DOI:
10.1039/C9RA09782A
(Paper)
RSC Adv., 2020,
10, 5040-5049
Two-dimensional square-grid iron(II) coordination polymers showing anion-dependent spin crossover behavior†
Received
22nd November 2019
, Accepted 24th January 2020
First published on 31st January 2020
Abstract
Two Fe(II)-based coordination polymers [Fe(tpmd)2(NCS)2]·5.5H2O (1) and [Fe(tpmd)2(NCSe)2]·7H2O (2) with the framework of square-grid type have been assembled from FeSO4·7H2O, N,N,N′,N′-tetrakis(pyridin-4-yl)methanediamine (tpmd), and KNCS/KNCSe in methanol and characterized. By utilizing two pyridine groups of a tpmd ligand, 1 and 2 are formed in two-dimensional layered structures through coordination of octahedral iron(II) ions with the tpmd to NCS−/NCSe− ligands in which they have a supramolecular isomorphous conformation. 1 shows a paramagnetic behavior between 2 and 300 K, while 2 exhibits two-step spin crossover (ca. 145 and 50 K) in the temperature range due to the coordination of NCSe− ligands. At 300 K 2 is fully high-spin state. However, at 100 K 2 becomes ca. 50% high spin and 50% low spin iron(II) ions, which is verified by magnetic moments. In the structural analysis of 2 at 100 K, two different layers are observed with different bond distances around iron(II) ions in which the layers are stacked alternately.
Introduction
Multi-dimensional coordination polymers with iron(II) ions have been intensively studied in magnetic systems, because of their potential functionalities such as spin crossover and molecular magnetism.1 Particularly, the spin crossover (SCO) of iron(II) ion is well-known behavior associated with spin state inter-change between S = 0 and S = 2. That is, iron(II) compounds with strong field ligands show low spin states (S = 0), while iron(II) compounds with weak field ligands exhibit high spin states (S = 2).2 In order to observe the SCO behaviors, it is very important to choose some ligands that coordinate to coordination compounds. Furthermore, this SCO phenomenon can be controlled by external stimuli, i.e., temperature, pressure, and light. As such, the SCO iron(II) coordination polymers have potential applications such as displays, optical sensors, and memory devices.3
A variety of coordination polymers with unique architecture and geometry have been constructed by using pyridine-based multidentate ligands and transition metal ions.4 For example, zinc(II) and cadmium(II) coordination polymers incorporating bis-9,10-(pyridine-4-yl)-anthracene unit have been synthesized and shown strong blue emission in the solid state. The polymers displayed 1-D and 2-D topologies depending on the anions and also porous structures with 2-D lattices.4e Another coordination framework [Fe(bipytz)(Au(CN)2)2] has been reported (bipytz = 3,6-bis(4-pyridyl)-1,2,4,5-tetrazine). This compound is topotactically changed to [Fe(bipydz)(Au(CN)2)2] by a single-crystal-to-single-crystal transformation (bipydz = 3,6-bis(4-pyridyl)-1,2-diazine). They show both 3-D Hofmann-types and exhibit significantly different SCO properties.4f
Many iron(II) coordination polymers containing NCS−, NCSe−, and NCBH3− as counter ions have been reported.5 Basically the anions are pseudohalide species and isomorphous, however, their electronic effects are quite different due to the electronegativities of S, Se and BH3. Therefore the anions can be influenced by the central iron(II) ion by coordination, especially, electronic structure. These ions can be applied for controlling the spin states of iron(II) ions in coordination compounds. The order of ligand field strength is increasing in the order of NCS−, NCSe−, and NCBH3−. Furthermore, the NCX− ligands have been proved to be good candidates to control the spin crossover temperature in coordination compounds. For example, mononuclear complexes [Fe(bpte)(NCX)2] (X = S, Se and BH3; bpte = S,S′-bis(2-pyridylmethyl)-1,2-thioethane) with the N4S2 coordination environment have been observed to have spin crossover behavior depending on the NCX− anions. That is, the spin crossover temperature for HS ↔ LS interconversion was gradually increased by increasing the ligand field strength of the NCX− ligands from NCS− to NCSe− to NCBH3−.6
Very recently, we have reported that porous coordination polymer [Fe(tpmd)2(NCBH3)2]·4H2O demonstrates spin crossover behavior depending on guest molecules, i.e., H2O and CO2 (tpmd = N,N,N′,N′-tetrakis(pyridin-4-yl)methanediamine). The functional polymer has been shown a three-dimensional porous framework. The structural characteristic and SCO behavior can be attributed to the counter anion (NCBH3−).7 Thus we have tried to make similar coordination polymers structurally but different SCO properties with NCX− ligands (X = S, Se). Remarkably, we have assembled two-dimensional coordination polymers exhibiting interesting SCO behaviors by NCS− and NCSe− ligands, contrary to the coordination polymer [Fe(tpmd)2(NCBH3)2]·4H2O. Herein, we report two coordination polymers [Fe(tpmd)2(NCS)2]·5.5H2O (1) and [Fe(tpmd)2(NCSe)2]·7H2O (2) that were prepared in methanol from Fe(II) salt, NCX− (X = S for 1, Se for 2), and tpmd ligand under an inert atmosphere. These coordination polymers show two-dimensional square-grid structures and spin crossover behavior depending on counter anions.
Experimental
General
All chemicals used in the synthesis were of reagent grade and used without further purification. N,N,N′,N′-Tetrakis(pyridin-4-yl)methanediamine (tpmd) was prepared according to the literature procedure.8 All syntheses of iron(II) species were carried out in a dry oxygen-free glove box by using deoxygenated solvents. Infrared spectra were recorded with a Thermo Fisher Scientific IR200 spectrophotometer (±1 cm−1) using KBr disk. Elemental analyses were carried out using a Fisons/Carlo Erba EA1108 instrument in air. Thermogravimetric analyses (TGA) were performed at a scan rate of 5 °C min−1 using a Seiko TG/DTA 320 & SSC 5200H Disk Station system. Magnetic susceptibilities were measured in applied field of 5000 Oe between 2 and 300 K on a Quantum Design MPMS superconducting quantum interference device (SQUID) magnetometer. Diamagnetic corrections were made [513.24 (1) and 585.04 × 10−6 (2) emu mol−1] by using Pascal's constants.
Syntheses of compounds
[Fe(tpmd)2(NCS)2]·5.5H2O (1). A solution of KNCS (0.194 g, 2.0 mmol) in MeOH (3 mL) was added to FeSO4·7H2O (0.280 g, 1.0 mmol) in MeOH (8 mL). The solution was stirred for 20 min and filtered to remove K2SO4. To the filtrate was slowly added a solution of tpmd (0.090 g, 0.25 mmol) in MeOH (3 mL) with stirring. From the mixture solution a microcrystalline yellow precipitate formed within 1 h, which was collected by filtration, washed with MeOH and dried. Yield: 101 mg (41%). Single crystals suitable for X-ray crystallography were obtained by layering of the MeOH solution of tpmd on the filtered MeOH solution of FeSO4·7H2O and KNCS for several days. IR (KBr, cm−1): 3453, 3036, 2957, 2060, 1583, 1499, 1364, 1220, 1069, 1007, 814. Anal. calcd for C44H47FeN14O5.5S2: C, 53.93; H, 4.83; N, 20.01; S, 6.54. Found: C, 53.57; H, 4.51; N, 20.44; S, 6.39.
[Fe(tpmd)2(NCSe)2]·7H2O (2). A solution of KNCSe (0.288 g, 2.0 mmol) in MeOH (3 mL) was added to FeSO4·7H2O (0.280 g, 1.0 mmol) in MeOH (8 mL). The solution was stirred for 20 min and filtered to remove K2SO4. To the filtrate was slowly added a solution of tpmd (0.090 g, 0.25 mmol) in MeOH (3 mL) with stirring. From the mixture solution a microcrystalline yellow precipitate formed within 1 h, which was collected by filtration, washed with MeOH and dried. Yield: 145 mg (51%). Single crystals suitable for X-ray crystallography were obtained by layering of the MeOH solution of tpmd on the filtered MeOH solution of FeSO4·7H2O and KNCSe for several days. IR (KBr, cm−1): 3435, 3061, 2973, 2930, 2059, 2187, 1592, 1499, 1364, 1220, 1055, 1011, 821. Anal. calcd for C44H50FeN14O7Se2: C, 48.01; H, 4.58; N, 17.81. Found: C, 48.34; H, 4.87; N, 17.52.
