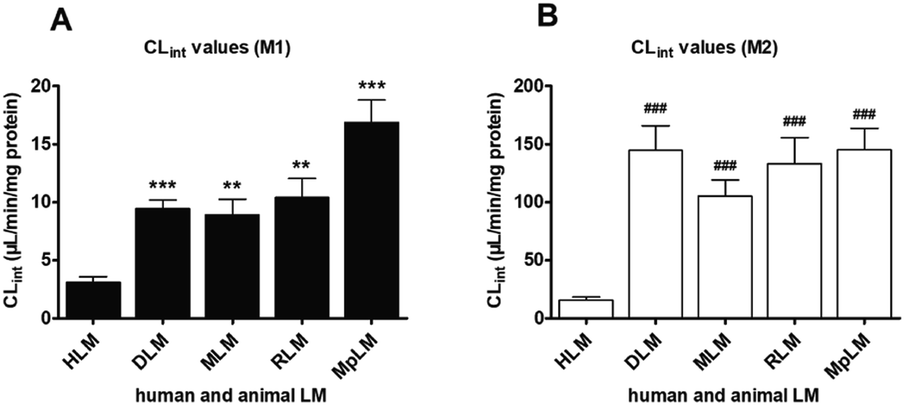 | ||
| Fig. 3 The intrinsic clearance (CLint) values of PI-103 for HLM and four animals microsomes. (A) Comparison of CLint values for oxidation; (B) comparison of CLint values for glucuronidation. All data represent the means ± SD of triplicate determinations (*, # compared with the CLint values of M1 and M2 in HLM, respectively. *, #p < 0.05, **, ##p < 0.01, ***, ###p < 0.001). | ||
As shown in Fig. S8,† the glucuronidation of PI-103 by DLM (Fig. S8A†), MLM (Fig. S8B†), RLM (Fig. S8C†) and MpLM (Fig. S8D†) all followed classical Michaelis–Menten kinetics. The Km values ranged from 0.71 to 2.99 μM, and the Vmax values ranged from 95.31 to 432.2 pmol min−1 mg−1 (Table 1). In addition, the CLint values for M2 were 145.23, 144.69, 133.39, 105.37 and 15.59 μL min−1 mg−1 by MpLM, DLM, RLM, MLM and HLM (Fig. 3B).
Marked species differences (reflected by CLint values, Fig. 3) were noted for the derived kinetic parameters (Table 1). Compared with the CLint values of M1 by HLM, about 5.4, 3.4, 3.0 and 2.9-fold of the CLint values by MpLM, RLM, DLM and MLM were obtained, respectively (Table 1). Likewise, up to 9.3, 9.3, 8.6 and 6.8-fold of the CLint values for M2 were calculated between HLM, and MpLM, DLM, RLM, MLM, respectively (Table 1). Therefore, there were significant species differences in the phase I metabolism (Fig. 3A) and glucuronidation (Fig. 3B) of PI-103. Neither dogs, rats, mice or mini-pigs were the most appropriate models for the in vivo metabolism of PI-103 in humans.
Concentration-dependent excretion of PI-103-O-glucuronide in HeLa1A9 cells
In this study, PI-103 has no significant toxicities in HeLa cells within the experimental concentrations (maximal 5 μM). As shown in Fig. S9A,† M2 was efficiently formed and excreted after incubation of PI-103 (2.5 and 5.0 μM) with HeLa1A9 cells. The excretion rates for M2 were 0.08 and 0.11 pmol min−1 when treated with PI-103 at 2.5 and 5.0 μM, respectively (Fig. S9B†). The excreted M2 and excretion rates exhibited concentration-dependency manner when PI-103 ranged from 2.5 to 5.0 μM within 120 min.The effects of efflux transporter silencing on the excretion of PI-103-O-glucuronide
As with the transfection method previously described, the single transporter silencing efficiency (reflected by mRNA and protein levels) in HeLa1A9 cells was verified to be approximately 30–50%, which indicated that BCRP-shRNA, MRP1-shRNA, MRP3-shRNA and MRP4-shRNA were successfully transfected into HeLa1A9 cells.We found that the knock-down of BCRP resulted in a significant reduction in excreted M2 (24.2–35.0%, p < 0.05, Fig. 4A), excretion rates of M2 (24.2%, p < 0.05, Fig. 4C) and the fraction of metabolized (fmet) PI-103 (27.6%, p < 0.05, Fig. 4E) but an increase in intracellular M2 (27.6%, p < 0.05, Fig. 4D). Furthermore, similar observations were also detected in the extracellular M2 (21.7–33.2%, p < 0.05, Fig. 4A), the efflux excretion rate of M2 (21.7%, p < 0.05, Fig. 4C), the intracellular M2 (29.9%, p < 0.05, Fig. 4D) and the fmet value (29.2%, p < 0.05, Fig. 4E) when MRP1 transporter was partially silenced. Likewise, silencing of the MRP4 transporter produced a significant decrease (25.4–38.4%, p < 0.05, Fig. 4B) in the excreted M2 as well as the efflux clearance of M2 (25.4%, p < 0.01, p < 0.05, Fig. 4C) and fmet value of PI-103 (33.8%, p < 0.01, Fig. 4E). On the contrary, a marked increase (34.7%, p < 0.05, Fig. 4D) was observed in the intracellular level of M2. However, there were almost no alterations (p > 0.05) in the efflux of extracellular M2 (5.1–11.6%, p > 0.05, Fig. 4B), the excretion rate of M2 (8.0%, p > 0.05, Fig. 4C), the intracellular level of M2 (9.4%, p > 0.05, Fig. 4D) and the fmet value of PI-103 (9.2%, p > 0.05, Fig. 4E) when MRP3 was silenced. Taken together, these findings clearly indicated that BCRP, MRP1 and MRP4 were the most important contributors to the efflux excretion of PI-103-related glucuronide.
