
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePossible scenario of forming a catalyst layer for proton exchange membrane fuel cells†
R. Zeng *abc,
H. Y. Zhangabc,
S. Z. Liangabc,
L. G. Wangac,
L. J. Jiangabc and
X. P. Liuabc
*abc,
H. Y. Zhangabc,
S. Z. Liangabc,
L. G. Wangac,
L. J. Jiangabc and
X. P. Liuabc
aGRINM Group Co. Ltd, No. 2 Xinjiekouwai Street, Xicheng Dist., Beijing, 100088, P. R. China. E-mail: zr_zengrong@163.com
bNational Engineering Research Center of Nonferrous Metals Materials and Products for New Energy, No. 2 Xinjiekouwai Street, Beijing, 100088, P. R. China
cGRIMAT Engineering Institute Co., Ltd., No. 11 Xingkedongda Street, Huairou Dist., Beijing, 101407, P. R. China
First published on 3rd February 2020
Abstract
Ionomer in the catalyst layer provides an ion transport channel which is essential for many electrochemical devices. As the ionomer and electrochemical catalyst are packed together in the catalyst layer, it is difficult to have a clear image of the ionomer distribution in the catalyst layer and how the ionomer is in contact with Pt or carbon. A highly dispersed catalyst was deposited on the TEM SiN grid directly using the same (ultrasonic spray) or a similar way as the catalyst was deposited on the membrane. By analyzing the distribution of various elements (C, F, S, Pt etc.), we found that the ionomer may coexist in the catalyst layer in three ways: ionomer covered Pt particles due to the relatively strong interaction between Pt and the ionomer; ionomer covered C particles; packed free ionomer in between the aggregated catalyst particles. The results show that the ionomer is prone to covering the surface of Pt particles as further evidenced by the accelerated degradation test (ADT).
1. Introduction
Proton Exchange Membrane Fuel Cells (PEMFCs) are at the beginning of commercial application and mass produced fuel cell vehicles were introduced into the market in Japan in 2014. However, their cost and durability are still remaining issues. To address these issues, it is the main concern to lower the Pt loading in the Membrane Electrode Assembly (MEA) and to improve the performance.MEA is the key part of PEMFCs and the ultra-thin catalyst layer of MEA is a hot topic of the research. The catalyst layer (CL) is a very complex chemical and geometric environment for electrochemical reactions in PEMFCs. This porous CL is composed of supported catalyst particles, ionomer etc. The reaction occurs at sites where various reacting species such as protons, electrons, and gases meet.
By using sum frequency generation spectroscopy (SFG), Noguchi's1 work showed that the interface between the PFSA (perfluorosulfonic acid) thin film and the Pt surface is different from the interface between PFSA and a HOPG surface. Water may be prone to accumulate at the interface between the PFSA thin film and the Pt surface. Based on our work on PEMFCs with dead-end anode, we also suggested the possibility of water accumulation at the interface between the PFSA ionomer thin film and Pt surface even when the fuel cell was running without external humidification.2 This highlights the need of further study on the properties of the interface between the ionomer and catalyst as well as the ionomer distribution in the catalyst layer.
The structure and properties of CLs, the interface between the ionomer and catalyst or ionomer distribution in CLs are always the main concerns in promoting PEMFC performance. A widely accepted model or assumption for the ionomer distribution in the catalyst layer is that the ionomer covered the aggregated Pt/C particles3–5 fully or partially. Eikerling's group has done the computational researches on this topic.6–8 Their work showed Nafion ionomer formed a thin adhesive film on the graphite sheet when only graphite carbon and ionomer were considered.7 The Nafion ionomer is predominantly adsorbed on the graphitized carbon sheet via the backbone, and the side chains face toward the pore space. The interfacial packing density of the sulfonic acid groups that face toward the pore space could have significant impact on the surface properties of pores in CLs. The surface properties of the graphite sheet affect the morphology of the Nafion ionomer and water. In the presence of Pt nanoparticles, on the other hand, the ionomer phase is more clustered and less connected with more anionic side chains pointing toward the Pt/C surface.8 Ionomer mostly covers the external surface of Pt/C aggregates (sizes < 50 nm) when hydrophobic carbon like Vulcan XC72 is applied. However, they did not discuss the interaction between the Pt and the ionomer. In order to manipulate the wetting properties of the micropores in the catalyst layer, Dowd Jr et al.9 suggested a heat treatment of the CLs to form hydrophobic pores in CLs. Gisu Doo et al.10 found that the CL structure changed as the size of ionomer aggregates in the catalyst dispersion ink changed. Park and his co-workers11 investigated four different carbon structure and their effects on the Pt and ionomer distribution. By N2 absorption analysis and TEM results, they suggested the blockage of the pore of carbon by Pt and ionomer. Yang et al.12 also discussed the interaction between carbon (XC-72, NH2-XC72, SO3H-XC-72) and Nafion ionomer in the catalyst ink dispersion. The size of the –NH2 functionalized carbon black aggregates increased significantly. This may significantly affect the CL structure.