X-ray crystallographic data collection and refinement
Single crystal of 1 was mounted on a CryoLoop® with Paratone® oil. Intensity data for 1 were collected with a Bruker APEX CCD-based diffractometer (Korea Basic Science Institute, Chonju branch) and using Mo Kα radiation (λ = 0.71073 Å, graphite monochromator) at 200(2) K. The raw data were processed to give structure factors using the Bruker SAINT program and corrected for Lorentz and polarization effects.9 Single crystal of 2 was coated with paratone-N oil. Single crystal diffraction data of 2 were collected with the same crystal at 298(2) and 104(2), respectively. The crystal diffraction data at each temperature were designated as 2a and 2b, respectively, with synchrotron radiation (λ = 0.80000 and 0.75000 Å) on an ADSC Quantum-210 detector at 2D SMC with a silicon (111) double crystal monochromator (DCM) at the Pohang Accelerator Laboratory, Korea. The ADSC Q210 ADX program10 was used for data collection (detector distance is 62 mm, omega scan; Δω = 1°, exposure time is 3 s per frame) and HKL3000sm (Ver. 703r)11 was used for cell refinement, reduction, and absorption correction. The crystal structures of 1, 2a, and 2b were solved by direct methods,12 and refined by full-matrix least-squares refinement using the SHELXL-2013 computer program.13 The positions of all non-hydrogen atoms were refined with anisotropic displacement factors. All hydrogen atoms were placed using a riding model, and their positions were constrained relative to their parent atoms using the appropriate HFIX command in SHELXL-2013, except the hydrogen atoms of water molecules. Even though the guest solvent molecules were found in the structure, those could not be well refined because of severe disorder. The final refinement was performed with modification of the structure factors for contribution of the disordered solvent electron densities using the SQUEEZE option of PLATON program.14 The crystallographic data and the results of refinements of 1, 2a, and 2b are summarized in Table 1.
Table 1 Summary of the crystallographic data for 1–2
| Compound |
1 |
2a |
2b |
| R1 = ∑||Fo| − |Fc||/∑|Fo|. wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. |
| Empirical formula |
C44H36FeN14S2 |
C46H45FeN14O3Se2 |
C44H36FeN14Se2 |
| Formula weight |
880.86 |
1055.72 |
974.64 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/c |
P21/c |
| a (Å) |
9.4607(5) |
9.6220(19) |
18.847(4) |
| b (Å) |
14.1404(8) |
14.689(3) |
14.005(3) |
| c (Å) |
19.0776(11) |
19.691(4) |
19.343(4) |
| β (°) |
99.5170(10) |
99.73(3) |
98.17(3) |
| V (Å3) |
2517.0(2) |
2743.1(10) |
5053.8(18) |
| Z |
2 |
2 |
4 |
| dcalc (g cm−3) |
1.162 |
1.278 |
1.281 |
| λ (Å) |
0.71073 |
0.80000 |
0.75000 |
| T (K) |
200(2) |
298(2) |
104(2) |
| μ (mm−1) |
0.426 |
2.252 |
2.039 |
| F(000) |
912 |
1074 |
1968 |
| Reflections collected |
18586 |
19072 |
34308 |
| Independent reflections |
6250 |
4563 |
10290 |
| Reflections with I > 2σ(I) |
3269 |
4871 |
5885 |
| Goodness-of-fit on F2 |
0.989 |
1.065 |
1.368 |
| R1a [I > 2σ(I)] |
0.0741 |
0.0806 |
0.1113 |
| wR2b [I > 2σ(I)] |
0.1874 |
0.2648 |
0.3629 |
| CCDC no. |
1946253 |
1946255 |
1946254 |
Results and discussion
Synthesis and characterization
From a solution of FeSO4·7H2O, tpmd, and KNCS/KNCSe at room temperature in deoxygenated methanol under an inert atmosphere of N2 gas, two yellow compounds [Fe(tpmd)2(NCS)2]·5.5H2O (1) and [Fe(tpmd)2(NCSe)2]·7H2O (2) are prepared in 41 and 51% yields, respectively. In order to exclude the influence of sulfate ions in the self-assembly, we have used KNCX (X = S, Se) salt instead of NaNCX in which K2SO4 precipitates were formed and thus removed by filtration. 1 and 2 are slightly soluble in water, but insoluble in acetone, methanol, and Me2SO. The IR spectrum of 1 in KBr pellet shows a strong band of the thiocyanate ion at 2060 cm−1, and the CH peaks of the pyridine and methylene moieties at 3036 and 2957 cm−1.15 The IR spectrum of 2 in KBr pellet displays strong bands of the selenocyanate ion at 2059 and 2187 cm−1, and the CH peaks of the pyridine and methylene groups at 3061 and 2973 cm−1.15 Although both compounds show similar patterns in the IR spectra, the main different peaks are attributed to NCS− and NCSe−, respectively. TGA of 1 showed a weight loss of 12.1% at 100 °C, which corresponds to the loss of all solvent molecules per unit formula (6.5H2O or 2MeOH·3H2O); no chemical decomposition was observed up to 250 °C. TGA of 2 revealed a weight loss of 14.6% at 100 °C, which corresponds to the loss of all solvent molecules per unit formula (9H2O or 2MeOH·6H2O); no chemical decomposition was observed up to 320 °C. The water contents of the polymers are very slightly higher than those that obtained by elemental analyses (+1H2O for 1 and +2H2O for 2), respectively. From the TGA traces, it was confirmed that compound 2 is more stable than 1 thermally. The compositions of 1 and 2 were determined by elemental analyses, infrared spectra, and single crystal X-ray diffraction analyses.