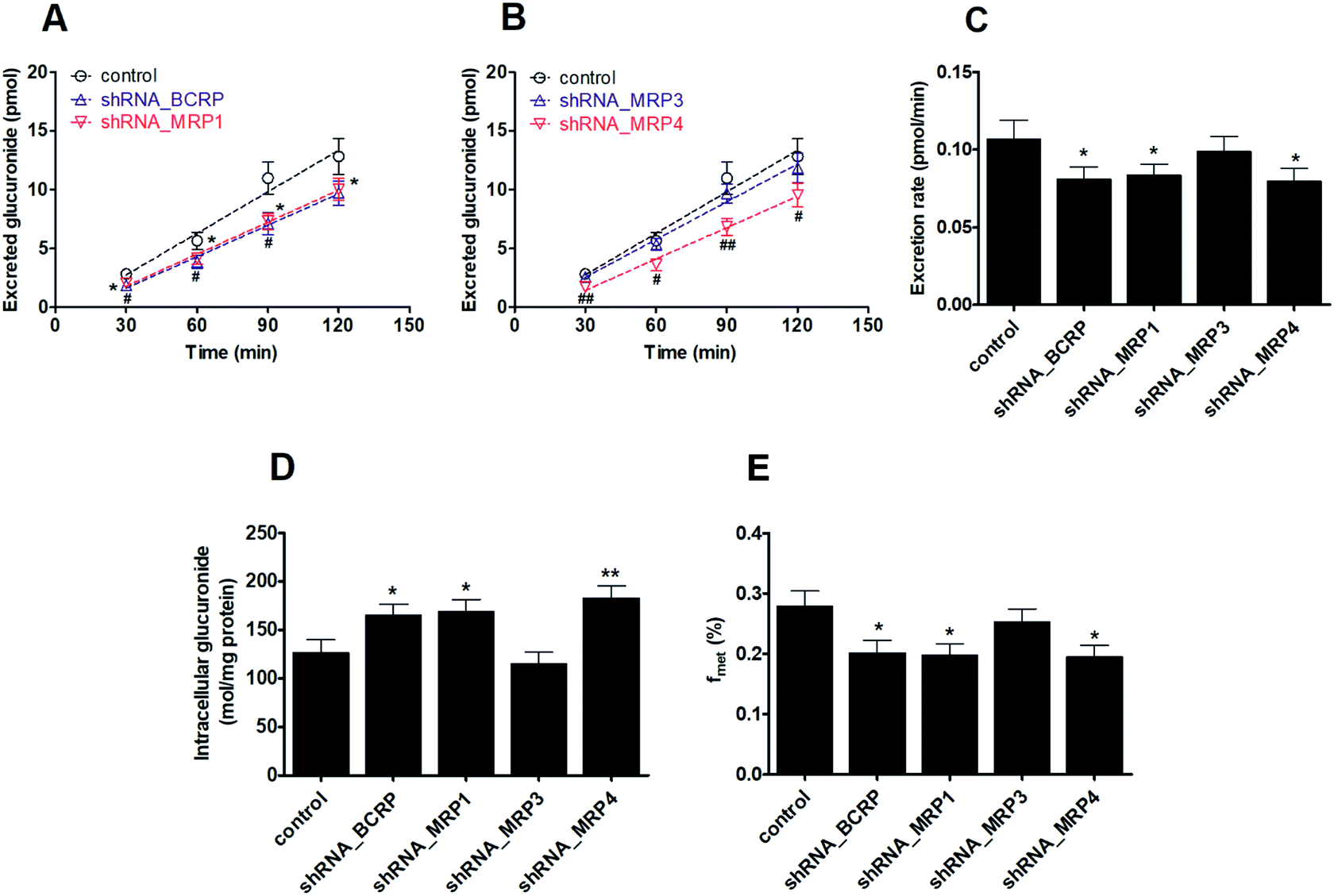 | ||
| Fig. 4 Effects of shRNA-mediated knock-down of efflux transporters for the excretion rates and intracellular levels of M2 in HeLa1A9 cells. (A) The accumulated M2 in extracellular medium when BCRP or MRP1 was silenced; (B) effects of MRP3 or MRP4 knock-down on the accumulation of M2 in extracellular solution; (C) excretion rates of M2 after BCRP, MRP1, MRP3, or MRP4 transporters were silenced; (D) the intracellular levels of M2 when inhibition of BCRP, MRP1, MRP3, or MRP4 transporters; (E) effects of BCRP, MRP1, MRP3, or MRP4 silencing on cellular glucuronidation (or fmet) of PI-103. All experiments were performed in triplicate. Data were presented as mean ± SD. (*, # compared with those of control, *, #p < 0.05, **, ##p < 0.01, ***, ###p < 0.001). | ||
Effects of PI-103 on the activities of human CYP and UGT isozymes
The inhibitory effects of PI-103 (1, 10 and 100 μM) towards human CYPs isozymes were first investigated, using the corresponding probe substrates of each CYP enzyme. It was evident from Fig. 5A that PI-103 exhibited negligible or weak inhibitory effects on CYP2A6, 2C8, 3A4 and 3A5. Notably, PI-103 displayed relatively strong inhibition of CYP1A1, 1A2, 2B6, 2C9, 2C19 and 2E1. Upon the addition of 100 μM PI-103, the residual activities of these six CYP isozymes were 21.39%, 8.63%, 29.70%, 14.22%, 14.67%, and 13.69% of the negative control, respectively. To further investigate the potential inhibitory effects of PI-103, the concentration-dependent inhibition curves of PI-103 against these six CYPs enzymes were investigated. As shown in Fig. S10† and Table 2, the inhibition of PI-103 towards these CYPs isoforms were dose-dependent, and their IC50 values were 16.76, 0.37, 2.25, 1.16, 0.59 and 0.33 μM for CYP1A1, 1A2, 2B6, 2C9, 2C19 and 2E1, respectively.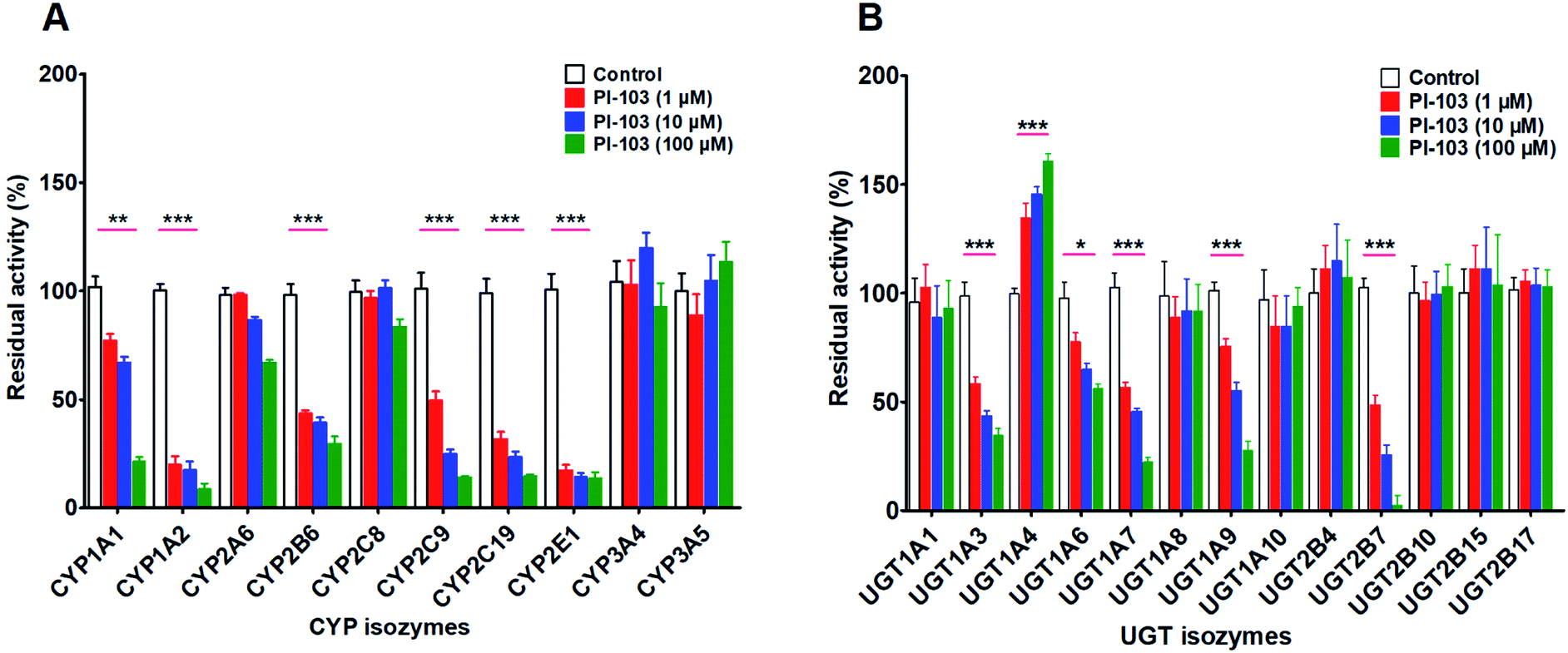 | ||
| Fig. 5 The effects of PI-103 against expressed isozymes. (A) Ten major CYPs isoforms; (B) thirteen main UGTs isozymes. The specific substrates were incubated at 37 °C in the absence (control) and presence of PI-103 (1, 10 and 100 μM). All experiments were performed in triplicate determinations. The data were presented as mean ± SD. (* compared with those of control, *p < 0.05, **p < 0.01, ***p < 0.001). | ||