The material-sensitive and conductive atomic force microscopy (AFM) was used to show the ionomer distribution in CL.13–21 The adhesion force image overlaid topography was used to distinguish the ionomer and the aggregated Pt/C. The ionically conductive regions, which implied the ionomer region in CL, could be recognized by their low adhesion signal. Xie et al.22 showed that the aggregation of catalyst decreased as the ionomer content increased by AFM and TEM. Cullen23 showed the ionomer coating film thickness is about 7 nm using AFM technology. Hiesgen's group15–21 had done a series of works on ionomer distribution in the CL by AFM. The AFM tip of nominal radius of less than 1 nm was used and an image pixel size was less than 1 nm (<0.36 nm) for high-resolution image. They found that the ionically conducting network with lamella structure was formed in the membrane15 and the cast ionomer thin film18 when forcing a current through the membrane or the thin film. The ionomer film, covering the catalyst aggregates in the catalyst layer, formed inhomogeneous structure15 in a thickness of 4–15 nm.20 The opposite effects of ionomer dimensional changes in membrane and catalyst layers were detected by AFM technology too.19 Big ionomer area about hundred nanometer size was shown in AFM images.17 The ionomer film thickness in CL was thinning and the ionomer was redistributed and aggregated in terms of operation time.21 This irreversible degradation of the ionomer in CL may result in performance degradation of MEAs. Stiffness mapping and current mapping were also used along with the adhesion force mapping to discern Pt, C and the ionomer. The adhesion mapping of AFM is a very good way to visualize the PFSA ionomer in CLs and deeper understanding of the effect of ionomer on MEAs was achieved. However, as the Pt particle size is only about 2–3 nm and carbon is about 30–50 nm in normal commercial Pt/C catalyst, how the ionomer covers within the Pt/C agglomerates is still unclear. The chemical information around Pt or C can't be achieved by AFM adhesion mapping too.
High resolution TEM coupled with energy dispersive X-ray spectroscopy (EDX) is a good way to give morphology and chemical information of nanomaterials. However, the damage of the PFSA ionomer film is the main concern. S. Yakovlev et al.24 discussed in detail about the morphology of Nafion membrane applying TEM technology. Damage of Nafion membrane structure was confirmed. In state-of-the-art to get chemical information of the CLs, reducing the incident flux and partial defocusing was applied to lower the radiation damage of the ionomer during the test at the cost of sacrificing the spatial resolution. Cullen23 addressed the E-beam damage of thin PFSA ionomer film. By lowering the temperature to −100 °C and electron dose (below 1 × 104 e− nm−2 s−1) at the cost of lowering the resolution, mass loss of F in the ionomer is below 10%. Multivariate statistical analysis (MVSA) was applied to EELS and EDX spectrum to reduce the noise.
Hitchcock's group25–28 had made the attempts to quantitatively map ionomer in catalyst layers too. Instead of STEM-EDX, they suggested scanning transmission X-ray microscopy (STXM) spectro-tomography at the C 1s and F 1s edges to keep the damage of thin ionomer film within an acceptable level.25 The electron beam spot size of STXM was about 50 nm. They further applied soft X-ray spectro-ptychography to quantitatively image PFSA ionomers in PEMFC cathode. The 2D spatial resolution better than 15 nm and a 3D spatial resolution better than 30 nm were achieved.28 The authors suggested further reducing the radiation dose and performing the test at cryogenic temperatures in order to lower the damage of PFSA ionomer and to improve the spatial resolution further.