Description of crystal structures
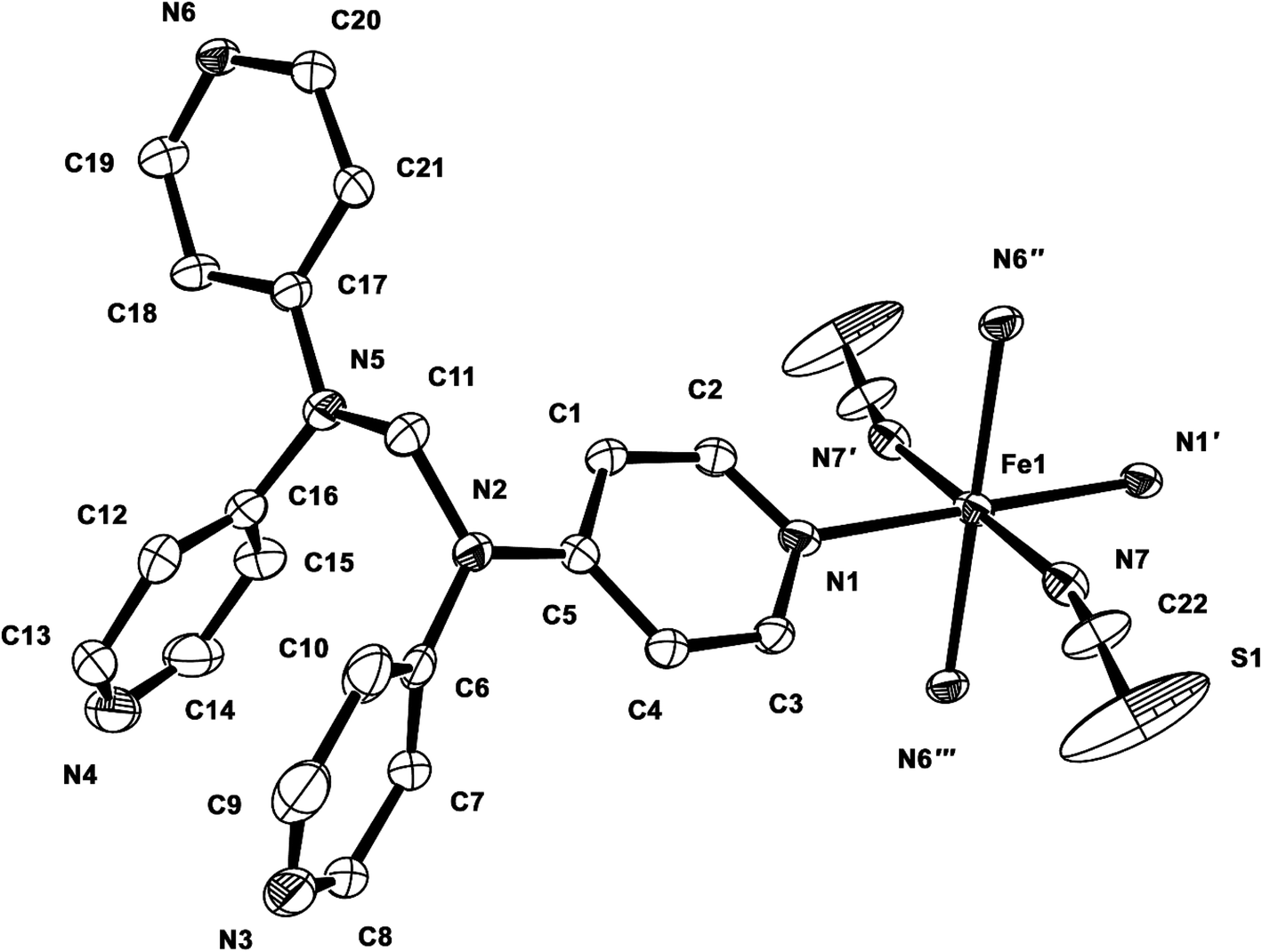
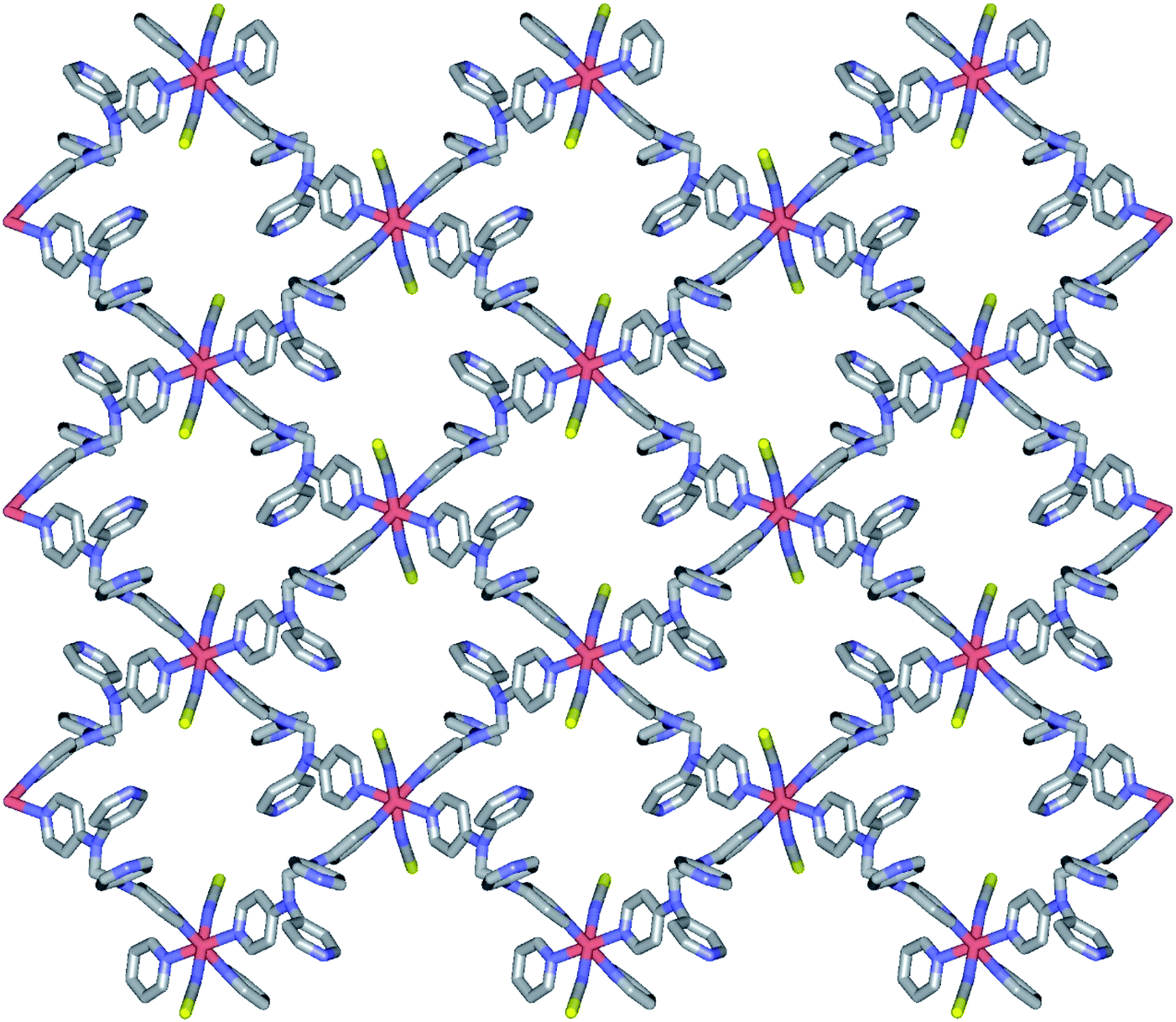
Structure of 1. Compound 1 crystallizes in the monoclinic space group P21/c, and the ORTEP view of 1 is shown in Fig. 1 and Table 2 lists the selected bond lengths and angles. The asymmetric unit of 1 includes a half iron ion, a tpmd ligand, and one thiocyanate ion. Thus, the iron ion is coordinated with four different pyridine groups of four tpmd ligands and two thiocyanate ions. The iron ion has a slightly distorted octahedral coordination geometry with four nitrogen atoms coordinated in the equatorial plane and two thiocyanate ions bonded at the axial positions. The average Fe–Neq and Fe–Nax bond distances are 2.232(2) and 2.129(3) Å, respectively. These bond distances indicate that the oxidation state of iron ion in 1 is 2+ and the electron configuration of the iron(II) ion is high spin state as shown in high spin iron(II) complexes, i.e., [Fe(tpa)(NCS)2], [(TPyA)FeII(THBQ2−)FeII(TPyA)](BF4)2, and [Fe(trz-py)2{Pt(CN)4}]·3H2O (tpa = TPyA = tris(2-pyridylmethyl)amine; THBQ2− = 2,3,5,6-tetrahydroxy-1,4-benzoquinonate; trz-py = 4-(2-pyridyl)-1,2,4,4H-triazole).16 Significantly, two pyridine groups in a tmpd ligand are not coordinated to iron(II) ion and they are positioned in a parallel mode with π–π interactions. Thus, through the linkage of four di-connected tpmd ligands and four di-connected iron(II) ions, a puckered square ring structure is formed and extended in a two-dimensional sheet along the bc plane (Fig. 2). The layers are stacked in a axis with thickness of 4.730 Å based on the high spin iron(II) ions. The bonding angles relating to the iron(II) ion lie from 84.76(10) to 180.00(7)°. Interestingly, the bonding angles of iron(II) ions with N atoms in equatorial and axial positions are very close to 90 and 180°, respectively. The two bpa groups in the tpmd ligand are connected by a methylene carbon at an angle of 112.4(3)°. The thiocyanato ligands are nearly linear and are slightly displayed bent coordination modes (∠N7C22S1; 177.0(5)°, ∠Fe1N7C22; 166.9(4)°). The N7–C22 and C22–S1 bond distances are 1.132(5) and 1.609(4) Å, respectively. The distances indicate that the NCS− ligand is composed of the NC triple bond and the CS single bond.17 The average Fe⋯Fe distance through the tpmd is 11.873(1) Å, while the shortest Fe⋯Fe separation is 9.461(1) Å. Calculations using PLATON indicate that 24.6% (619.6 Å3) of the void volume is occupied by guest molecules.18 Due to the severe disorder of solvent molecules, the guest molecules are determined by IR spectra, elemental analyses, and TGA data (Fig. S1†).
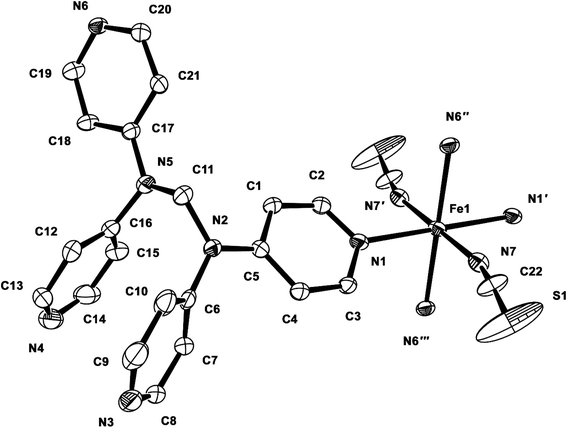 |
| | Fig. 1 ORTEP view of 1. The atoms are represented by 30% probable thermal ellipsoid. Hydrogen atoms are omitted for clarity. (′) −x + 1, −y + 1, −z; (′′) −x + 1, y + 1/2, −z + 1/2; (′′′) x, −y + 1/2, z − 1/2. | |
Table 2 Selected bond distances (Å) and angles (°) of 1a
| 1 |
| Symmetry transformations used to generate equivalent atoms: (′) −x + 1, −y + 1, −z; (′′) −x + 1, y + 1/2, −z + 1/2; (′′′) x, −y + 1/2, z − 1/2. |
| Fe1–N1 |
2.214(3) |
Fe1–N6′′′ |
2.250(3) |
| Fe1–N1′ |
2.214(3) |
Fe1–N7 |
2.129(3) |
| Fe1–N6′′ |
2.250(3) |
Fe1–N7′ |
2.129(3) |
| N7–C22 |
1.132(5) |
C22–S1 |
1.609(4) |
| N1–Fe1–N7 |
89.67(11) |
N1′–Fe1–N7′ |
89.67(11) |
| N1–Fe1–N7′ |
90.33(11) |
N7–Fe1–N6′′ |
90.32(11) |
| N1–Fe1–N6′′ |
95.24(10) |
N7–Fe1–N6′′′ |
89.68(11) |
| N1–Fe1–N6′′′ |
84.76(10) |
N7′–Fe1–N6′′ |
89.68(11) |
| N1–Fe1–N1′ |
180.0 |
N7′-Fe1-N6′′′ |
90.32(11) |
| N1′–Fe1–N6′′ |
84.76(10) |
N6′′–Fe1–N6′′′ |
180.00(7) |
| N1′–Fe1–N6′′′ |
95.24(10) |
N7–Fe1–N7′ |
180.0 |
| N1′–Fe1–N7 |
90.33(11) |
N7–C22–S1 |
177.0(5) |
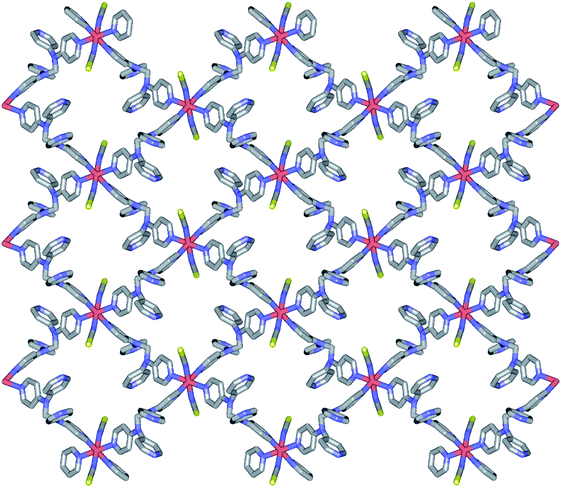 |
| | Fig. 2 Schematic view of 1, showing an extended 2-D layered structure via the coordination of iron(II) ions and tpmd ligands. All atoms are displayed as sticks and hydrogen atoms are omitted for clarity. | |
Yellow crystals of 2 suitable for X-ray analysis were obtained by layering of the methanol solution of tpmd ligand on the methanol solution of FeSO4·7H2O and KNCSe. The X-ray diffraction data of 2 were collected at 298 K (2a) and 104 K (2b). The unit cell parameters of 2a are very similar to those of 1 that measured at 200 K but different to those of 2b. Especially, the length of a axis in 2b is almost double for that of a axis in 2a. Thus the volume of unit cell of 2b is approximately double (5053.8 Å3/2743.1 Å3 ≅ 1.84), although the space groups of 2a and 2b are same.