| Isozymes | Substrate | IC50 (μM) | Ki (μM) | α | Type of inhibition | Goodness of fit (R2) |
|---|---|---|---|---|---|---|
| a —: not available. | ||||||
| UGT1A3 | 4-MU | 6.89 ± 0.73 | 2.58 ± 0.84 | 2.97 ± 1.61 | Mixed-type | 0.9757 |
| UGT1A6 | 4-MU | 167.60 ± 15.28 | — | — | — | — |
| UGT1A7 | 4-MU | 6.25 ± 0.84 | 5.64 ± 0.60 | — | Noncompetitive | 0.9611 |
| UGT1A9 | Propofol | 16.58 ± 1.29 | — | — | — | — |
| UGT2B7 | 4-MU | 1.59 ± 0.21 | 1.35 ± 0.15 | — | Noncompetitive | 0.9721 |
| CYP1A1 | Ethoxyresorufin | 16.76 ± 1.76 | — | — | — | — |
| CYP1A2 | Phenacetin | 0.37 ± 0.04 | 0.38 ± 0.03 | — | Noncompetitive | 0.9779 |
| CYP2B6 | Bupropion | 2.25 ± 0.28 | 0.64 ± 0.22 | 5.09 ± 4.48 | Mixed-type | 0.9673 |
| CYP2C9 | Tolbutamide | 1.16 ± 0.15 | 0.56 ± 0.12 | 2.45 ± 0.94 | Mixed-type | 0.9901 |
| CYP2C19 | Mephenytoin | 0.59 ± 0.06 | 0.53 ± 0.03 | — | Noncompetitive | 0.9848 |
| CYP2E1 | Chlorzoxazone | 0.33 ± 0.04 | 0.25 ± 0.01 | — | Noncompetitive | 0.9949 |
As shown in Fig. 5B, PI-103 displayed strong inhibitory effects on the catalytic activities of UGT1A3, 1A6, 1A7, 1A9 and 2B7, and the inhibitory effects against the activities of UGT1A1, 1A8, 1A10, 2B4, 2B10, 2B15 and 2B17 were negligible. At 100 μM for PI-103, the activities of UGT1A3, 1A6, 1A7, 1A9 and 2B7 were inhibited to 34.80%, 56.03%, 22.32%, 27.78% and 2.56% of their control activities, respectively. Furthermore, the dose-dependent inhibition curves of PI-103 against these UGTs isoforms were constructed to investigate its inhibitory potential. As shown in Fig. S11† and Table 2, PI-103 dose-dependently inhibited these tested UGTs enzyme, and the IC50 values for UGT1A3, 1A6, 1A7, 1A9 and 2B7, were calculated to be 6.89, 167.60, 6.25, 16.58 and 1.59 μM, respectively. These findings prompted us to further investigate the types of inhibition kinetics and the corresponding inhibition parameters of PI-103, specifically for CYP1A2, 2B6, 2C9, 2C19, 2E1, and UGT1A3, 1A7 and 2B7 with IC50 values less than 10 μM.
Inhibition or activation kinetics of PI-103 on human CYP and UGT isozymes
