
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of new N,N′-Pd(Pt) complexes based on sulfanyl pyrazoles, and investigation of their in vitro anticancer activity†
Vnira R. Akhmetova *a,
Nail S. Akhmadieva,
Marat F. Abdullinb,
Lilya U. Dzhemilevaa and
Vladimir A. D'yakonova
*a,
Nail S. Akhmadieva,
Marat F. Abdullinb,
Lilya U. Dzhemilevaa and
Vladimir A. D'yakonova
aInstitute of Petrochemistry and Catalysis, Russian Academy of Sciences, 141 Prospekt Oktyabrya, 450075 Ufa, Russian Federation. E-mail: vnirara@mail.ru; Fax: +7 3472 842750; Tel: +7 3472 842750
bUfa Institute of Chemistry, Russian Academy of Sciences, 71 Prospekt Oktyabrya, 450054 Ufa, Russian Federation
First published on 17th April 2020
Abstract
The synthesis of new N,N′-mononuclear bi-ligand Pd(II) and tri-ligand Pt(II)complexes bearing sulfanyl(phenyl, benzyl, cyclohexyl, 4-hydroxyphenyl)3,5-dimethyl-1H-pyrazole ligands has been carried out. The obtained compounds were studied for apoptosis-inducing activity and effect on the cell cycle for Jurkat, K562, and U937 neoplastic cell cultures and conditionally normal human embryonic kidney HEK293 cells. The cells showed the highest sensitivity to platinum and palladium complexes in comparison with ligands and cisplatin. The cytotoxic properties are enhanced for compounds with cyclohexyl substituents at the S-atom in sulfanyl pyrazoles and complexes.
Introduction
Virtually all antitumor agents are very potent cellular poisons or toxins, which are detrimental for the rapidly dividing cells of malignant tumors. The key or preferable concept of the chemotherapeutic action is possibly the most complete eradication and/or retardation of the growth, division, and metastasis of the malignant cell clone accompanied by conventionally acceptable damaging action on healthy tissues. This indicates that it is conceptually impossible to administer chemotherapy without accompanying destruction of healthy cells with high proliferative capacity (hematopoietic and immunocompetent cells; keratinocytes of skin and appendages (hair, nails); and epithelia of the gastrointestinal tract and respiratory and urinary systems).1Currently, most of chemotherapy protocols include drugs affecting the DNA synthesis in the cell. For example, widely used drugs are platinum agents (cisplatin, carboplatin, and oxaliplatin), which are efficient against solid tumors, including hereditary cancer syndromes such as breast and ovarian cancer syndromes caused by gene mutations of the DNA repair system.2,3 However, because of high rates of clonal selection in the tumor, neoplastic cells quickly acquire drug resistance to the agents that damage their genome. The p53-deficient tumors are even less responsive to damage of their genetic apparatus by platinum compounds.4 Therefore, many researchers engaged in the search for new anticancer compounds containing a metal in the molecule are looking for the most efficient, targeting compounds with selective toxicity or multi-targeting ones with cytostatic and antioxidant functions that could efficiently act on malignant tumors.5 Sulfur- and nitrogen-containing molecules and their metal(Pt, Pd)complexes are among such promising compounds.6 Sulfur atoms in organic molecules can be oxidized by various free radicals (OH˙, O˙, R˙), thus suppressing the development of malignant neoplasms under potentiation of oxidative stress in mitochondria.7 In addition, the antioxidant properties for the sulfur-containing palladium(II) complex were established by in vitro and in vivo methods.8,9
A known efficient sulfur-containing drug used to treat hematological malignancies and autoimmune diseases is treosulfan,10 which has sulfur atoms in the molecule and which is used, in some cases, in combination with cisplatin containing N–Pt bonds (Fig. 1). The combination of these drugs provides alkylation of the nucleophilic centers, thus disturbing DNA synthesis, which, in turn, arrests the cell cycle in the G1 and G2 phases and induces apoptosis.11 Structural analogues of treosulfan (methanesulfonic acid derivatives), busulfan and mannosulfan, are also used as alkylating drugs (Fig. 1).12,13
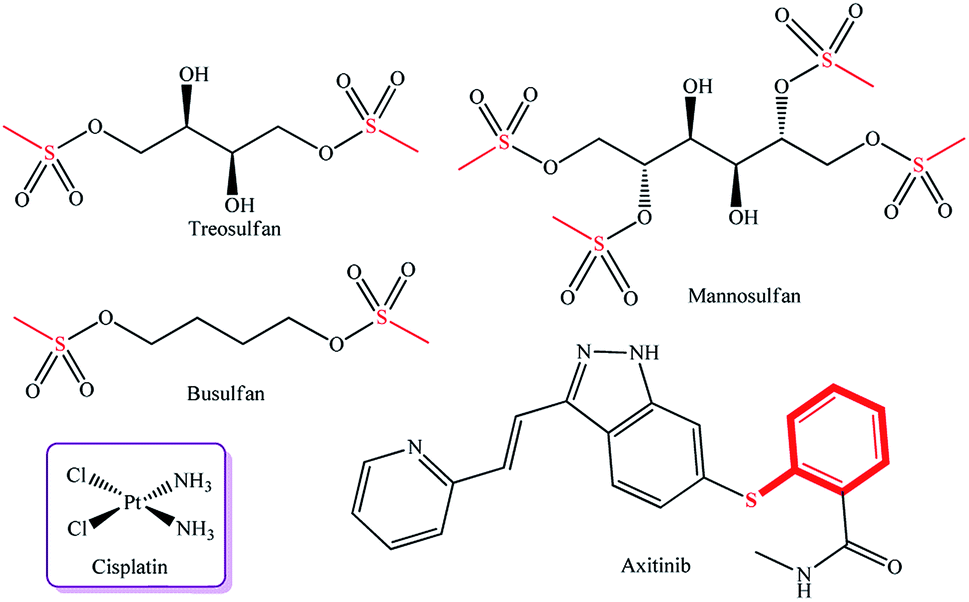 | ||
| Fig. 1 Commercially available metal complex drug cisplatin and cytotoxic drugs containing a sulfur atom. | ||
One more innovative drug, Axitinib,14 containing a pyrazole ring and a sulfur atom in the molecule, is a highly selective inhibitor of tyrosine kinases (VEGFR-1, VEGFR-2, and VEGFR-3) involved in pathological angiogenesis, tumor growth, and metastasis mechanisms. Many examples of pyrazole complexes of palladium and platinum as efficient anticancer agents are presented in the literature.15–20 There are published data on dichloro-bis(pyrazole)palladium(II) and dichloro-bis(pyrazole)platinum(II) complexes, which showed antitumor activity against the CaSki, HeLa, and p53 Jurkat T cell lines,21 and on palladium(II) trans-complexes with 5-hydrazine-1,3-dimethyl-4-nitro-1H-pyrazole ligands, which exhibited the cytotoxic effect against basal cell carcinoma cells.22 It was established that methyl groups fixed in the 3 and 5-positions of the pyrazole rings in platinum complexes enhance anticancer activity against MCF-7 and MDA-MB-231 mammary cell lines.23 It is noteworthy that sulfanyl-substituted pyrazoles were not tested previously as ligands for Pd and Pt complexes and were not evaluated for antitumor activity. Thus, a relevant task is to develop new palladium and platinum complexes with sulfanyl-substituted pyrazole ligands for the formation of a library of compounds with high antitumor potential.