2a is isostructural to 1. Thus, the geometric structure of 2a is almost same to that of 1 as shown in Fig. S2.† Due to the selenocyanate ions of 2a, the bond lengths and angles are slightly different to 1 (Table 3). The average Fe–Neq/Nax bond distances are 2.208(1) Å, which is consistent with high spin state of the iron(II) ion as shown in the structure of 1.16,19 2a is formed two dimensional network structure with square grid motifs along the bc plane (Fig. 2 and 3). The layers are stacked in the a axis with a thickness of 4.811 Å, which is slightly increased for 1 by the size of the Se atom. The Se atoms of selenocyanate ligands have some disorder and divide into two selenium atoms with site occupancy factors.20 The selenocyanate ligands are almost linear and show a bent coordination mode with iron(II) ions (∠N7C22Se1; 174.0(5)°, ∠C22N7Fe1; 160.7(4)°). The NC bond distance (1.136(7) Å) in the coordinated NCSe− ion is very similar to that of NC (1.132(5) Å) in NCS− of 1, which is indicative of the triple bond of CN. The average Fe⋯Fe distance through the tpmd are 12.831(2) Å, while the shortest Fe⋯Fe separation between the layers is 9.622(2) Å. Calculations using PLATON indicate that 18.7% (512.5 Å3) of the void volume is occupied by guest molecules.18
Table 3 Selected bond distances (Å) and angles (°) of 2aa
| 2a |
| Symmetry transformations used to generate equivalent atoms: (′) −x + 1, −y, −z; (′′) −x + 1, y + 1/2, −z + 1/2; (′′′) x, −y − 1/2, z − 1/2. |
| Fe1–N1 |
2.219(3) |
Fe1–N6′′′ |
2.236(3) |
| Fe1–N1′ |
2.219(3) |
Fe1–N7 |
2.138(4) |
| Fe1–N6′′ |
2.236(3) |
Fe1–N7′ |
2.138(4) |
| N7–C22 |
1.136(7) |
C22–Se1 |
1.796(5) |
| N1–Fe1–N7 |
89.54(15) |
N1′–Fe1–N7′ |
89.54(15) |
| N1–Fe1–N7′ |
90.46(15) |
N7–Fe1–N6′′ |
90.67(14) |
| N1–Fe1–N6′′ |
93.96(13) |
N7–Fe1–N6′′′ |
89.33(14) |
| N1–Fe1–N6′′′ |
86.04(13) |
N7′–Fe1–N6′′ |
89.33(14) |
| N1–Fe1–N1′ |
180.0 |
N7′–Fe1–N6′′′ |
90.67(14) |
| N1′–Fe1–N6′′ |
86.04(13) |
N6′′–Fe1–N6′′′ |
180.00(16) |
| N1′–Fe1–N6′′′ |
93.96(13) |
N7–Fe1–N7′ |
180.0 |
| N1′–Fe1–N7 |
90.46(15) |
N7–C22–Se1 |
174.0(5) |
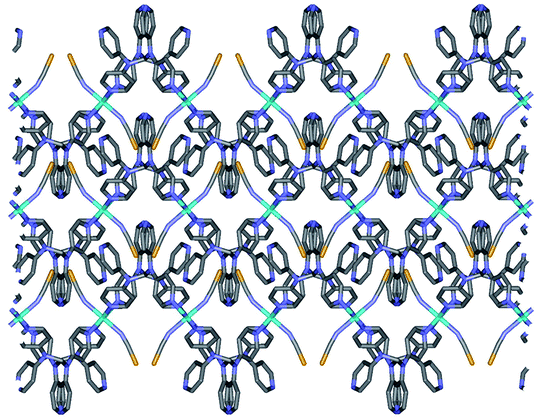 |
| | Fig. 3 Schematic packing view of 2a, displaying extended 2D layers via the coordination of Fe(II) ions and tpmds (Fe1, sky blue, N, blue, C, grey, Se, yellow). Hydrogen atoms are omitted for clarity. Each layered structure is similar to that of 1 as shown in Fig. 2. | |
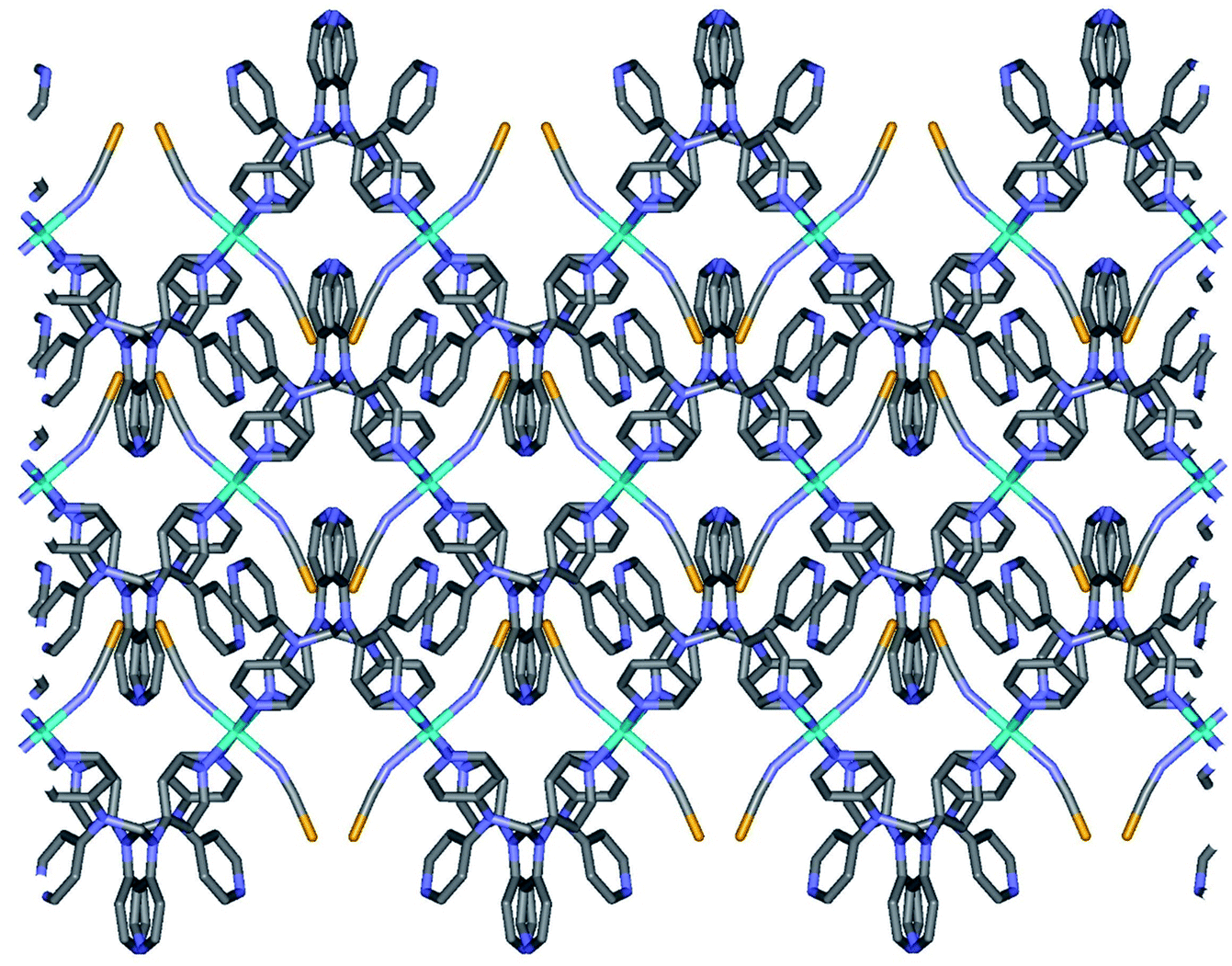
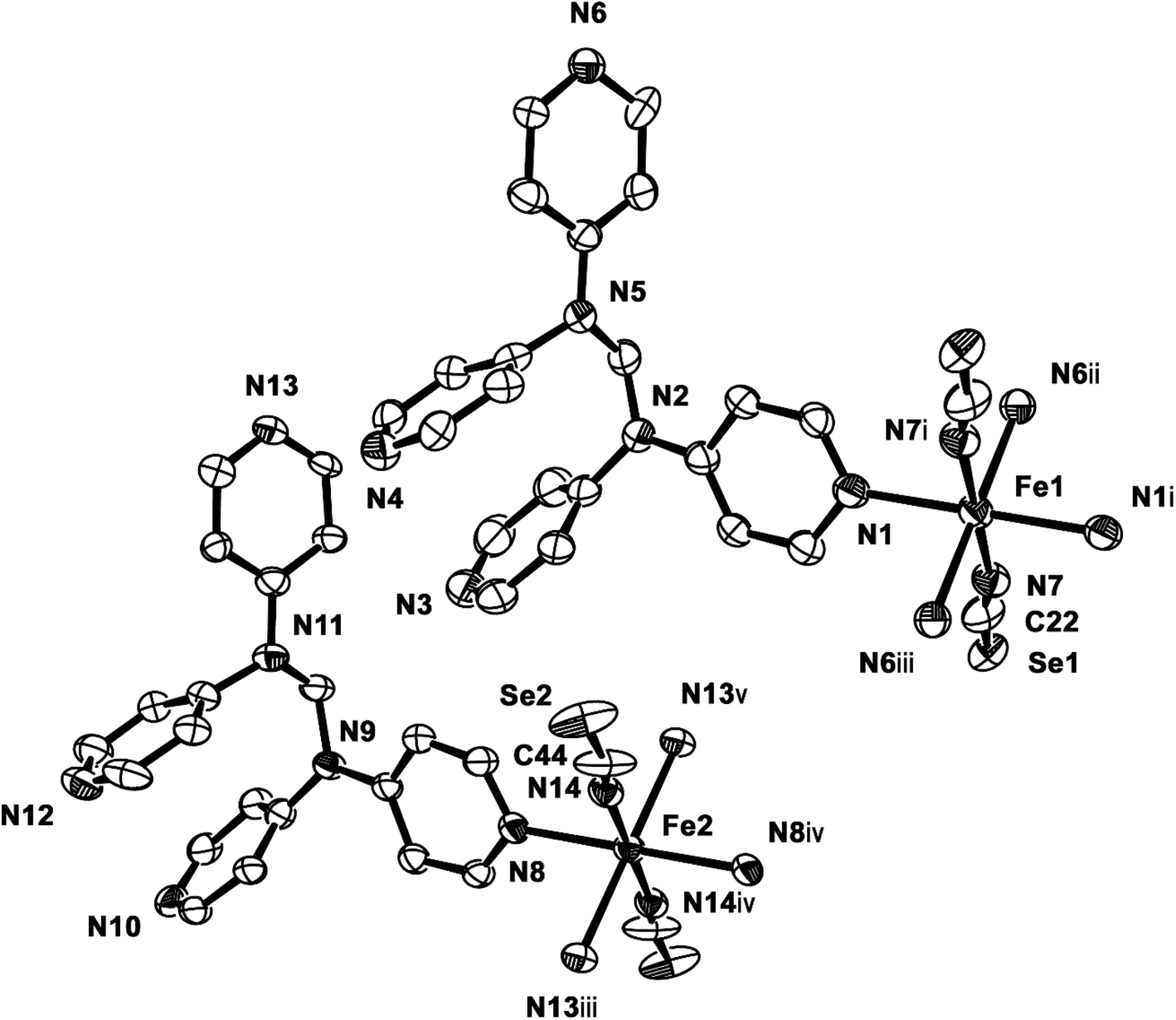
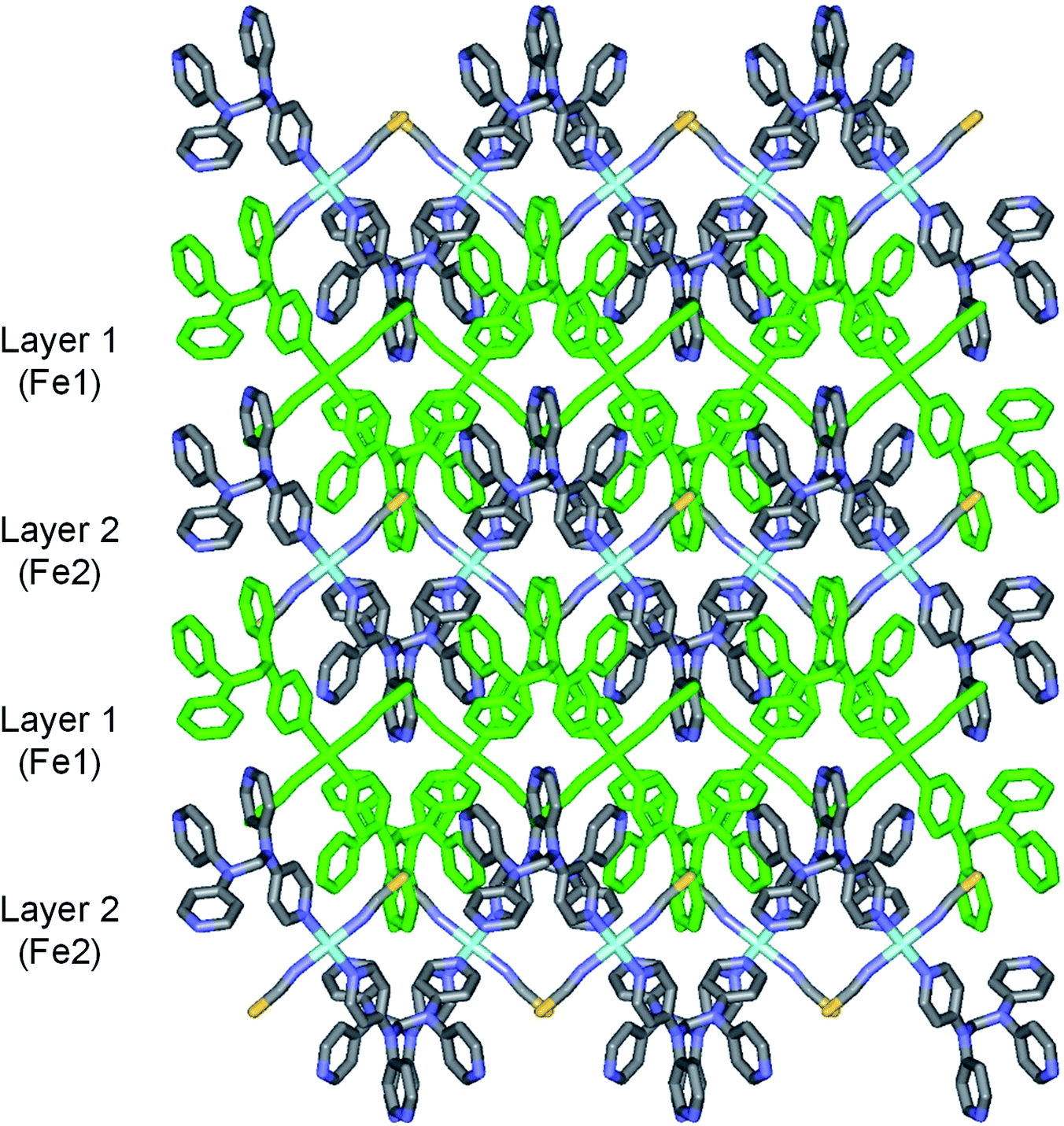
2b crystallizes in the monoclinic space group P21/c, and the ORTEP view of 2b is shown in Fig. 4 and Table 4 lists the selected bond lengths and angles. Very interestingly, in the asymmetric unit of 2b, there are two crystallographically distinct iron(II) sites Fe1 and Fe2. This means that 2b includes two kinds of iron(II) ions displaying different structural and electronic properties, compared to the structure of 2a that measured at 298 K. Each iron(II) ion in 2b is coordinated with a tpmd ligand and one selenocyanate ion and has also inversion centers. Compared to 2a, the cell parameters of 2b increase by 9.225 Å in the crystallographic a direction and decrease by 0.684 and 0.348 Å in the b and c directions, respectively. Thus, considered Z values (2 for 2a, 4 for 2b), the unit cell volume of 2b decreases by 216.2 Å3 or 7.9% for 2a, which is typical phenomena for spin crossover behavior in iron(II) complexes.21 Both iron(II) ions Fe1 and