Inhibition kinetic analyses were further conducted to characterize the inhibition types and the inhibition kinetic constants of PI-103 towards CYP1A2, 2B6, 2C9, 2C19 and 2E1. Three conventional inhibition models (i.e., competitive, noncompetitive and mixed-type) were used to describe the inhibition data of PI-103. PI-103 exerted noncompetitive inhibition against CYP1A2, 2C19 and 2E1, as well as mixed inhibition against CYP2B6 and 2C9 according to the AIC and SC values (Table S2†). Further, the Dixon plots (Fig. 6) also provided strong evidences to support this judgment. The Ki values of PI-103 were 0.38, 0.64, 0.56, 0.53 and 0.25 μM for CYP1A2, 2B6, 2C9, 2C19 and 2E1, respectively (Table 2). The α values for CYP2B6 and 2C9 were 5.09 and 2.45, respectively. These results indicated that PI-103 displayed relatively strong inhibitory effects on CYP1A2, 2B6, 2C9, 2C19 and 2E1.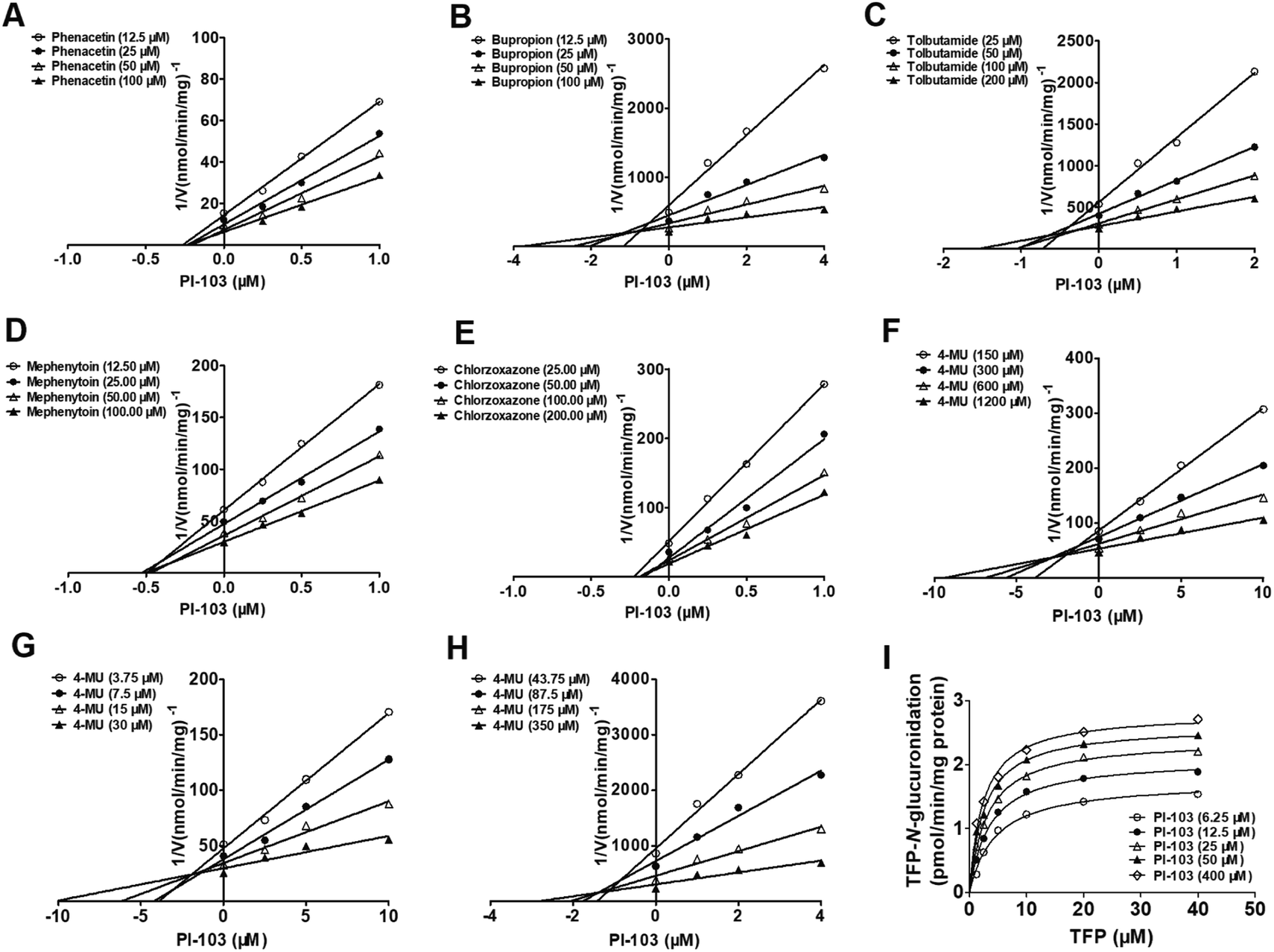 | ||
| Fig. 6 The Dixon plots for the effects of PI-103 towards five CYPs and four UGTs isoforms. (A) Phenacetin-N-deacetylation for CYP1A2; (B) bupropion-hydroxylation for CYP2B6; (C) tolbutamide-4-hydroxylation for CYP2C9; (D) mephenytoin-4-hydroxylation for CYP2C19; (E) chlorzoxazone-6-hydroxylation for CYP2E1; (F) 4-MU-glucuronidation for UGT1A3; (G) 4-MU-glucuronidation for UGT1A7; (H) 4-MU-glucuronidation for UGT2B7; (I) TFP-N-glucuronidation for UGT1A4. All data represent the means ± SD of triplicate determinations. | ||
Similarly, noncompetitive and mixed-type inhibition models best fitted the inhibition data of PI-103 towards UGT1A3, 1A7 and 2B7 according to the AIC and SC values (Table S2†). The best fitting of this model to the data was also justified by the Dixon plots (Fig. 6). The Dixon plots showed that PI-103 exhibited mixed reversible inhibition behaviors towards UGT1A3, whereas it acted as a noncompetitive inhibitor against UGT1A7 and 2B7. Further, the Ki values of PI-103 against 4-MU-glucuronidation by UGT1A3, 1A7 and 2B7 were also determined as 2.58, 5.64 and 1.35 μM, respectively (Table 2). The α values for UGT1A3 was 2.97. In contrast, it was noted that PI-103 could significantly activate UGT1A4, and the maximal rate change was 1.6-fold (Fig. 6I). The binding affinity of PI-103 toward UGT1A4 was calculated as 13.76 μM. These findings suggested that PI-103 was a potent non-selective inhibitor of many human CYP and UGT isozymes with low Ki values ranging from 0.25–0.64 μM for CYPs isozymes and 1.35–5.64 μM for UGT enzymes.