Results and discussion
Chemistry
Recently, we have proposed a four-component one-pot synthesis of sulfanyl pyrazoles,24 which are promising ligands for the synthesis of potential metal-containing cytostatics.In this publication, we report the syntheses of 4-(sulfanylmethyl)-3,5-dimethyl-1H-pyrazoles as precursors for Pd and Pt complexes.
Sulfanyl pyrazoles 4a–d were prepared using the multicomponent reaction of acetylacetone 1 with formaldehyde, mercaptans 2a–d (phenyl, benzyl, cyclohexyl, 4-hydroxyphenyl), and hydrazine in the presence of catalytic amounts of BuONa. This strategy was developed, implementing the classical one-pot synthesis approach, in which the domino reaction “thiomethylation-Paal–Knorr cyclization”25 occurs via intermediate sulfanylmethyl 1,3-diketone 3. The thiomethylation and heterocyclization steps give water as a by-product.
The reaction pathway can be represented in the following way (Scheme 1).
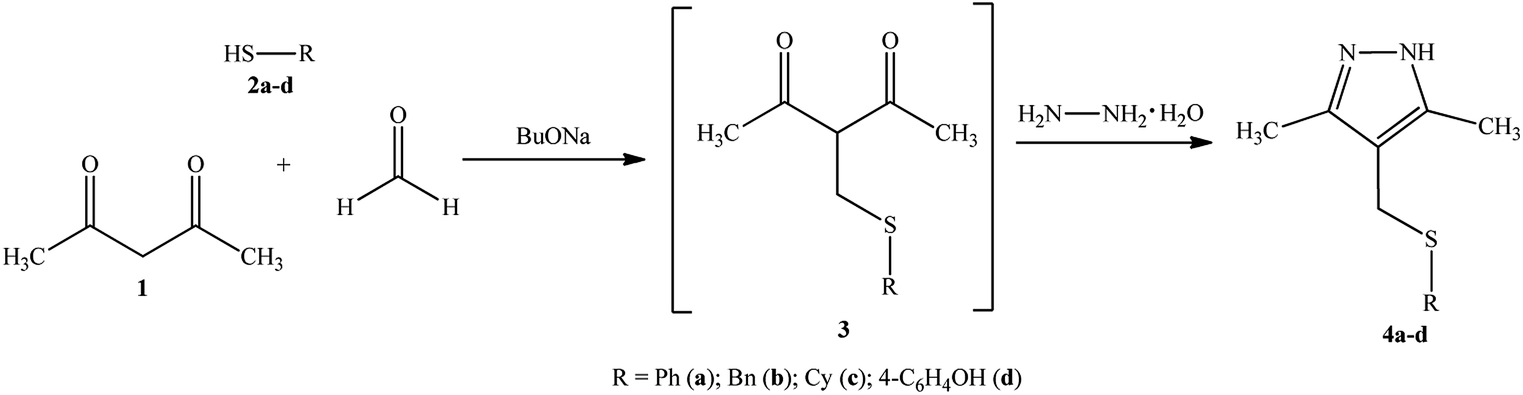 | ||
| Scheme 1 Synthesis of the sulfanyl pyrazole ligands 4a–d. | ||
The structure of compounds 4a–d was reliably confirmed by spectral methods.24 For compound 4d, a single crystal was grown and studied by X-ray diffraction (Fig. 2).
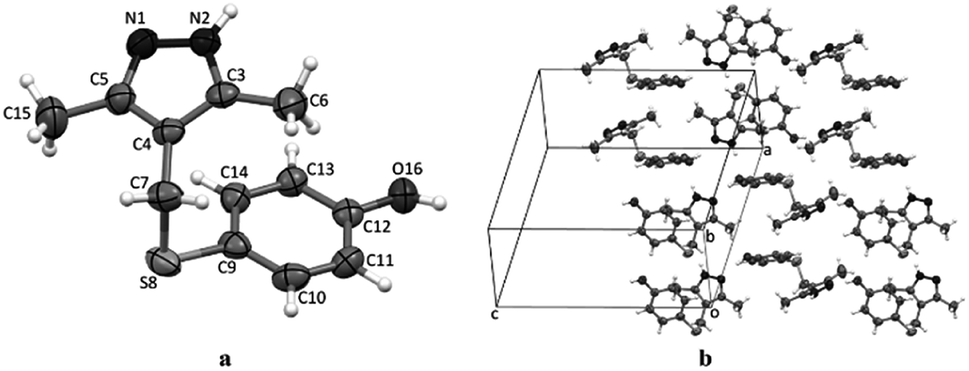 | ||
| Fig. 2 The geometry of molecules 4d in a crystal (a). Atoms are represented by thermal ellipsoids (p = 50%). Packing of the molecules 4d in their respective unit cell (b). | ||
Since cyclohexyl- and benzyl-containing ligands 4b and 4c are more efficient cytostatics than phenylsulfanylpyrazole 4a (Table 1), we used ligands based on 4-[(benzylsulfanyl)methyl]-3,5-dimethyl-1H-pyrazole 4b and 4-[(cyclohexylsulfanyl)methyl]-3,5-dimethyl-1H-pyrazole 4c.
| Compounds | Jurkat | K562 | U937 | HEK293 |
|---|---|---|---|---|
| 4a | 2.47 ± 0.03 | 2.89 ± 0.03 | 2.94 ± 0.03 | 3.80 ± 0.03 |
| 4b | 1.24 ± 0.05 | 1.98 ± 0.06 | 2.15 ± 0.06 | 2.98 ± 0.06 |
| 4c | 1.49 ± 0.06 | 2.16 ± 0.06 | 1.98 ± 0.07 | 3.15 ± 0.05 |
| 4d | 1.89 ± 0.02 | 2.34 ± 0.01 | 2.27 ± 0.02 | 3.34 ± 0.08 |
| 5a | 0.42 ± 0.03 | 0.52 ± 0.04 | 0.60 ± 0.04 | 0.92 ± 0.09 |
| 5b | 0.57 ± 0.02 | 0.68 ± 0.04 | 0.71 ± 0.04 | 0.98 ± 0.02 |
 |
0.11 ± 0.01 |  |
 |
 |
| 6b | 0.19 ± 0.02 | 0.23 ± 0.04 | 0.28 ± 0.04 | 0.85 ± 0.07 |
 |
 |
0.30 ± 0.03 | 0.31 ± 0.02 | 0.46 ± 0.01 |
 |
 |
 |
 |
 |
| Cisplatin | 0.69 ± 0.02 | 0.54 ± 0.06 | 0.96 ± 0.08 | 0.99 ± 0.09 |
The lipophilicity of benzyl- and cyclohexylsulfanylpyrazoles 4b and 4c, when evaluating by reversed phase HPLC,26 showed a greater value for 4c (see Table S2, ESI†).