Fe2 are coordinated with four nitrogens (N1, N1i, N6ii, N6iii for Fe1 and N8, N8iv, N13iii, N13v for Fe2) and two selocyanate ions (N7, N7i for Fe1 and N14, N14iv for Fe2). Likewise to 1 and 2a, each iron(II) has a slightly distorted octahedral coordination geometry with four pyridyl nitrogen atoms and two selonocyanate ions. The Fe–Neq bond distances, in the range 2.170(4)–2.192(4) Å for Fe1 and 2.228(4)–2.261(4) Å for Fe2, slightly longer than Fe–Nax bond distances; 2.120(4) and 2.136(4) Å for Fe1 and Fe2, respectively. The average Fe–Neq/Nax bond distances are 2.161(2) and 2.208(2) Å for Fe1 and Fe2, respectively. That is, the average bond distance related to Fe1 is slightly shorter than that of Fe2. This means that the electron configurations between Fe1 and Fe2 can be different, i.e., low spin for Fe1 and high spin for Fe2, even though the iron ions are maintained 2+ oxidation states. As shown in 1 and 2a, the extended 2D layered structure of 2b shows basically same pattern. However, the neighboring layers have different electronic properties, that is, one layer is high spin, the other is low spin (Fig. 5). Some examples with an intermediate spin state (50% HS/50% LS) such as mononuclear [Fe(tpa)(NCS)2], dinuclear [(TPyA)FeII(THBQ2−)FeII(TPyA)](BF4)2, and three-dimensional [Fe(NCS)2(tppm)] solvent have been known (tppm = 4,4′,4′′,4′′′-tetrakis(4-pyridylethen-2-yl)tetraphenylmethane).5c,16a,b Generally, the first two compounds are shown that high- and low-spin units arrange alternately. The last one contains alternating high- and low-spin metal centers in the three-dimensional framework. Likewise, 2b includes alternating high- and low-spin Fe(II) units. However, each Fe(II) unit is belonged to two different layers unexpectedly (Fig. 5). As far as we know, we have not found any examples that includes individual HS and LS layers alternately. The bonding angles relating to the iron(II) ions in Fe1 and Fe2 lie from 86.60(16) to 180.0(3)° and 83.74(14) to 180.0(3)°, respectively. Interestingly, the bonding angles of iron(II) ions with N atoms in equatorial and axial positions are very close to 90 and 180°, respectively. The two bpa groups in the ligands are connected by a methylene carbon at an angle of 110.5(5) and 112.5(4)° for Fe1 and Fe2, respectively. The Se atoms of the selenocyanate groups have slightly thermal disorder because selenium atoms are heavy atoms having large electron densities and thus divide into three selenium atoms with different site occupancy factors as linear modes.20 Thus the selenocyanate groups show more bent modes than 1 (∠N7C22Se1; 161.5(6)°, ∠N7C22Se2; 169.5(6)°, ∠N7C22Se3; 152.5(6)°, ∠C22N7Fe1; 166.5(5)°, related with Fe1, ∠N14C44Se4; 152.4(8)°, ∠N14C44Se5; 169.2(9)°, ∠N14C44Se6; 156.5(9)°, ∠C44N14Fe2; 168.9(6)°, related with Fe2). The NC bond distances of NCSe− ligands in Fe1 and Fe2 are 1.179(7) and 1.133(8) Å, respectively. The former shows slightly longer than that of the latter (Δd = 0.046 Å). This difference can be attributed to the electronic states, i.e. low spin for Fe1 and high spin for Fe2. That is, the low spin Fe1 can interact to N7 more strongly with π back bonding character, in which the bond distance of N7–C22 can be weaken and elongated. Contrary to this, the interaction of Fe2 and N14 decreases and that of N14–C44 increases due to the high spin state of Fe2. Thus the weak π back bonding of Fe2–N14 can be induced more strong bond of N14–C44 as compared to that of N7–C22.22 The average Fe⋯Fe distance through the tpmds is 11.940(2) Å, while the shortest Fe⋯Fe separation between the layers is 9.424(2) Å. Calculations using PLATON indicate that 22.8% (1152.8 Å3) of the void volume is occupied by guest molecules.18