In this paper, we still apply the high resolution TEM and high-angle annular dark-field scanning TEM (HAADF-STEM) to get high resolution images and try to rebuild the ionomer distribution based on the trace elemental distribution. The electron dose is lower than 1 × 104 e− nm−2 s−1. We chose the bottom up way to deposit the catalyst on the TEM grid instead of making the TEM samples from the formed catalyst layer. This helps us to image the ionomer distribution when the catalyst layer is formed.
2. Experiments
The experiments were performed on Tecnai G2F20 (FEI, USA). As F is very sensitive to electron dose in TEM tests, we try to detect carbon and sulphur instead. Carbon can indicate where the ionomer is, while sulphur indicates where the sulfonic acid group is. SiN grid was used to prepare TEM samples so that carbon distribution can be shown. The fresh catalyst ink was prepared and ultrasonically sprayed on the SiN grid once to three times or dropped on the SiN grid. The aged catalyst was scratched from the aged catalyst layer in membrane electrode assemblies (MEAs) after 30k cycles of 0.6–1.0 V, redistributed in ethanol and deposited on the TEM grid. The fresh and aged catalysts were compared with each other. In order to improve the stability of the ionomer under TEM test, the solvent in the catalyst ink was evaporated off and formed free standing CL which was similar to the ultrasonic way to form CLs. The given CL was immersed in the 1.0 mol L−1 CuSO4 or Al2(SO4)3 for 30 min 3 times to transfer H+ in ionomer to Cu2+ or Al3+. Then the CL was cleaned with water and re-dispersed in iso-propanol. The prepared ink was dropped on the SiN grid for TEM test.3. Results and discussion
The elemental content of the original SiN grid is shown in Table 1. Only Si, N, O and very low content of Cl (0.06 at%) are detected, which means that the SiN grid is free of C, F, S. The contents of Si, N, O are 42.82 at%, 44.41 at% and 12.71 at%, respectively. HAADF images and EDX maps of SiN grid is shown in Fig. S1 (ESI†). Si, N and O are homogenously distributed.| SiN | Catalyst | Away catalyst | |
|---|---|---|---|
| C (at%) | — | 62.83 | 48.51 |
| Pt (at%) | — | 1.54 | −0.05 |
| S (at%) | — | 0.18 | 0.01 |
| F (at%) | — | −0.21 | −0.27 |
| O (at%) | 12.71 | 6.43 | 4.80 |
| Si (at%) | 42.82 | 18.52 | 22.09 |
| N (at%) | 44.41 | 10.71 | 24.92 |
| Cl (at%) | 0.06 |
The TEM of the fresh catalyst covered with the PFSA ionomer in H+, Cu2+ and Al3+ is shown in Fig. S2 (ESI†). As the ionomer film in Cu2+ or Al3+ are more robust than the film in H+ when exposed to the electron beam, the ionomer film in Cu2+ or Al3+ is clearly discerned from the SiN background. The ionomer film in Cu2+ or Al3+ doesn't cover the catalyst in an aggregated state as expected, but forms a thin film in between the aggregated catalysts. Although there is no clear thin film among the aggregated Pt/C catalysts when using ionomer in H+, HAADF images and EDX maps of the ionomer in H+ (Fig. 1) confirm that the thin film of ionomer in H+ does exist among the aggregated Pt/C catalyst particles. The atom concentration of C, Pt, S, F, O, Si and N are shown in Table 1. As we know, PFSA ionomer is sensitive to the electron beam energy and dose, and the mass loss of the ionomer, especially the F mass loss is significant. No detectable F is found in the area of catalyst and away from the catalyst. No detectable S is found in the area away from the catalyst. The atom concentration of S on the catalyst is very low (0.18 at%) but shows similar distribution as that of Pt, namely S appears at the same position where Pt is. This means that S is close to and interacts with Pt. It is possible that sulfonic acid functional group of the ionomer covers Pt surface. O is shown at all the detected area. As the oxygen could be from the SiN surface, it is hard to determine whether O comes from the decomposed ionomer. However, carbon appears on the SiN surface in the area away from the catalyst, where