Palladium and platinum N,N′-complexes were prepared by mixing sulfanyl-substituted pyrazole ligands with metal salts (PdCl2, PdBr2, K2PtCl4) in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at room temperature in acetonitrile or aqueous acetone using general procedures described previously.27 The isolated compounds were air-stable powders, readily soluble in DMSO and DMF and insoluble in water, ethers or chlorinated solvents (Scheme 2).
:1 ratio at room temperature in acetonitrile or aqueous acetone using general procedures described previously.27 The isolated compounds were air-stable powders, readily soluble in DMSO and DMF and insoluble in water, ethers or chlorinated solvents (Scheme 2).
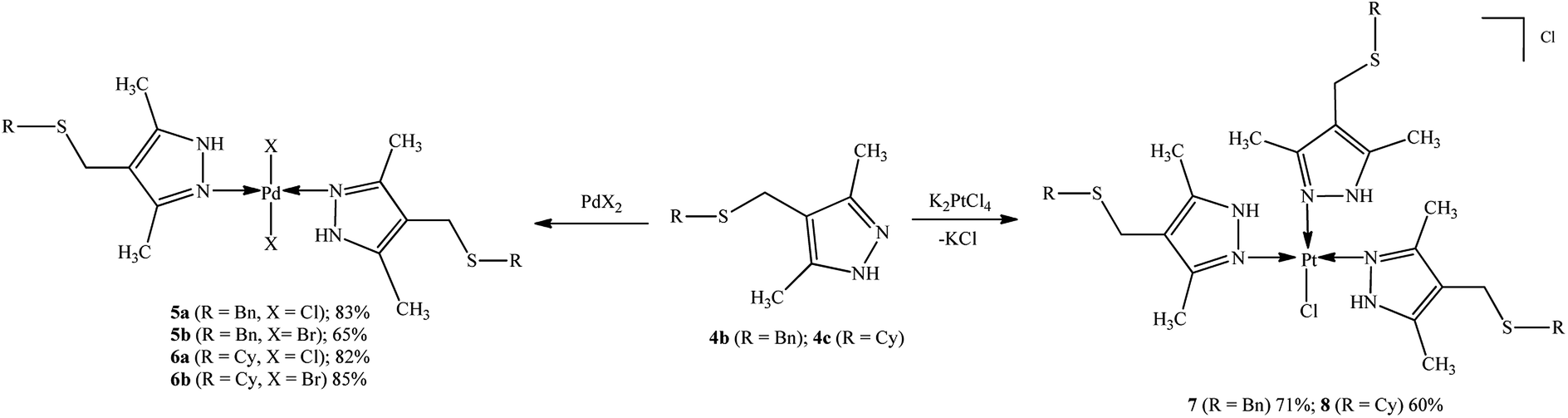 | ||
| Scheme 2 Synthesis and expected structure of palladium(II) and platinum(II) complexes 5a,b, 6a,b, 7, and 8. | ||
The structures of products were confirmed by spectral data (see ESI†). Although the molecule contains soft28 sulfur atoms in the Bn(Cy)–S–CH2 sulfide group of the ligand, the coordination involves imine nitrogen atoms N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of the pyrazole group. Evidently, this is due to higher steric accessibility of the nitrogen lone pair29 as compared with the sulfur atoms, which are sterically hindered (Fig. 2).
C of the pyrazole group. Evidently, this is due to higher steric accessibility of the nitrogen lone pair29 as compared with the sulfur atoms, which are sterically hindered (Fig. 2).
The 1H NMR spectra of palladium(II) complexes 5a and 5b and platinum(II) complex 7 exhibit two characteristic signals for the methyl groups of pyrazole rings at δH 2.06–2.13 ppm and δH 2.15–2.52 ppm with equal integrated intensities. Meanwhile, the initial ligand 4b exhibits one singlet signal at δH ∼ 2.19 ppm, which confirms metal coordination to pyrazole nitrogen atoms. The signals of the methylene groups of the –CH2SCH2– moiety occur at δH 2.31–3.48 ppm and δH 3.50–3.69 ppm, respectively. In the 1H NMR spectra of the complexes, the NH signal (δH ∼ 13 ppm) is shifted downfield with respect to this signal of the free ligand (δH ∼ 10 ppm). The 13C NMR spectrum of platinum complex 7, unlike the spectra of dichloro palladium and dibromo palladium complexes, shows a double set of signals for the –CH2SCH2– moiety at δC 23.89 and 24.47 ppm, δC 35.54 and 35.97 ppm. The strong absorption band at ν 477 cm−1 in the IR spectrum of platinum complex 7 corresponds to Pt–N stretching vibrations, which confirms the structure attributed to complex 7.30
In the positive ion ESI mass spectra of the compounds 5a and 5b groups of peaks with m/z 643–652 Da and 688–696 Da, correspondingly, relate to an isotopic distribution of the [M–Cl + CH3CN]+ and the [M–Br + CH3CN]+ ion with elemental composition of C28H35ClN5PdS2 and C28H35BrN5PdS2 formed by the chlorine (bromine) elimination from the molecule and association with the solvent molecule (CH3CN). In case of the compound 6a the positive ion mass spectrum contains two groups of peaks with m/z 588–594 and 628–636 Da corresponding to ions [M–Cl]+ and [M–Cl + CH3CN]+. Positive ion ESI mass spectrum of 6b contains peaks of the ion [M–Br + CH3CN]+ with m/z 672–681 Da whereas its negative ion spectrum contains peaks with m/z 791–799 Da corresponding to [M + Br]− ion. The isotopic distribution of the peaks with m/z 925–931 Da in the positive ion ESI mass spectrum of 7 corresponds to [M–Cl]+ ion formed from the molecule consisting of three ligand molecules with PtCl2.31 In the positive ion mass spectrum of the compound 8 group of peaks with m/z 900–907 Da relates to [M–Cl]+ ion with monoisotopic mass 902 Da formed from the compound also consisting of three ligand molecules with PtCl2.
Thus, new trans-dichloro (5a, 6a) and trans-dibromo (5b, 6b) palladium bi-ligand complexes with N–Pd–N bonds and dichloro platinum tri-ligand complexes with three N–Pt bonds (7, 8) have been synthesized.