Discussion
PI-103 is a promising phosphatidylinositol 3-kinase inhibitor which is likely to provide an additional cancer therapeutic option. However, no published reports to date have investigated its metabolic fates. In our study, the in vitro metabolism in pooled HLM, HIM, expressed CYPs and UGTs, were investigated for the first time, and the results demonstrate that PI-103 was subjected to glucuronidation rather than oxidative metabolism in pooled HLM and HIM since higher CLint values of glucuronidation were obtained (Table 1). Notably, CYP1A1 and UGT1A10 in extrahepatic tissues (mainly the intestine) as well as CYP1A2 and UGT1A1 mainly in the liver all, accounted for the significant contributions to the metabolism of PI-103.23 Apart from the metabolism in the liver and the intestine, the lung and kidney may also participate in its metabolic fate because CYP1A1 is also present in these tissues, and UGT1A9 is mainly expressed in the kidney.24 Therefore, the metabolic roles of the lung and kidney should not be underestimated. Furthermore, the reaction phenotyping results (Fig. S2†) combined with chemical inhibition assays (Fig. S5†) and activity correlation analyses assays (Fig. S6†) all provided sufficient evidence to support these results.Characterization of PI-103-O-glucuronidation also assumed a significant role in the understanding of its systemic exposure and disposition. Soluble PI-103-O-glucuronides were transported out of the cell through high affinity efflux transporters, and became sequestered in the water compartment of the tissues that eventually led to elimination.21 In this study, a previously developed HeLa1A9 cell model combined with a gene silencing approach was applied to evaluate the efflux transport of PI-103-O-glucuronides.21 BCRP, MRP1 and MRP4 were identified as the key contributors (Fig. 4). However, there is a significant limitation regarding the unexplained role of MRP2 in glucuronide excretion due to the absence of MRP2 in HeLa1A9 cells.21 Fortunately, a developed MDCKII-MRP2-UGT1A1 cell model,25 provides an appropriate means to determine how the over-expression of MRP2 and UGT1A9 (or UGT1A10) influences the cellular kinetics of the PI-103-O-glucuronidation processes.