S and F are hardly detected. The C concentration in the area of catalyst and away from the catalyst is 62.83 at% and 48.51 at%, respectively. However, the scenario of C distribution in the area of catalyst and away from the catalyst is quite different. As shown in Fig. 1, the EDX mapping image of C in the area of catalyst clearly shows C is centralized as the carbon particle while C in the area away from the catalyst is distributed homogenously. The elemental EDX mapping in the area of the catalyst and away from the catalyst with ionomer in Cu2+ and Al3+ are shown in Fig. S3 and S4 (ESI†) respectively. The elemental contents in the area of the catalyst, away from the catalyst with ionomer in Cu2+ and Al3+ are listed in Table S1 (ESI†). The distribution and the content of S, Pt, C, F, O, and Si are similar to that of the catalyst with ionomer in H+. The content of C is as high as 48 at%, 83.24 at% and 83.88 at% on the surface of SiN in the area away from the catalyst with ionomer in H+, Cu2+ and Al3+ respectively. It is sensible that C content of ionomer in Cu2+ and Al3+ is higher than that of ionomer in H+. As carbon can only be from the ionomer in this area, it supports the hypothesis that part of the ionomer in the catalyst ink is deposited away from the catalyst. This implies that the ionomer in H+ does form thin film among the aggregated catalyst. The ionomer itself may pack together when the catalyst ink is deposited layer by layer. If the ionomer itself packs together during the first few deposited catalyst layers, the formed thin catalyst layer next to the membrane could be denser than the catalyst layer away from the membrane. This is consistent with our finding before that the catalyst layer formed a condensed layer about 1 μm next to the membrane when ultrasonically deposited the catalyst ink on the membrane.29 The packed ionomer may also form the aggregated ionomer in the CLs away from the membrane too. This is consistent with the work before17 that there was big aggregated ionomer up to several hundred nanometer size in the catalyst layer.
 | ||
| Fig. 1 HAADF images (where the red rectangle show the mapping area) and EDX maps with colored pixels indicating where the element was detected above background in the area of the catalyst (the upper images) and away from the catalyst (the lower images). C is red; S is purple; F is blue; Pt is yellow; O is cyan; Si is grey; N is green. | ||
The drift corrected spectrum of EDX linear scan of the selected catalyst particle in STEM is applied to show more details about the distribution of C, F, S, Pt and Al or Cu. S, Al, Cu would indicate where the sulfonic acid hydrophilic functional group is. The results are shown in Fig. 2. The trace line of the EDX scan which can be clearly seen in Fig. 2(a) gives a hint that the ionomer actually covers on the carbon surface but disappears during the test. S or F from the damaged ionomer may disseminate and escape away from the high vacuum chamber of TEM equipment. However, the content of S changes coincident with the content of Pt as shown in Fig. 2(a–c) no matter the ionomer in the catalyst is in H+, Al3+ or Cu2+ form. It is sensible because the theoretical absorption energy of S on fcc and hcp site of Pt (111) is about 5.95 eV and 5.75 eV (ref. 30) respectively which can cause strong absorption of S on Pt. As the interaction distance of two atoms is short, this absorbed S on Pt implies that the Pt surface is covered by S which is from sulfonic acid functional group of the decomposed ionomer. It is consistent with the elemental mapping results in Fig. 1 where S appears at the same position where Pt is. It is also consistent with Noguchi's1 work and Eikerling's theoretical simulation8 which suggested that the sulfonic acid group of the ionomer was prone to cover the Pt surface. Besides S, the content of F, Al and Cu also change coincident with the content of Pt although the peaks are much weaker than S and Pt peak.