Biology studies
A known metal complex-based cytotoxic agent with a broad spectrum of activity, cisplatin manufactured by Bayer Pharma AG (Germany), was chosen as the reference agent.When measured by flow cytometry, the cytotoxic effect of compounds 4a–d, 5a,b, 6a,b, 7, and 8 against three tumor cell lines, Jurkat, K562, and U937 and conditionally normal human embryonic kidney cells HEK293, shows a clear dose-dependent behavior, specific to each compound (Table 1). It can be seen from Table 1 that, in general, the inhibitory concentrations IC50, determined by exposure of all prepared compounds to the above mentioned cell lines followed by cell staining with the 7AAD dye, vary depending on the cell culture. It can be seen that palladium and platinum complexes are more active against Jurkat, K562, and U937 cells than free ligands. When compared with HEK293 cell culture, higher IC50 values for human kidney embryonic cells were noted. As a rule, conditionally normal HEK293 cells react worse to damaging agents than a Jurkat neoplastic cells. In addition, dichloro palladium complexes 5a and 6a are more active than the corresponding dibromo complexes 5b and 6b. A typical feature is the increase in the cytotoxic activity for cyclohexyl derivatives 4c, 6a,b, and 8 as compared with phenyl (4a,d) and benzyl (4b, 5a,b, and 7) derivatives. The lowest IC50 values were found for Jurkat cell culture; for platinum complexes 7 and 8, these values were 0.07 and 0.02 μM, respectively. In the case of cisplatin, the IC50 value of Jurkat cells is 0.69 μM; this is an order of magnitude lower cytotoxic activity compared to compound 7 and 30 times lower activity than that of compound 8 (Table 1). Palladium complexes 5a,b and 6a,b are somewhat less active than platinum complexes 7 and 8 against Jurkat cells (marked red in Table 1). However, palladium 6a and platinum 8 complexes are more active against K562 and U937 cells (marked red in Table 1). At that, all these compounds show higher cytotoxic effect than cisplatin.
An activity series for compounds 4–8 was composed on the basis of the determined IC50 values (Table 2).
|
|
|---|---|
| Neoplastic cell lines | Correlations between structure and cytotoxicity of compounds 4–8 |
| Jurkat | 8 (CyPzPtCl2) > 7 (BnPzPtCl2) > 6a (CyPzPdCl2) > 6b (CyPzPdBr2) > 5a (BnPzPdCl2) > 5b (BnPzPdBr2) > (cisplatin) > 4b (BnPz) > 4c (CyPz) > 4d (4-OHPhPz) > 4a (PhPz) |
| K562 | 8 (CyPzPtCl2) > 6a (CyPzPdCl2) > 6b (CyPzPdBr2) > 7 (BnPzPtCl2) > 5a (BnPzPdCl2) > (cisplatin) > 5b (BnPzPdBr2) > 4b (BnPz) > 4c (CyPz) > 4d (4-OHPhPz) > 4a (PhPz) |
| U937 | 8 (CyPzPtCl2) > 6a (CyPzPdCl2) > 6b (CyPzPdBr2) > 7 (BnPzPtCl2) > 5a (BnPzPdCl2) > 5b (BnPzPdBr2) > (cisplatin) > 4c (CyPz) > 4b (BnPz) > 4d (4-OHPhPz) > 4a (PhPz) |
| HEK293 | 6a (CyPzPdCl2) > 8 (CyPzPtCl2) > 7 (BnPzPtCl2) > 6b (CyPzPdBr2) > 5a (BnPzPdCl2) > 5b (BnPzPdBr2) > (cisplatin) > 4b (BnPz) > 4c (CyPz) > 4d (4-OHPhPz) > 4a (PhPz) |
The determination of apoptosis induced by the action of compound 7 using the AnnexinV/7AAD intravital double staining method was carried out on Jurkat neoplastic cell lines at a concentration of the test compound from 0.5 μM to 1.5 μM. It was found that Pt complex 7 causes the death of neoplastic cells through the apoptotic pathway (see Fig. 3). As seen, there is exactly the dose-dependent nature of the death along the apoptotic pathway. Thus, at a concentration of 1 μM of compound 7, we observe 23.65% of cells in the early and late stages of apoptosis, and at a concentration of 1.5 μM, the percentage of apoptosis of neoplastic cells was already 37.63% (Fig. 3).
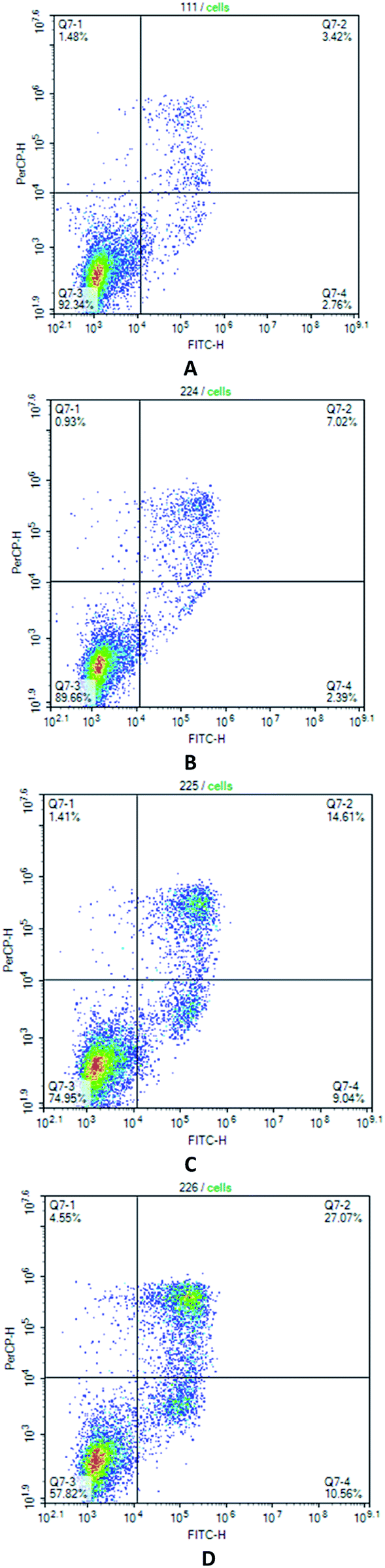 | ||
| Fig. 3 Jurcat cells treated with compound 7 in various concentrations, stained with Annexin V/7AAD, and analyzed by flow cytometry. (A) control; (B) complex 7 (0.5 μM); (C) complex 7 (1.0 μM); (D) complex 7 (1.5 μM). The histogram shows the apoptosis rate (%); *p < 0.05 when compared with the control group (n = 5). | ||
Thereby, Pd complexes 6a,b and Pt complexes 7, 8 are candidate compounds for studying the apoptosis-inducing activity.
Well known,32,33 that some compounds containing a metal atom in their structure, especially platinum and ruthenium atoms, bind with DNA of cancer cells. In our experiment (Fig. 4), we observed a change in the electrophoretic mobility of DNA molecules, which may be a consequence of the interaction of complexes 6a and 8 directly with desoxyribonucleic acid, and, as a result, can be one of the mechanisms of cytotoxicity of the synthesized by us organometallic compounds.