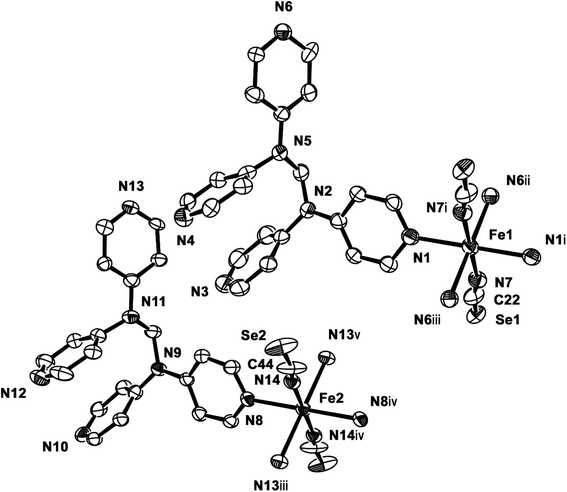 |
| | Fig. 4 ORTEP view of 2b. The atoms are represented by 30% probable thermal ellipsoid. Hydrogen atoms are omitted for clarity. (i) −x, −y, −z; (ii) −x, y − 1/2, −z + 1/2; (iii) x, −y + 1/2, z − 1/2; (iv) −x + 1, −y, −z; (v) −x + 1, y − 1/2, −z + 1/2. | |
Table 4 Selected bond distances (Å) and angles (°) of 2ba
| 2b |
| Symmetry transformations used to generate equivalent atoms: (i) −x, −y, −z; (ii) −x, y − 1/2, −z + 1/2; (iii) x, −y + 1/2, z − 1/2; (iv) −x + 1, −y, −z; (v) −x + 1, y − 1/2, −z + 1/2. |
| Fe1–N1 |
2.170(4) |
Fe2–N8 |
2.228(4) |
| Fe1–N1i |
2.170(4) |
Fe2–N8iv |
2.228(4) |
| Fe1–N6ii |
2.192(4) |
Fe2–N13iii |
2.261(4) |
| Fe1–N6iii |
2.192(4) |
Fe2–N13v |
2.261(4) |
| Fe1–N7 |
2.120(4) |
Fe2–N14 |
2.136(4) |
| Fe1–N7i |
2.120(4) |
Fe2–N14iv |
2.136(4) |
| N7–C22 |
1.179(7) |
N14–C44 |
1.133(8) |
| C22–Se1 |
1.824(6) |
C44–Se2 |
1.841(7) |
| N1–Fe1–N1i |
180.0 |
N8–Fe2–N8iv |
180.0 |
| N1–Fe1–N6ii |
93.40(16) |
N8–Fe2–N13iii |
83.74(14) |
| N1–Fe1–N6iii |
86.60(16) |
N8–Fe2–N13v |
96.26(14) |
| N1–Fe1–N7 |
90.60(16) |
N8–Fe2–N14 |
91.10(15) |
| N1–Fe1–N7i |
89.40(16) |
N8–Fe2–N14iv |
88.90(15) |
| N1i–Fe1–N6ii |
86.60(16) |
N8iv–Fe2–N13iii |
96.26(14) |
| N1i–Fe1–N6iii |
93.40(16) |
N8iv–Fe2–N13v |
83.74(14) |
| N1i–Fe1–N7 |
89.40(16) |
N8iv–Fe2–N14 |
88.90(15) |
| N1i–Fe1–N7i |
90.60(16) |
N8iv–Fe2–N14iv |
91.10(15) |
| N7–Fe1–N6ii |
89.76(16) |
N13iii–Fe2–N14 |
90.52(15) |
| N7i–Fe1–N6ii |
90.24(16) |
N13iii–Fe2–N14iv |
89.48(15) |
| N7–Fe1–N6iii |
90.24(16) |
N13v–Fe2–N14 |
89.48(15) |
| N7i–Fe1–N6iii |
89.76(16) |
N13v–Fe2–N14iv |
90.52(15) |
| N6ii–Fe1–N6iii |
180.0(3) |
N13iii–Fe2–N13v |
180.0(3) |
| N7–Fe1–N7i |
180.0 |
N14–Fe2–N14iv |
180.0 |
| N7–C22–Se1 |
169.5(6) |
N14–C44–Se2 |
169.2(9) |
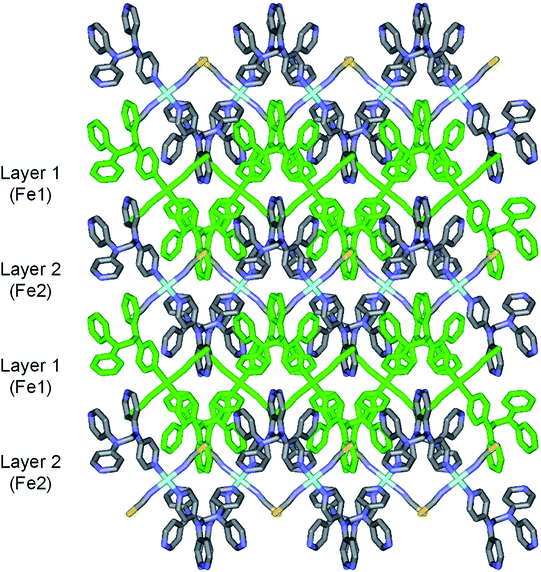 |
| | Fig. 5 Schematic view of 2b, showing extended 2D layered arrangements via the coordination of Fe(II) ions and tpmds (all atoms displayed as stick diagram. Layer 1 is all green, while layer 2 shows as colorful diagram; Fe, sky blue, N, blue, C, grey, Se, yellow). Layer 1 (Fe1) and layer 2 (Fe2) indicate low spin and high spin states, respectively. | |
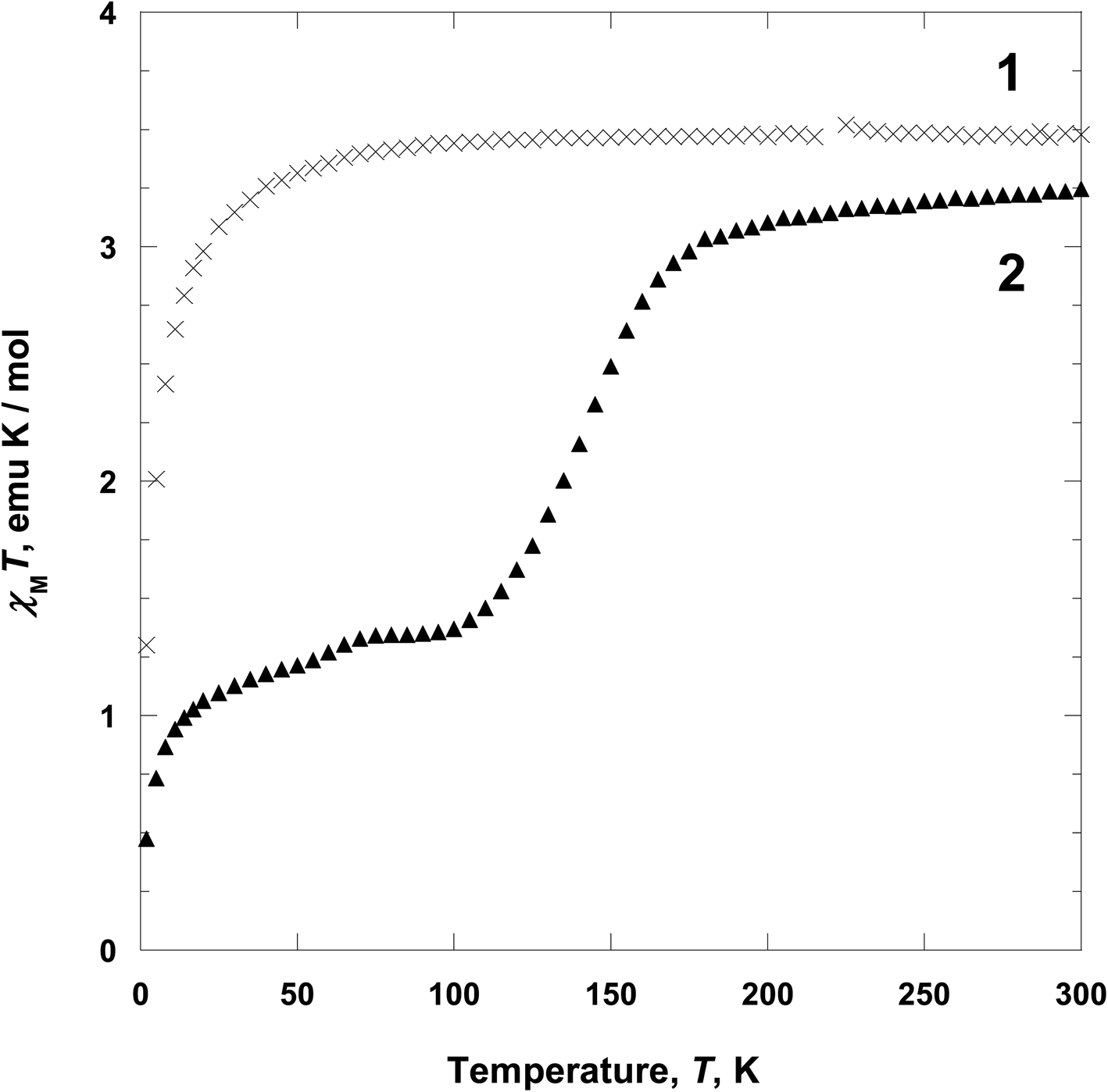
Magnetic properties. Variable-temperature 2 to 300 K magnetic susceptibility, χ, measurements on polycrystalline samples of 1 and 2 have been performed at 5000 Oe on a SQUID magnetometer (Fig. 6). For 1, at 300 K, the χMT value is ca. 3.45 emu K mol−1, which is the expected value for high spin iron(II) ion (S = 2)2,23 and it is approximately constant with decreasing temperature until 80 K. Below 80 K, χMT value decrease to 1.27 emu K mol−1 at 2 K. The decrease of χMT in the low temperature may be due to the zero-field splitting of iron(II) in the high spin state and/or intermolecular interactions.24 Above 2 K, χ−1(T) can be fit to the Curie–Weiss expression χM = C/(T − θ) with θ = −2.968 K (C = 3.523 emu K mol−1) (Fig. S3†).25 The θ value indicates that 1 has a weak intermolecular magnetic interaction. For 2, from 300 K to 190 K, the χMT value remains nearly constant ca. 3.10 emu K mol−1, which is the expected value for high spin iron(II) ion (S = 2).2,23 On further cooling, the χMT value decreases