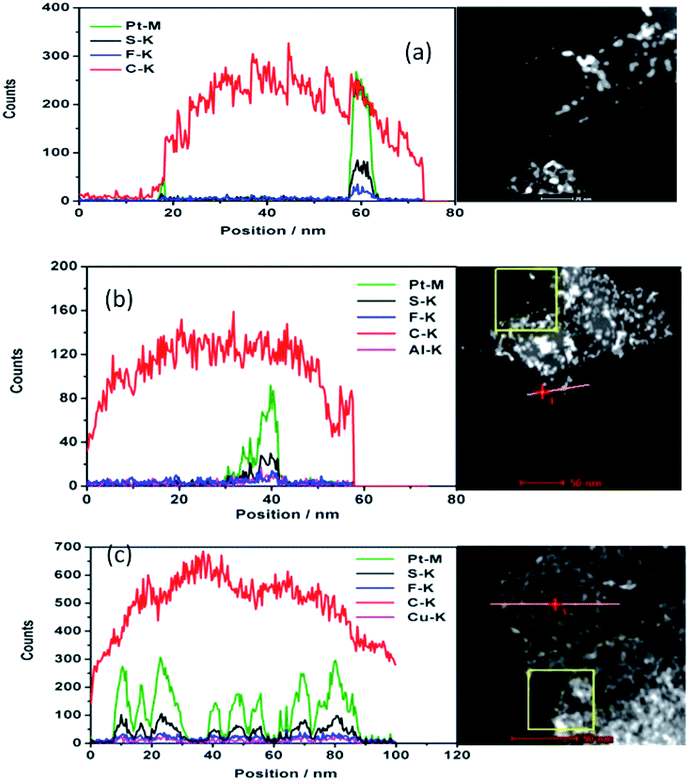 | ||
| Fig. 2 Characterization of the ionomer covered nanoparticles. HAADF (right) and typical drift corrected spectrum (left) of EDX linear scan of the selected particle in STEM for deposited catalyst with ionomer in H+ (a), Al3+ (b), Cu2+ (c). The red line in (a) where the line scan performed was removed so that the track of the line scan can be seen (Pt: green; S: black; F: blue; C: red; Al: purple; Cu: purple). | ||
Combined with the information from the elemental linear scan and mapping results, we suggest that the ionomer cover the surface of the catalyst including the surface of carbon and Pt, and the sulfonic acid functional group is near the surface of Pt which shows a stronger interaction between Pt and the ionomer than that of carbon and the ionomer. The ionomer can also be deposited on the surface away from the catalyst. We prone to think that thin ionomer film covers primary carbon particle (∼50 nm) surface based on our test and Eikerling's simulation8 although we did not scan every Pt/C particle and it is impossible to discern the ionomer thin film attached on carbon surface from the free ionomer film deposited on carbon surface. The scenario of forming the catalyst layer could be: the catalyst including the carbon and Pt surface covered by the ionomer and the free ionomer deposited layer by layer coexist. The free ionomer may pack together to form the ion exchange channel among the ionomer covered catalyst. As S is left beside Pt, the hydrophilic side chain of the ionomer, which contains the sulfonic acid functional group, covers the Pt surface. Eikerling suggested that the ionomer covers the carbon surface by the CF backbone and the sulfonic acid group faces the pores in CLs.7 The ionomer covering Pt surface may be beneficial to Pt ions (Pt2+) by oxidizing Pt particles during fuel cell operation to transfer through the ion exchange channels in CLs. It causes the Pt depletion zone of CLs near the membrane and formation of the Pt band in the membrane, or results in the growth of Pt particles.31,32
The aged catalysts in cathode catalyst layer, cycled 30k cycles from 0.6 V to 1.0 V at a scan rate of 50 mV s−1, are re-dispersed in ethanol and deposited on the TEM Cu grid with carbon ultrathin film. The elemental linear scan of catalyst particle is shown in Fig. 3. Similar to the fresh catalyst, the content of S and Pt change coincidently at the detached Pt particles and the Pt on C. It shows there is no S or F on carbon. As the relatively strong interaction between S and Pt, the ionomer covered Pt surface may be kept during the ageing process.
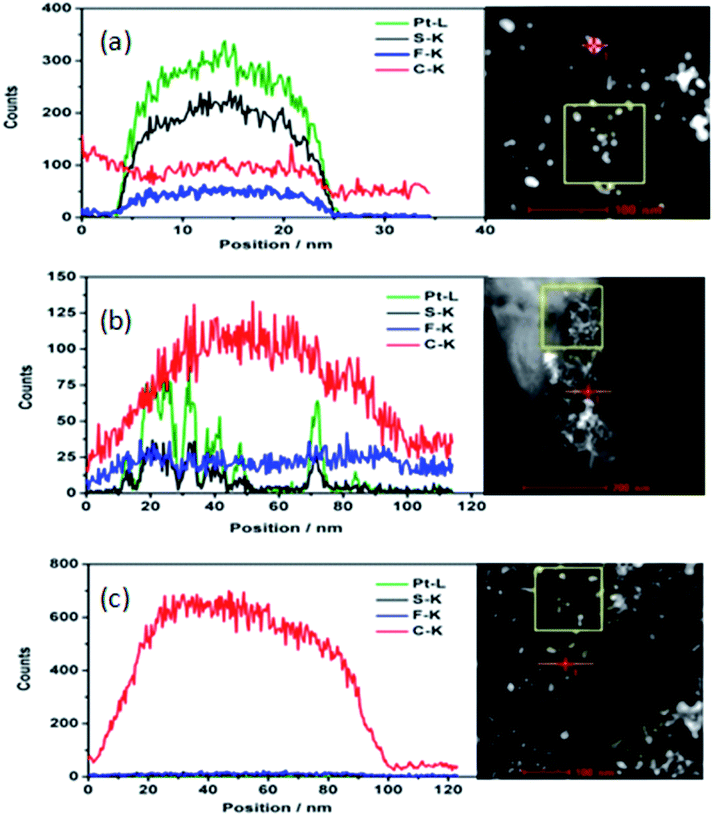 | ||
| Fig. 3 HAADF (right) and typical drift corrected spectrum (left) of EDX linear scan of the selected aged particle in STEM of detached Pt particle (a); C with Pt (b); C with no Pt (c) along with the scan line (Pt: green; S: black; F: blue; C: red). | ||