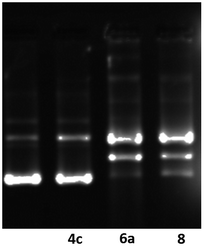 | ||
| Fig. 4 Modification of gel electrophoretic mobility of pHOT1 plasmid DNA, when incubated 1 h with various concentrations of cyclohexyl-containing compounds 4c, 6a and 8. Concentrations are as follows: (line 1) untreated pHOT1 plasmid DNA; (line 2) compound 4c 1 μM + plasmid DNA; (line 3) compound 6a 1 μM + plasmid DNA; (line 4) compound 8 1 μM + plasmid DNA. | ||
The N,N′-dichloro-tri[(4-(cyclohexylsulfanyl)methyl)-3,5-dimethyl-1H-pyrazole]platinum(II) complex 8 was identified as the lead compound with respect to the Jurkat, K562, and U937 cell lines, with its cytotoxic action being three times higher than that of the N,N′-dichloro-tri[(4-(benzylsulfanyl)methyl)-3,5-dimethyl-1H-pyrazole]platinum complex 7 (IC50, Tables 1, 2 and ESI† (selectivity index), Table S3, ESI†).
Conclusions
The cytotoxic activity of metal complexes bearing sulfanyl-substituted pyrazole ligands has been studied for the first time. The introduction of sulfide sulfur into the ligands bearing a pyrazole moiety had a beneficial effect on increasing the cytotoxic activity of Pt(II) and Pd(II) complexes by in vitro evolution.Cyclohexylsulfanyl(dichloro)platinate was found to be more active than benzyllsulfanyl(dichloro)platinate. Cyclohexyl substituted pyrazole ligand have greater ability to penetrate into neoplastic cells as compared to benzyl and phenyl substituted compounds, which follows from the assessment of their lipophilicity. When testing on K562 and U937 cell lines, cyclohexylsulfanyl-containing Pd(II) complexes demonstrated greater cytotoxic activity than benzylsulfanyl-containing Pt(II) complex. Dichloro-palladium complexes were found to be more active than their dibromo analogues.
Dichloro Pd and dichloro Pt complexes with cyclohexylsulfanylpyrazole ligands have been shown to have affinity for the DNA of neoplastic cells.
Taking into account the IC50 and SI values of the leader complex – dichlorotri-[(4-(cyclohexylsulfanyl)methyl)-3,5-dimethyl-1H-pyrazole]platinate, which far exceeds the activity of cisplatin (a cytotoxic drug in chemotherapy), the obtained new complex may serve as the basis for the development of more efficient antitumor agents.
Experimental
Materials and methods
All synthesized compounds were characterized by 1H and 13C, 15N NMR spectral data that were recorded on spectrometers Bruker Avance 400 NMR (400.13 MHz and 100.62 MHz) and Bruker Ascend III HD 500 (500.17 MHz and 125.78 MHz), internal standard TMS, solvent DMSO-D6. IR spectra were recorded on a Bruker Vertex-70V FTIR and Specord M80 spectrometers. Matrix-assisted laser desorption/ionization (MALDI) mass spectrum was recorded on a Bruker's device MALDI TOF Autoflex III (compounds 7 and 8 in DMSO) with sinapinic acid as a matrix. Electrospray ionization (ESI) mass spectra were obtained on a HPLC mass spectrometer LCMS-2010EV (Shimadzu) in the positive and negative ions mode at the ionizing electrode potential of 4.5 kV and −3.5 kV, respectively. Sample solution (direct syringe sample inlet) was in methanol (acetonitrile), mobile phase (acetonitrile/water, 95:5) flow rate was 0.1 mL min−1. The heater's and the desolvation line's temperature was 200 and 230 °C, respectively. The nebulizer gas (nitrogen) flow rate was 1.5 L min−1. Elemental analysis was performed on a Carlo Erba 1106 elemental analyzer. Melting points were determined on a Kofler hot-stage microscope and utilized uncorrected. Individuality and purity of synthesized compounds were controlled by means of TLC on Sorbfil plates; I2 was used as developer. A known pharmaceutical drug, cisplatin, manufactured by Bayer Pharma AG (Germany), with cis-diaminedichloridoplatinum(II) as the active substance, was used as the reference drug.
3,5-Dimethyl-4-[(phenylsulfanyl)methyl]-1H-pyrazole (4a). White powder, 1H NMR spectrum (400 MHz, CDCl3), δ, ppm: 2.17 (6H, s, CH3); 3.94 (2H, s, CH2); 7.24–7.37 (5H, m, H Ph); 10.93 (1H, br. s, NH).
4-[(Benzylsulfanyl)methyl]-3,5-dimethyl-1H-pyrazole (4b). White powder, 1H NMR spectrum (400 MHz, CDCl3), δ, ppm: 2.19 (6H, s, CH3); 3.49 (2H, s, PhCH2S); 3.69 (2H, s, SCH2); 7.25–7.35 (5H, m, H Ph); 10.32 (1H, br. s, NH).
4-[(Cyclohexylsulfanyl)methyl]-3,5-dimethyl-1H-pyrazole (4c). White powder, 1H NMR spectrum (400 MHz, CDCl3), δ, ppm: 1.26–1.40 (4H, m, CH2); 1.60–1.64 (2H, m, CH2); 1.76–1.79 (2H, m, CH2); 1.97–2.00 (2H, m, CH2); 2.28 (6H, s, CH3); 2.52–2.60 (1H, m, CHS); 3.57 (2H, s, SCH2); 10.63 (1H, br. s, NH).
4-{[(3,5-Dimethyl-1H-pyrazol-4-yl)methyl]sulfanyl}-phenol (4d). White powder, 1H NMR spectrum (400 MHz, DMSO-D6), δ, ppm (J, Hz): 1.95 (6H, s, CH3); 3.76 (2H, s, SCH2); 6.69 (2H, d, J = 8.0, H Ar), 7.12 (2H, d, J = 8.0, H Ar); 9.60 (1H, s, OH); 11.99 (1H, br. s, NH).
Dichlorodi-[(4-(benzylsulfanyl)methyl)-3,5-dimethyl-1H-pyrazole]palladium complex (5a). Yellow powder, yield, 0.15 g (83%). Mp 176–178 °C. Found: C, 48.79; H, 5.23; Cl, 11.13; N, 8.62; S, 9.83. Anal. calc. for C26H32Cl2N4PdS2: C, 48.64; H, 5.02; Cl, 11.04; N, 8.73; Pd, 16.58; S, 9.99. IR (Nujol, cm−1): ν = 3470 (s), 1584 (s), 1299 (s), 1198 (m), 724 (m), 477 (s), 362 (s), 341 (s). 1H-NMR (ppm): δ = 13.24 (s, NH), 7.25–7.33 (m, Ar), 3.69 (s, SCH2Ph), 3.48 (s, PzCH2S), 2.52 (s, CH3), 2.13 (s, CH3). 13C-NMR (ppm): δ = 9.5, 13.3, 23.9, 36.0, 113.3, 127.3, 128.9, 129.2, 138.6, 141.6, 148.8. 15N-NMR (ppm): δ = 195.8 and 200.7 (NH). MS (ESI), m/z (I, %): 646 (65) [M-Cl + CH3CN]+.