in two steps, to a minimum of 0.46 emu K mol−1 at 2 K, corresponding to low spin iron(II) (S = 0). The first spin crossover step, centered at 145 K, shows gradual decrease to a plateau and reaches a χMT value of 1.45 emu K mol−1 at 100 K, indicative of a spin state conversion in ca. 50% of iron(II) ions. Indeed, there are two crystallographically distinct iron(II) sites Fe1 and Fe2 at 104 K as shown in Fig. 4 and the spin state of Fe1 is close to low spin as compared to Fe2. That is, one layer including Fe1 becomes a low spin state, while the other layer with Fe2 is a high spin state. The second spin crossover step, centered at 50 K, is also gradual decreased and reached a χMT value of 0.46 emu K mol−1 at 2 K, indicative of the existence of a relatively small residual amount of high spin iron(II) ion (ca. 30%, g = 2) in 2. In fact, this decrease in magnetic moment can be attributed to spin transition (Fe2) and/or intermolecular interactions.
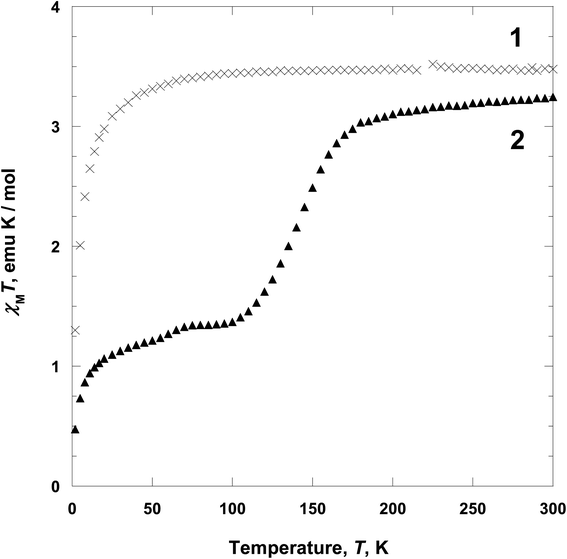 |
| | Fig. 6 Temperature dependence of χMT for 1 (x) and 2 (▲) at 5000 Oe upon heating. | |
From the above data, we have observed that the magnetic behaviors of 1 and 2 are anion-dependent. Generally NCS− anion can induce spin crossover behavior, even though low temperature comparatively. However, in 1, any spin crossover behavior was not occurred due to the weak ligand field. Contrary to 1, compound 2 shows unusual two-step spin crossover due to the strong ligand field of NCSe−. This means that NCSe− ion is more strong donor than NCS− ion.5b,c,22 In the cases of 1 and 2, NCS− ion is not enough for inducing spin crossover, however, NCSe− ion generates spin crossover due to the strong ligand field. Thus we have demonstrated that the anions (NCS− and NCSe−) can be used for controlling the spin crossover behavior from paramagnetic to diamagnetic.5d,e,26
Conclusions
Two iron(II) coordination polymers 1 and 2 including tpmd and NCS− or NCSe− ligands were synthesized and characterized. Polymer 1 shows a square grid geometry that linked iron(II) ion and tpmd ligand each other, which gives rise to a 2-D layered structure. Furthermore, two pyridine groups of a tpmd are free and the NCS− anions are coordinated in axial positions. Polymers 1 and 2 are isomorphous structures, even though the anions are different. However, 2 shows slightly different geometry at different temperatures (104 and 298 K), due to the spin state change. That is, at low temperature, two distinct iron(II) ions exist by different spin states as verified by X-ray data, i.e., one layer is high spin, the other is low spin, in which the layers are packed alternately. Finally, interestingly, 2 display two-step spin-crossover behavior at 145 K and below 10 K. The spin crossover between 100 and 180 K indicates that a half of iron(II) ions changed from high spin to low spin that the SCO behavior is in good agreement with X-ray crystal data. 1 shows a paramagnetic behavior and did not exhibit a spin crossover in the temperature range. The magnetic behaviors are quite sensitive to pseudo-halide anions, i.e. NCS− and NCSe−.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by Basic Science Research Program (No. 2015R1D1A1A01059872 and 2018R1D1A1B07043305) and under the framework of international cooperation program (No. 2016K2A9A2A08003735) through the National Research Foundation of Korea (NRF). X-ray crystallography at the PLS-II 2D-SMC beamline was supported in part by MEST and POSTECH.