Based on the above observation, we suggest the ionomer distribution in the catalyst layer shown in Fig. 4 instead of the traditional images.3–5 There are three possible kinds of ionomer coverage with different interaction with the catalyst: relatively strong interaction between Pt and the ionomer where the ionomer covers Pt particles; interaction between C and the ionomer where the ionomer covers C particles; packed free ionomer in between the Pt/C aggregated catalyst particles. This challenges the traditional understanding of the structure of the catalyst layer. It may have profound effects on the structure changing of catalyst layer when fuel cell runs for long time or reversal voltage happens. The structure change of the CLs when reversal voltage happens will be discussed in the near future. The new model of the composition distribution or structure of the catalyst layer may also cause profound effects on the simulation of ion, water and gases transportation in the catalyst layer.
 | ||
| Fig. 4 Schematic of the ionomer distribution in the catalyst layer. | ||
4. Conclusions
Highly dispersed catalyst is deposited by ultrasonic spray or dropped on the TEM SiN grid directly the same or similar way as the catalyst is deposited on the membrane. Although TEM technology can't give the real image of the ionomer distribution in the catalyst layer due to the mass loss of the ionomer especially F, the trace S and C elemental distribution reveal the ionomer distribution near and away from the catalyst by using SiN grid for high resolution TEM. The results show that the ionomer may coexist in the catalyst layer in three ways: the ionomer covers Pt particles due to the relatively strong interaction between Pt and the ionomer; ionomer covers C particles; packed free ionomer among the catalyst particles possibly forming ionomer agglomerates in CLs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the foundation (Z171100000917011) of Beijing Municipal Science and Technology Commission and the foundation (52218) of GRINM Group Co. Ltd. for financial support of this work. We also thank Jie Bai and Siran Liu for preparing the MEAs.References
- H. Noguchi, K. Taneda, H. Naohara and K. Uosaki, Electrochem. Commun., 2013, 27, 5 CrossRef CAS.
- D. Jiang, R. Zeng, S. M. Wang, L. J. Jiang and J. R. Varcoe, J. Power Sources, 2014, 265, 45 CrossRef CAS.
- D. Lee and S. Hwang, Int. J. Hydrogen Energy, 2008, 33, 2790 CrossRef CAS.
- S. Shukla, K. Domican, K. Karan, S. Bhattacharjee and M. Secanell, Electrochim. Acta, 2015, 156, 289 CrossRef CAS.
- R. Shimizu, Y.-C. Park, K. Kakinuma, A. Iiyama and M. Uchida, J. Electrochem. Soc., 2018, 165(6), F3063 CrossRef CAS.
- J. F. Liu and M. Eikerling, Electrochim. Acta, 2008, 53, 4435 CrossRef CAS.
- T. Mashio, K. Malek, M. Eikerling, A. Ohma, H. Kanesaka and K. Shinohara, J. Phys. Chem. C, 2010, 114, 13739 CrossRef CAS.
- K. Malek, T. Mashio and M. Eikerling, Electrocatal, 2011, 2, 141 CrossRef CAS.
- R. P. Dowd Jr, C. S. Day and T. V. Nguyen, J. Electrochem. Soc., 2017, 164(2), F138 CrossRef.
- G. Doo, J. H. Lee, S. Yuk, S. Choi, D.-H. Lee, D. W. Lee, H. G. Kim, S. H. Kwon, S. G. Lee and H.-T. Kim, ACS Appl. Mater. Interfaces, 2018, 10, 17835 CrossRef CAS PubMed.