Dibromodi-[(4-(benzylsulfanyl)methyl)-3,5-dimethyl-1H-pyrazole]palladium complex (5b). Yellow powder, yield, 0.13 g (65%). Mp 166–168 °C. Found: C, 42.87; H, 4.46; N, 7.48; S, 8.91; Br, 21.73. Anal. calc. for C26H32Br2N4PdS2: C, 42.73; H, 4.41; N, 7.67; Pd, 14.56; S, 8.77; Br, 21.86. IR (Nujol, cm−1): ν = 3469 (s), 3079 (m), 1295 (s), 710 (m), 477 (m), 382 (s), 359 (s), 278 (m). 1H-NMR (ppm): δ = 13.21 (s, NH), 7.21–7.29 (m, Ar), 3.62 (s, SCH2Ph), 3.44 (s, PzCH2S), 2.48 (s, CH3), 2.09 (s, CH3). 13C-NMR (ppm): δ = 9.5, 13.9, 23.9, 35.8, 113.5, 127.3, 128.9; 129.2, 138.6, 141.9, 149.1. 15N-NMR (ppm): δ = 177.0 and 205.0 (NH). MS (ESI), m/z (I, %): 690 (52) [M-Br + CH3CN]+.
Dichlorodi-[(4-(cyclohexylsulfanyl)methyl)-3,5-dimethyl-1H-pyrazole]palladium complex (6a). Yellow powder, yield, 0.14 g (82%). Mp 192–194 °C. Found: C, 46.15; H, 6.39; N, 8.89; S, 10.31; Cl, 11.42. Anal. calc. for C24H40Cl2N4PdS2: C, 46.04; H, 6.44; N, 8.95; Pd, 17.00; S, 10.24; Cl, 11.33. IR (cm−1): ν = 3468 (s), 3161 (m), 2714 (m), 1587 (s), 1308 (s), 1204 (m), 846 (m), 773 (s). 1H-NMR (ppm): δ = 13.20 (s, NH), 3.56 (s, PzCH2S), 2.58 (s, CHS), 2.21 and 2.18 (s, CH3), 1.90–1.93, 1.68–1.69, 1.54–1.56, 1.23–1.28 (m, CH2). 13C-NMR (ppm): δ = 9.6, 13.4, 22.3, 25.8, 33.5, 43.3, 114.0, 141.3, 148.7. MS (ESI), m/z (I, %): 589 (41) [M–Cl]+, 630 (59) [M–Cl + CH3CN]+.
Dibromodi-[(4-(cyclohexylsulfanyl)methyl)-3,5-dimethyl-1H-pyrazole]palladium complex (6b). Yellow powder, yield, 0.17 g (85%). Mp (dec.) 218–220 °C. Found: C, 40.24; H, 5.69; N, 7.91; S, 9.15; Br, 22.43. Anal. calc. for C24H40Br2N4PdS2: C, 40.32; H, 5.64; N, 7.84; Pd, 14.88; S, 8.97; Br, 22.35. IR (Nujol, cm−1): ν = 3244 (s), 1578 (s), 1196 (s), 820 (s), 658 (s), 580 (s), 508 (s), 321 (s). 1H-NMR (ppm): δ = 13.19 (s, NH), 3.56 (s, PzCH2S), 2.58 (s, CHS), 2.22 (s, CH3), 1.89–1.93, 1.68–1.69, 1.54–1.56, 1.23–1.25 (m, CH2). 13C-NMR (ppm): δ = 9.6, 13.9, 22.3, 25.8, 33.6, 43.2, 114.1, 141.6, 148.9. MS (ESI), m/z (I, %): 674 (56) [M–Br + CH3CN]+, 791 (48) [M + Br]−.
Dichlorotri-[(4-(benzylsulfanyl)methyl)-3,5-dimethyl-1H-pyrazole]platinum complex (7). Light yellow powder, yield, 0.14 g (71%). Mp (dec.) 86–88 °C. IR (Nujol, cm−1): ν = 3446 (s), 3162 (s), 1580 (s), 1206 (m), 1071 (s), 701 (m), 504 (s), 477 (s), 342 (s), 310 (m). 1H-NMR (ppm): δ = 13.81 (s, NH), 7.23–7.32 (m, Ar), 3.65 and 3.69 (s, SCH2Ph), 3.48 and 3.53 (s, PzCH2S), 2.31 and 2.34 (s, CH3), 2.06 and 2.15 (s, CH3). 13C-NMR (ppm): δ = 9.6, 10.7, 13.1, 23.9, 24.5, 35.5, 36.0, 113.5, 127.2, 127.3, 128.8, 128.9, 129.2, 138.6, 138.9, 141.3, 147.9. 15N-NMR (ppm): δ = 170.3 and 192.6 (NH), 179.6 and 206.1 (NH). MS (MALDI TOF) 926.6899 [M–Cl]+; MS (ESI), m/z (I, %): 926 (87) [M–Cl]+.
Dichlorotri-[(4-(cyclohexylsulfanyl)methyl)-3,5-dimethyl-1H-pyrazole]platinum complex (8). Light yellow powder, yield, 0.12 g (60%). Mp 102–104 °C. IR (Nujol, cm−1): ν = 3445 (s), 3191 (m), 1585 (s), 1203 (m), 1037 (s), 722 (m). 1H-NMR (ppm): δ = 13.79 (s, NH), 3.53 (s, PzCH2S), 2.54 (s, CHS), 2.12 (s, CH3), 1.90–1.94, 1.68–1.69, 1.54–1.55, 1.24–1.28 (m, CH2). 13C-NMR (ppm): δ = 9.7; 13.1, 22.8, 26.0, 33.6, 42.9, 111.6, 141.8, 147.8. MS (MALDI TOF) 903.3284 [M–Cl]+; MS (ESI), m/z (I, %): 902 (76) [M–Cl]+, 819 (19) [M–Cl–C6H11]+.