Additionally, the multidrug resistance of cancer cells towards chemotherapeutic drugs is a major factor in cancer therapy, accounting for treatment failure in over 90% of human patients with metastatic or recurrent cancer.26 In this study, another important shortcoming was that this HeLa1A9 cell model is not suitable for the efflux transport of parent compounds (e.g., PI-103), because efflux transporters requires initially high intracellular concentrations of substrates (e.g., PI-103). Unfortunately, we cannot guarantee initially higher intracellular concentrations of the substrates in this model. Recently, a basal membrane vesicle model,27 has provided a practical tool to investigate the function of BCRP and MRPs in the efflux transport of PI-103. As a chemotherapeutic drug candidate, the efflux transport mechanism of PI-103 must be further elucidated.
It is well accepted that the coordinated interplay between efflux transporters and UGT enzymes indicated an interdependence between efflux transporters (i.e., BCRP, MRPs) and glucuronidation, which was determined as the “glucuronidation-transport interplay”, limiting the systemic exposure of clinical drugs.28 In this study, the reduced expression of BCRP, MRP1 and MRP4 all led to decreased glucuronidation and an increase of intracellular glucuronides (Fig. 4). This finding was attributed to the fact that the glucuronides can be hydrolyzed to the parent compound (e.g., PI-103) by β-glucuronidase within HeLa1A9 cells as previously reported.28,29 De-glucuronidation mediated by β-glucuronidase represents one important aspect of glucuronidation-efflux interplay. In addition, the decreased metabolized fraction of PI-103 was also most likely the result of the elevated impact of β-glucuronidase activity when efflux transporters were knocked-down. Therefore, de-glucuronidation is a critical determinant for the intracellular exposure of parent drug or its glucuronides.
In clinics, greater attention should be given to avoiding the potential risks of drug–drug interactions by considering the inhibitory effects of bioactive compounds on several human CYPs and UGTs.30,31 Therefore, the inhibitory effects of PI-103 against human CYP and UGT isozymes were investigated for the first time (Fig. 6), and our findings suggest that PI-103 is a potent and broad-spectrum inhibitor of human CYPs and UGTs (Table 2). Notably, PI-103 displayed potent inhibitory effects against CYP1A2, 2B6, 2C9, 2C19 and 2E1 with low Ki values ranging from 0.25 to 0.64 μM, while the Ki values for UGT1A3, 1A7 and 2B7 ranged between 1.35 and 5.64 μM. These five CYPs metabolize approximately 40% of clinically used drugs.32 CYP1A2 could metabolize many commonly used therapeutic drugs, such as phenacetin, theophylline, and caffeine.33 Furthermore, CYP2B6 is mainly involved in the metabolism of bupropion and efavirenz, whereas CYP2E1 mainly metabolizes chlorzoxazone.33 In addition, CYP2C9 participates in the metabolism of tolbutamide, phenytoin, S-warfarin, among others.33 Furthermore, CYP2C19 is an important enzyme that is responsible for the metabolism and elimination of proton pump inhibitors, benzodiazepines and antidepressants.33 The low Ki values (below 1 μM) suggest that much more caution should be exercised when PI-103 was co-administered with these CYPs substrates.
Similarly, UGT isozymes participate in metabolic elimination of endogenous compounds, and occupy more than 35% of phase II drug metabolism.34 UGT1A3 is an important enzyme that participates in the clearance of bile acids.18 Meanwhile, UGT1A3 appear to be of particular importance in the metabolism of many clinical drugs (e.g., alizarin, cyproheptadine).18 Inhibition of UGT1A3 function may lead to abnormal bile acid levels and altered systemic exposure of substrate drugs. UGT1A7, as an extra-hepatic UGT, plays a key role in the first biochemical protective barrier, and impaired UGT1A7 activity could result in increased risks for developing several types of cancers.35 UGT2B7 is an abundant UGT enzyme in the liver, kidney and intestine, and is responsible for the glucuronidation of many clinically used drugs, such as zidovudine and morphine, among others.18 It is noteworthy that UGT2B7 shows a lower catalytic capability in children below 11 years old which was approximately 25% of that observed in adults.36 Therefore, UGT2B7 substrates may undergo a slow clearance and reach a relatively high exposure in children when co-administrated with PI-103. In most cases, PI-103 is intravenously or intraperitoneally administered, and its exposure levels against these UGT enzymes may exceed the Ki values (Table 2). It can be expected that some undesired effect from low or impaired activities of these UGT isozymes could be observed in those with co-administration of PI-103.
Notably, the inhibition of PI-103 towards human CYP and UGT isozymes could also be influenced by genetic polymorphisms.35 This is because the protein expression and functional activity of several enzymes could be dramatically altered by polymorphic expression.37 For example, CYP1A1/CYP1A2 rs2472297C>T is a potential genetic marker associated with variability in CYP1A2-mediated drug metabolism.38 In addition, UGT1A1 polymorphisms (1A1*6, 1A1*28) should be investigated to reduce or avoid the dose-limiting toxicities of chemotherapy drugs.23 In contrast, two-fold higher glucuronidation toward several carcinogenic environmental contaminants was observed in UGT1A9*22/*22 livers compared with UGT1A9*1/*1 and UGT1A9*1/*22 livers.39 UGT1A10 codon 139 (Glu > Lys) polymorphism exhibited higher catalytic activities against polycyclic aromatic hydrocarbons, and this genotype is most likely an important determinant in the risk for tobacco-related cancers.40 Therefore, it is necessary to detect the genetic polymorphisms of active CYP and UGT isozymes for several substrate drugs with a narrow therapeutic window.