- Y.-C. Park, H. Tokiwa, K. Kakinuma, M. Watanabe and M. Uchida, J. Power Sources, 2016, 315, 179 CrossRef CAS.
- F. Yang, L. Xin, A. Uzunoglu, Y. Qiu, L. Stanciu, J. Ilavsky, W. Z. Li and J. Xie, ACS Appl. Mater. Interfaces, 2017, 9, 6530 CrossRef CAS PubMed.
- J. Xie, F. Xu, D. L. Wood, K. L. More, T. A. Zawodzinski and W. H. Smith, Electrochim. Acta, 2010, 55, 7404 CrossRef CAS.
- S. Ma, C.-H. Solterbeck, M. Odgaard and E. Skou, Appl. Phys. A, 2009, 96, 581 CrossRef CAS.
- R. Hiesgen, T. Morawietz, M. Handl, M. Corasaniti and K. A. Friedrich, Electrochim. Acta, 2015, 162, 86 CrossRef CAS.
- T. Morawietz, M. Handl, M. Simolka, K. A. Friedrich and R. Hiesgen, ECS Trans., 2015, 68(3), 3 CrossRef CAS.
- T. Morawietz, M. Handl, C. Oldani, K. A. Friedrich and R. Hiesgen, ACS Appl. Mater. Interfaces, 2016, 8(40), 27044 CrossRef CAS PubMed.
- R. Hiesgen, T. Morawietz, M. Handl and K. A. Friedrich, Mater. Res. Soc. Symp. Proc., 2015, 1774, 19 CrossRef.
- T. Morawietz, M. Handl, C. Oldani, K. A. Friedrich and R. Hiesgen, Fuel Cells, 2018, 18(3), 239 CrossRef CAS.
- T. Morawietz, M. Handl, K. A. Friedrich and R. Hiesgen, ECS Trans., 2018, 86(13), 179 CrossRef CAS.
- T. Morawietz, M. Handl, C. Oldani, P. Gazdzicki, J. Hunger, F. Wilhelm, J. Blake, K. A. Friedrich and R. Hiesgen, J. Electrochem. Soc., 2018, 165(6), F3139 CrossRef CAS.
- J. Xie, F. Xua, D. L. Wood, K. L. More, T. A. Zawodzinski and W. H. Smith, Electrochim. Acta, 2010, 55, 7404 CrossRef CAS.
- D. A. Cullen, R. Koestner, R. S. Kukreja, Z. Y. Liu, S. Minko, O. Trotsenko, A. Tokarev, L. Guetaz, H. M. Meyer, C. M. Parish and K. L. More, J. Electrochem. Soc., 2014, 161(10), F1111 CrossRef CAS.
- S. Yakovlev, N. P. Balsara and K. H. Downing, Membranes, 2013, 3, 424 CrossRef PubMed.
- L. G. A. Melo, A. P. Hitchcock, J. Jankovic, J. Stumper, D. Susac and V. Berejnov, ECS Trans., 2017, 80(8), 275 CrossRef CAS.
- J. Wu, L. G. A. Melo, X. H. Zhu, M. M. West, V. Berejnov, D. Susac, J. Stumper and A. P. Hitchcocka, J. Power Sources, 2018, 381, 72 CrossRef CAS.
- L. G. A. Melo and A. P. Hitchcock, Microanal, 2018, 24(suppl. 2), 460 CrossRef.
- J. Wu, X. H. Zhu, M. M. West, T. Tyliszczak, H.-W. Shiu, D. Shapiro, V. Berejnov, D. Susac, J. Stumper and A. P. Hitchcock, J. Phys. Chem. C, 2018, 122, 11709 CrossRef CAS.
- R. Zeng, H. Y. Zhang, L. J. Jiang and X. P. Liu, ECS Trans., 2017, 80(8), 215 CrossRef CAS.
- K. Shen, Y. X. Zhang, J. L. Wang, Z. S. Lu and Z. X. Yang, J. At. Mol. Phys., 2011, 28(1), 156 CAS.
- C. Takei, K. Kakinuma, K. Kawashima, K. Tashiro, M. Watanabe and M. Uchida, J. Power Sources, 2016, 324, 729 CrossRef CAS.
- H. R. Yu, A. Baricci, A. Casalegno, L. Guetaz, L. Bonville and R. Maric, Electrochim. Acta, 2017, 247, 1169 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09864j |
| This journal is © The Royal Society of Chemistry 2020 |