X-ray crystallography
All diffraction measurements were performed at room temperature (298 K) using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) on a Agilent Xcalibur (Eos, Gemini) single-crystal diffractometer. Collection and processing of data performed with using the program CrysAlisPro Oxford Diffraction Ltd. The structures were solved with the ShelXS34 structure solution program using Direct Methods and refined with the ShelXL35 refinement package using Least Squares minimization. The structure was refined by a full-matrix least-square technique using anisotropic thermal parameters for non-hydrogen atoms. The H16 atom in structure 1 was located in a difference Fourier map and refined isotropically, and the other H atoms were positioned geometrically and treated using a riding model, fixing the bond lengths at 0.960, 0.970, 0.930 and 0.860 Å for CH3, CH2, CH and NH atoms, respectively. Atomic coordinates, bond lengths, bond angles, and thermal parameters. The crystallographic data for compound 4d are collected in Table 3.| CCDC | 1954607 |
| Empirical formula | C12H14N2OS |
| Formula weight | 234.31 |
| Temperature/K | 293(2) |
| Crystal system | Monoclinic |
| Space group | C2/c |
| a/Å | 19.1997(11) |
| b/Å | 7.7319(4) |
| c/Å | 16.5888(10) |
| α/° | 90 |
| β/° | 95.914(5) |
| γ/° | 90 |
| Volume/Å3 | 2449.5(2) |
| Z | 8 |
| ρcalc/g cm−3 | 1.271 |
| μ/mm−1 | 0.245 |
| F(000) | 992.0 |
| Radiation | MoKα (λ = 0.71073) |
| 2Θ range for data collection/° | 4.266 to 58.278 |
| Index ranges | −26 ≤ h ≤ 24, −6 ≤ k ≤ 10, −19 ≤ l ≤ 22 |
| Reflections collected | 5838 |
| Independent reflections | 2802 [Rint = 0.0215] |
| Data/restraints/parameters | 2802/0/151 |
| Goodness-of-fit on F2 | 1.041 |
| Final R indexes [I ≥ 2σ(I)] | R1 = 0.0461, wR2 = 0.1180 |
| Final R indexes [all data] | R1 = 0.0644, wR2 = 0.1323 |
| Largest diff. peak/hole/e Å−3 | 0.22/−0.26 |
Cell culturing
Jurkat, K562, U937 and HEK293 cells were received from the Russian Collection of Cell Cultures (Institute of Cytology, Russian Academy of Sciences, St.-Petersburg). The cells were cultured in the RPMI 1640 complete medium (Biolot) containing 10% of fetal calf serum; 2 mM of L-glutamine; 50 U mL−1 of penicillin; and 50 μg mL−1 of streptomycin. HEK293 cell line was cultured as monolayers and maintained in Dulbecco's modified Eagle's medium (DMEM, Gibco BRL) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin solution. The cells were cultured in vials in a CO2 incubator at a temperature of 37 °C, a 5% CO2 atmosphere, and saturation humidity. The cell viability was studied for 5th to 10th passage cells. For experiments, the suspension cell cultures that have reached the logarithmic growth phase were subcultured into 24-well plates (100 thousand cells per well). After addition of test compounds, the cells were incubated for 24 h and the results were analyzed by flow cytometry.Evaluation of cell viability
The cell viability was evaluated by flow cytometry using 7-AAD (7-aminoactinomycin D), a fluorescent DNA dye (Sony Biotechnology Inc.). This dye cannot penetrate living cells with undamaged, intact membranes.The intensity of 7-AAD fluorescence in the BL-4 channel (PerCP) was evaluated on a NovoCyteTM 2000 flow cytometer (ACEA). The instrument was adjusted against control samples including living unstained cells (estimation of autofluorescence), living cells stained with 7AAD with no dye uptake and low fluorescence level (gating of living cells), and cells incubated under the same conditions with compounds inducing cell death (gating of dead cells). In each sample, at least 1 × 104 cells were measured. If dead cells are present, the cell population is cytometrically subdivided into two groups: living cells show a low fluorescence level, whereas dead cells have high fluorescence intensity.
Study of apoptosis
The quantitative analysis of apoptosis-inducing activity was performed using Alexa Fluor® 488 Annexin V/Dead Cell Apoptosis Kit, with fluorescence being recorded on a NovoCyteTM 2000 flow cytometer (ACEA). Phosphatidylserine externalization on the plasmatic membrane surface is a reliable sign of cell apoptosis or necrosis. Normally functioning living cells have a minor amount of phosphatidylserine on the outer cell membrane and, hence, the interaction with Alexa Fluor® 488 annexin V is insignificant. In addition, undamaged cell membrane is impermeable for propidium iodide. In early apoptosis, phosphatidylserine molecules appear on the cell surface; however, the membrane is still impermeable for dyes that bind to DNA (such as propidium iodide). The membrane integrity is damaged at later stages of cell death. Thus, when apoptosis is detected, four types of cells are distinguished: living cells (annexin V−/PI−), early apoptotic cells (annexin V+/PI−), late apoptotic cells (annexin V+/PI+), and necrotic cells (annexin V−/PI+).Interaction between DNA plasmid pHOT and tested compounds
The reaction mixture (20 μL) containing 0.25 μg of the DNA plasmid pHOT (TopoGen, USA), and the tested compound was incubated in the buffer (35 mM Tris–HCl, pH 8.0; 72 mM KCl, 5 mM MgCl, 5 mM dithiothreitol, 5 mM spermidine, and 0.01% bovine serum albumin) for 30 min at 37 °C using a Biosan thermostat (Latvia). The reaction was terminated by adding sodium dodecyl sulfate up to a concentration of 1%. A 0.1% solution of bromophenol blue (1:10) was added and the samples were electrophoresed in the presence of ethidium bromide. The reaction products were separated in a 1% agarose gel (3 V cm−1) for 4–6 h. After the electrophoresis without ethidium bromide, the gels were treated with a solution of ethidium bromide (0.5 μg mL−1). The gels were visualized in the UV light in a Infinity VX2 1120/Blue X-Press gel documentation system (Vilber Lourmat, France).
Statistical analysis
All numerical data are presented as mean ± standard deviation from at least three independent experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially partly supported by the Russian Foundation for Basic Research (Project numbers 17-43-020292 r_a, 18-29-09068), Grants Council of the President of Russian Federation (grant NSh-5240.2018.3) and within the framework of the Project part of the State Assignment AAAA-A19-119022290010-9 and AAAA-A20-120012090029-0. The structural data were obtained with the financial support of the Russian Ministry of Education and Science (project no. 2019-05-595-000-058) on unique equipment at the “Agidel” Collective Usage Center (Ufa Federal Research Center, Russian Academy of Sciences).References
- Z. Darzynkiewicz, F. Traganos and D. Wlodkowic, Eur. J. Pharmacol., 2009, 25(1–3), 625, DOI:10.1016/j.ejphar.2009.05.032 , 143–150.
- X. Wang and Z. Guo, Chem. Soc. Rev., 2013, 42, 202–224, 10.1039/c2cs35259a.
- A. Salim Abu-Surrah and M. Kettunen, Curr. Med. Chem., 2006, 13, 1337–1357, DOI:10.2174/092986706776872970.
- S. Niazi, M. Purohit and J. H. Niazi, Eur. J. Med. Chem., 2018, 5(158), 7–24, DOI:10.1016/j.ejmech.2018.08.099.