Conclusions
In summary, this is the first study to characterize the metabolic pathways of PI-103. PI-103 was prone to undergo mono-oxidation and glucuronidation in human liver and intestine (Fig. S1†). Additionally, CYP1A1, 1A2, and UGT1A1, 1A9, 1A10 were identified as the most important contributors for metabolism (Table 1). Meanwhile, chemical inhibition assays (Fig. S5†) and the activity correlation analyses results (Fig. S6†) both indicated that CYP1A2 and UGT1A1, 1A9 participate in the metabolic elimination of PI-103. In addition, there were marked species differences in the metabolism of PI-103 (Fig. 3). Furthermore, efflux transport assays combined with a gene silencing approach suggested that BCRP, MRP1, MRP4 were the most critical transporters for the transport of PI-103-O-glucuronide (Fig. 4). Moreover, PI-103 demonstrated broad-spectrum inhibition towards human CYPs and UGTs isozymes with IC50 values ranging from 0.33 to 6.89 μM (Table 2). Among them, PI-103 showed potent non-competitive inhibition against CYP1A2, 2C19, 2E1 with IC50 and Ki values of less than 1 μM (Table 2). For the UGTs isozymes, PI-103 exhibited moderate non-competitive inhibition against UGT1A3, 1A7, 2B7 with Ki values between 1.35 and 5.64 μM (Table 2). In contrast, PI-103 activated UGT1A4 in a mechanistic two-site model with a Ki value of 13.76 μM (Fig. 6I). Taken together, PI-103 undergoes significant hepatic and intestinal metabolism, and many efflux transporters are involved in the excretion of its glucuronide. Furthermore, PI-103 exhibited potent and broad-spectrum inhibition against human CYP and UGT isozymes (Fig. 5). These findings may increase understanding of the metabolic fates of PI-103, and the possible harmful DDIs when PI-103 is co-administered with these CYP and UGT substrates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Key R & D and promotion projects of Henan Province (182102310164, 182102310146), National Natural Science Foundation of China (81803638, 81903704), Foundation of He’nan Educational Committee (20A350012) and Hospital Youth Foundation of the First Affiliated Hospital of Zhengzhou University (YNQN2017200).References
- B. Vanhaesebroeck, J. Guillermet-Guibert, M. Graupera and B. Bilanges, Nat. Rev. Mol. Cell Biol., 2010, 11, 329 CrossRef CAS PubMed
.
- P. Liu, H. Cheng, T. M. Roberts and J. J. Zhao, Nat. Rev. Drug Discovery, 2009, 8, 627–644 CrossRef CAS PubMed
- S. J. Leevers, B. Vanhaesebroeck and M. D. Waterfield, Curr. Opin. Cell Biol., 1999, 11, 219–225 CrossRef CAS
- J. A. Engelman, J. Luo and L. C. Cantley, Nat. Rev. Genet., 2006, 7, 606 CrossRef CAS PubMed
- Z. A. Knight, B. Gonzalez, M. E. Feldman, E. R. Zunder, D. D. Goldenberg, O. Williams, R. Loewith, D. Stokoe, A. Balla, B. Toth, T. Balla, W. A. Weiss, R. L. Williams and K. M. Shokat, Cell, 2006, 125, 733–747 CrossRef CAS PubMed
- S. Park, N. Chapuis, V. Bardet, J. Tamburini, N. Gallay, L. Willems, Z. A. Knight, K. M. Shokat, N. Azar, F. Viguie, N. Ifrah, F. Dreyfus, P. Mayeux, C. Lacombe and D. Bouscary, Leukemia, 2008, 22, 1698–1706 CrossRef CAS PubMed
- D. B. Gursel, Y. S. Connell-Albert, R. G. Tuskan, T. Anastassiadis, J. C. Walrath, J. J. Hawes, J. C. Amlin-Van Schaick and K. M. Reilly, Neuro Oncol., 2011, 13, 610–621 CrossRef CAS PubMed
- R. Hill, R. K. Kalathur, S. Callejas, L. Colaco, R. Brandao, B. Serelde, A. Cebria, C. Blanco-Aparicio, J. Pastor, M. Futschik, A. Dopazo and W. Link, Breast Canc. Res., 2014, 16, 482 CrossRef PubMed
- Q. W. Fan, Z. A. Knight, D. D. Goldenberg, W. Yu, K. E. Mostov, D. Stokoe, K. M. Shokat and W. A. Weiss, Cancer Cell, 2006, 9, 341–349 CrossRef CAS PubMed
- N. Yang, R. Sun, X. Liao, J. Aa and G. Wang, Pharmacol. Res., 2017, 121, 169–183 CrossRef CAS PubMed
- V. Burkina, M. K. Rasmussen, N. Pilipenko and G. Zamaratskaia, Toxicology, 2017, 375, 10–27 CrossRef CAS PubMed
- X. Lv, J. B. Zhang, J. Hou, T. Y. Dou, G. B. Ge, W. Z. Hu and L. Yang, Biotechnol. J., 2019, 14, e1800002 CrossRef PubMed
- W. He, J. J. Wu, J. Ning, J. Hou, H. Xin, Y. Q. He, G. B. Ge and W. Xu, Toxicol. In Vitro, 2015, 29, 1569–1576 CrossRef CAS PubMed
- Z. Yao, S. Li, Z. Qin, X. Hong, Y. Dai, B. Wu, W. Ye, F. J. Gonzalez and X. Yao, RSC Adv., 2017, 7, 52661–52671 RSC
- X. Lv, Y. Xia, M. Finel, J. Wu, G. Ge and L. Yang, Acta Pharm. Sin. B, 2019, 9, 258–278 CrossRef PubMed
- L. Wang, X. Hong, Z. Yao, Y. Dai, G. Zhao, Z. Qin, B. Wu, F. J. Gonzalez and X. Yao, Xenobiotica, 2018, 48, 357–367 CrossRef CAS PubMed
- FDA Draft Guidance for Industry on Drug Interaction Studies-Study design, data analysis, and implication for dosing and labeling, 2017, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/ Search PubMed.