- S. P. Basourakos, L. Li, A. M. Aparicio, P. G. Corn, J. Kim and T. C. Thompson, Curr. Med. Chem., 2017, 24(15), 1586–1606, DOI:10.2174/0929867323666161214114948.
- Kh. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. N. Mabkhot, F. A. Al-aizari and M. Ansar, Molecules, 2018, 23(1), 134–219, DOI:10.3390/molecules23010134.
- L. A. Pham-Huy, H. He and C. Pham-Huy, Indian J. Clin. Biochem., 2015, 30, 11–26, DOI:10.1007/s12291-014-0446-0.
- V. R. Akhmetova, R. A. Galimova, N. S. Akhmadiev, A. M. Galimova, R. A. Khisamutdinov, G. M. Nurtdinova, E. F. Agletdinov and V. A. Kataev, Adv. Pharm. Bull., 2018, 8, 267–275, DOI:10.15171/apb.2018.031.
- N. S. Akhmadiev, A. M. Galimova, V. R. Akhmetova, V. R. Khairullina, R. A. Galimova, E. F. Agletdinov, A. G. Ibragimov and V. A. Kataev, Adv. Pharm. Bull., 2019, 9, 674–684, DOI:10.15171/apb.2019.079.
- A. Schmittel, M. Schmidt-Hieber, P. Martus, N. E. Bechrakis, R. Schuster, J. M. Siehl, M. H. Foerster, E. Thiel and U. Keilholz, Ann. Oncol., 2006, 17, 1826–1829, DOI:10.1093/annonc/mdl309.
- S. Puyo, D. Montaudon and P. Pourquier, Crit. Rev. Oncol. Hemat., 2014, 89, 43–61, DOI:10.1016/j.critrevonc.2013.07.006.
- A. Galaup and A. Paci, Expert Opin. Drug Metab. Toxicol., 2013, 9, 333–347, DOI:10.1517/17425255.2013.737319.
- L. Falzone, S. Salomone and M. Libra, Front. Pharmacol., 2018, 9, 1300, DOI:10.3389/fphar.2018.01300 , 1–26.
- Y. Zhang, D. Xu, X. Wang, M. Lu, B. Gao and X. Giao, Mol. Med. Rep., 2014, 9, 83–90, DOI:10.3892/mmr.2013.1781.
- F. K. Keter and J. Darkwa, BioMetals, 2012, 25, 9–21, DOI:10.1007/s10534-011-9496-4.
- E. Budzisz, I.-P. Lorenz, P. Mayer, P. Paneth, L. Szatkowski, U. Krajewska, M. Rozalskid and M. Miernicka, New J. Chem., 2008, 32, 2238–2244, 10.1039/b808301k.
- J. Quirante, D. Ruiz, A. Gonzalez, C. Lopez, M. Cascante, R. Cortes, R. Messeguer, C. Calvis, L. Baldoma, A. Pascual, Y. Guerardel, B. Pradines, M. Font-Bardia, T. Calvet and C. Biot, J. Inorg. Biochem., 2011, 105, 1720–1728, DOI:10.1016/j.jinorgbio.2011.09.021.
- T. Lazarevića, A. Rilak and Ž. D. Bugarčić, Eur. J. Med. Chem., 2017, 142, 8–31, DOI:10.1016/j.ejmech.2017.04.007.
- A. R. Kapdi and I. J. S. Fairlam, Chem. Soc. Rev., 2014, 43, 4751–4777, 10.1039/C4CS00063C.
- U. Ndagi, N. Mhlongo and M. E. Soliman, Drug Des., Dev. Ther., 2017, 11, 599–616, DOI:10.2147/DDDT.S119488.
- F. K. Keter, S. Kanyanda, S. S. L. Lyantagaye, J. Darkwa, D. J. G. Rees and M. Meyer, Cancer Chemother. Pharmacol., 2008, 63, 127–138, DOI:10.1007/s00280-008-0721-y.
- A. S. Abu-Surrah, K. A. A. Safieh, I. M. Ahmad, M. Y. Abdalla, M. T. Ayou, A. K. Qaroush and A. M. Abu-Mahtheieh, Eur. J. Med. Chem., 2010, 45, 471–475, DOI:10.1016/j.ejmech.2009.10.029.
- R. Czarnomysy, A. Surazynski, A. Muszynska, A. Gornowicz, A. Bielawska and K. Bielawski, J. Enzyme Inhib. Med. Chem., 2018, 33(1), 1006–1023, DOI:10.1080/14756366.2018.1471687.
- N. S. Akhmadiev, V. R. Akhmetova, T. F. Boiko and A. G. Ibragimov, Chem. Heterocycl. Compd., 2018, 54, 344–350, DOI:10.1007/s10593-018-2271-5.
- C. A. M. Afonso, N. R. Candeias, D. P. Simão, A. F. Trindade, A. S. Jaime, J. A. S. Coelho, B. Tan and R. Franzén, Comprehensive organic chemistry experiments for the laboratory classroom, The Royal Society of Chemistry, 2016, p. 366 Search PubMed.
- OECD Guidelines for the testing of chemicals Test 117: Partition coefficient (n-octanol/water), High performance liquid chromatography (HPLC) method, Paris, 2004 Search PubMed.
- N. S. Akhmadiev, E. S. Mescheryakova, R. A. Khisamutdinov, A. N. Lobov, M. F. Abdullin, A. G. Ibragimov, R. V. Kunakova and V. R. Akhmetova, J. Organomet. Chem., 2018, 872, 54–62, DOI:10.1016/j.jorganchem.2018.07.026.
- J. L. Reed, Inorg. Chem., 2008, 47, 5591–5600, DOI:10.1021/ic701377n.
- R. Mukherjee, Coord. Chem. Rev., 2000, 203, 151–218, DOI:10.1016/S0010-8545(99)00144-7.
- K. Nakamoto, Infrared and Raman spectra of inorganic and coordination compounds, Mir, Moscow, 1991, p. 363 Search PubMed.
- A. V. Khripun, V. Yu. Kukushkin, S. I. Selivanov, M. Haukka and A. J. L. Pombeiro, Inorg. Chem., 2006, 45, 5073–5083, DOI:10.1021/ic0602300.
- C. A. Rabik and M. E. Dolan, Cancer Treat. Rev., 2007, 33, 9–23, DOI:10.1016/j.ctrv.2006.09.006.
- A. Grau-Campistany, A. Massaguer, D. Carrion-Salip, F. Barragán, G. Artigas, P. López-Senín, V. Moreno and V. Marchán, Mol. Pharm., 2013, 10, 1964–1976, DOI:10.1021/mp300723b.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122, DOI:10.1107/S0108767307043930.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8, DOI:10.1107/S205322961402421.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1954607. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra09783j |
| This journal is © The Royal Society of Chemistry 2020 |