- V. Uchaipichat, P. I. Mackenzie, D. J. Elliot and J. O. Miners, Drug Metab. Dispos., 2006, 34, 449–456 CrossRef CAS PubMed
- L. Zhu, G. Ge, Y. Liu, Z. Guo, C. Peng, F. Zhang, Y. Cao, J. Wu, Z. Fang, X. Liang and L. Yang, Chem. Res. Toxicol., 2012, 25, 2663–2669 Search PubMed
- X. Hong, Y. Zheng, Z. Qin, B. Wu, Y. Dai, H. Gao, Z. Yao, F. J. Gonzalez and X. Yao, Int. J. Mol. Sci., 2017, 18, 1983 Search PubMed
- Z. Qin, B. Zhang, J. Yang, S. Li, J. Xu, Z. Yao, X. Zhang, F. J. Gonzalez and X. Yao, Front. Pharmacol., 2019, 10, 496 Search PubMed
- H. Sun, T. Zhang, Z. Wu and B. Wu, J. Pharm. Sci., 2015, 104, 244–256 CrossRef CAS PubMed
- Z. Qin, S. Li, Z. Yao, X. Hong, J. Xu, P. Lin, G. Zhao, F. J. Gonzalez and X. Yao, J. Pharm. Biomed. Anal., 2018, 155, 157–168 CrossRef CAS PubMed
- U. M. Zanger and M. Schwab, Pharmacol. Ther., 2013, 138, 103–141 CrossRef CAS PubMed
- M. Wang, G. Yang, Y. He, B. Xu, M. Zeng, S. Ge, T. Yin, S. Gao and M. Hu, Mol. Nutr. Food Res., 2016, 60, 1967–1983 CrossRef CAS PubMed
- M. Paskeviciute and V. Petrikaite, Drug Delivery Transl. Res., 2019, 9, 379–393 CrossRef CAS PubMed
- M. Afrouzian, R. Al-Lahham, S. Patrikeeva, M. Xu, V. Fokina, W. G. Fischer, S. Z. Abdel-Rahman, M. Costantine, M. S. Ahmed and T. Nanovskaya, Biochem. Pharmacol., 2018, 156, 467–478 CrossRef CAS PubMed
- E. Quan, H. Wang, D. Dong, X. Zhang and B. Wu, Drug Metab. Dispos., 2015, 43, 433–443 CrossRef CAS PubMed
- Z. Qin, S. Li, Z. Yao, X. Hong, B. Wu, K. W. Krausz, F. J. Gonzalez, H. Gao and X. Yao, Food Funct., 2018, 9, 1410–1423 RSC
- G. B. Ge, Chin. J. Nat. Med., 2019, 17, 801–802 Search PubMed
- Q. H. Zhou, Y. D. Zhu, F. Zhang, Y. Q. Song, S. N. Jia, L. Zhu, S. Q. Fang and G. B. Ge, Chin. J. Nat. Med., 2019, 17, 858–870 Search PubMed
- J. A. Williams, R. Hyland, B. C. Jones, D. A. Smith, S. Hurst, T. C. Goosen, V. Peterkin, J. R. Koup and S. E. Ball, Drug Metab. Dispos., 2004, 32, 1201–1208 CrossRef CAS PubMed
- R. L. Walsky and R. S. Obach, Drug Metab. Dispos., 2004, 32, 647–660 CrossRef CAS PubMed
- W. E. Evans and M. V. Relling, Science, 1999, 286, 487–491 CrossRef CAS PubMed
- S. Nagar and R. P. Remmel, Oncogene, 2006, 25, 1659–1672 CrossRef CAS PubMed
- M. J. Zaya, R. N. Hines and J. C. Stevens, Drug Metab. Dispos., 2006, 34, 2097–2101 CrossRef CAS PubMed
- C. Guillemette, E. Levesque and M. Rouleau, Clin. Pharmacol. Ther., 2014, 96, 324–339 CrossRef CAS PubMed
- M. M. Soderberg, T. Haslemo, E. Molden and M. L. Dahl, Pharmacogenet. Genomics, 2013, 23, 279–285 CrossRef PubMed
- C. M. Street, Z. Zhu, M. Finel and M. H. Court, Xenobiotica, 2017, 47, 1–10 CrossRef CAS PubMed
- R. W. Dellinger, J. L. Fang, G. Chen, R. Weinberg and P. Lazarus, Drug Metab. Dispos., 2006, 34, 943–949 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09906a |
| This journal is © The Royal Society of Chemistry 2020 |