References
-
(a) D. A. Reed, B. K. Keitz, J. Oktawiec, J. A. Mason, T. Runčevski, D. J. Xiao, L. E. Darago, V. Crocellà, S. Bordiga and J. R. Long, Nature, 2017, 550, 96–100 CrossRef CAS PubMed;
(b) N. C. Galve, E. Coronado, M. Giménez-Marqués and G. M. Espallargas, Inorg. Chem., 2014, 53, 4482–4490 CrossRef;
(c) B. Drahoš, R. Herchel and Z. Trávníček, Inorg. Chem., 2018, 57, 12718–12726 CrossRef;
(d) Y.-Z. Zheng, W. Xue, M.-L. Tong, X.-M. Chen, F. Grandjean and G. J. Long, Inorg. Chem., 2008, 47, 4077–4087 CrossRef CAS PubMed;
(e) P.-F. Yao, Y. Tao, H.-Y. Li, X.-H. Qin, D.-W. Shi, F.-P. Huang, Q. Yu, X.-X. Qin and H.-D. Bian, Cryst. Growth Des., 2015, 15, 4394–4405 CrossRef CAS;
(f) C. Lochenie, K. Schötz, F. Panzer, H. Kurz, B. Maier, F. Puchtler, S. Agarwal, A. Köhler and B. Weber, J. Am. Chem. Soc., 2018, 140, 700–709 CrossRef CAS PubMed;
(g) J. López-Cabrelles, M. Giménez-Marqués, G. M. Espallargas and E. Coronado, Inorg. Chem., 2015, 54, 10490–10496 CrossRef PubMed;
(h) S. A. Sahadevan, A. Abhervé, N. Monni, C. S. de Pipaón, J. R. Galán-Mascarós, J. C. Waerenborgh, B. J. C. Vieira, P. Auban-Senzier, S. Pillet, E.-E. Bendeif, P. Alemany, E. Canadell, M. L. Mercuri and N. Avarvari, J. Am. Chem. Soc., 2018, 140, 12611–12621 CrossRef CAS PubMed;
(i) N. F. Sciortino and S. M. Neville, Aust. J. Chem., 2014, 67, 1553 CrossRef CAS.
- P. Gütlich and H. A. Goodwin, in Spin Crossover in Transition Metal Compounds I, Springer-Verlag, 2004, vol. 1 Search PubMed.
-
(a) I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC;
(b) C. Sánchez-Sánchez, C. Desplanches, J. M. Clemente-Juan, M. Clemente-León and E. Coronado, Dalton Trans., 2017, 46, 2680–2689 RSC.
-
(a) F.-J. Yazigi, C. Wilson, D.-L. Long and R. S. Forgan, Cryst. Growth Des., 2017, 17, 4739–4748 CrossRef CAS;
(b) I. A. Bhat, D. Samanta and P. S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 9497–9502 CrossRef CAS PubMed;
(c) B. Ding, J. Wu, X. X. Wu, J. Z. Huo, Z. Z. Zhu, Y. Y. Liu and F. X. Shi, RSC Adv., 2017, 7, 9704–9718 RSC;
(d) W. Chen, R. Fan, H. Zhang, Y. Dong, P. Wang and Y. Yang, Dalton Trans., 2017, 46, 4265–4277 RSC;
(e) S. I. Vasylevskyi, D. M. Bassani and K. M. Fromm, Inorg. Chem., 2019, 58, 5646–5653 CrossRef CAS PubMed;
(f) J. E. Clements, J. R. Price, S. M. Neville and C. J. Kepert, Angew. Chem., Int. Ed., 2014, 53, 10164–10168 CrossRef CAS PubMed.
-
(a) K. Nebbali, C. D. Mekuimemba, C. Charles, S. Yefsah, G. Chastanet, A. J. Mota, E. Colacio and S. Triki, Inorg. Chem., 2018, 57, 12338–12346 CrossRef CAS;
(b) X.-R. Wu, H.-Y. Shi, R.-J. Wei, J. Li, L.-S. Zheng and J. Tao, Inorg. Chem., 2015, 54, 3773–3780 CrossRef CAS;
(c) X.-Y. Chen, R.-B. Huang, L.-S. Zheng and J. Tao, Inorg. Chem., 2014, 53, 5246–5252 CrossRef CAS PubMed;
(d) E. Milin, V. Patinec, S. Triki, E.-E. Bendeif, S. Pillet, M. Marchivie, G. Chastanet and K. Boukheddaden, Inorg. Chem., 2016, 55, 11652–11661 CrossRef CAS PubMed;
(e) J. Klingele, D. Kaase, M. Schmucker, Y. Lan, G. Chastanet and J.-F. Létard, Inorg. Chem., 2013, 52, 6000–6010 CrossRef CAS PubMed.
- A. Arroyave, A. Lennartson, A. Dragulescu-Andrasi, K. S. Pedersen, S. Piligkos, S. A. Stoian, S. M. Greer, C. Pak, O. Hietsoi, H. Phan, S. Hill, C. J. McKenzie and M. Shatruk, Inorg. Chem., 2016, 55, 5904–5913 CrossRef CAS PubMed.
- J. W. Shin, A. R. Jeong, S. Jeoung, H. R. Moon, Y. Komatsumaru, S. Hayami, D. Moon and K. S. Min, Chem. Commun., 2018, 54, 4262–4265 RSC.
- J. W. Shin, J. M. Bae, C. Kim and K. S. Min, Inorg. Chem., 2013, 52, 2265–2267 CrossRef CAS.
- Saint Plus, Version 6.02, Bruker Analytical X-ray, Madison, WI, 1999 Search PubMed.
- A. J. Arvai and C. Nielsen, ADSC Quantum-210 ADX Program, Area Detector System Corporation, Poway, CA, USA, 1983 Search PubMed.
- Z. Otwinowski and W. Minor, in Methods in enzymology, ed. C. W. Carter Jr and R. M. Sweet, Academic Press, New York, 1997, vol. 276, part A, p. 307 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS.
- PLATON program and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, 6th edn, John Wiley and Sons, Inc., New Jersey, 2009, part B, pp. 120–131 Search PubMed.
-
(a) B. Li, R.-J. Wei, J. Tao, R.-B. Huang, L.-S. Zheng and Z. Zheng, J. Am. Chem. Soc., 2010, 132, 1558–1566 CrossRef CAS;
(b) K. S. Min, K. Swierczek, A. G. DiPasquale, A. L. Rheingold, W. M. Reiff, A. M. Arif and J. S. Miller, Chem. Commun., 2008, 317–319 RSC;
(c) E. Milin, V. Patinec, S. Triki, E.-E. Bendeif, S. Pillet, M. Marchivie, G. Chastanet and K. Boukheddaden, Inorg. Chem., 2016, 55, 11652–11661 CrossRef CAS.
- G. L. Miessler, P. J. Fischer and D. A. Tarr, Inorganic Chemistry, 5th edn, Pearson, New York, 2014, pp. 47–49 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
-
(a) V. M. Hiiuk, S. Shova, A. Rotaru, V. Ksenofontov, I. O. Fritsky and I. A. Gural’skiy, Chem. Commun., 2019, 55, 3359–3362 RSC;
(b) A. E. Thorarinsdottir, A. I. Gaudette and T. D. Harris, Chem. Sci., 2017, 8, 2448–2456 RSC;
(c) G. J. Halder, C. J. Kepert, B. Moubaraki, K. S. Murray and J. D. Cashion, Science, 2002, 298, 1762–1765 CrossRef CAS.
- N. A. Barnes, S. M. Godfrey, J. Hughes, E. Z. Khan, I. Mushtaq, R. T. A. Ollerenshaw, R. G. Pritchard and S. Sarwar, Dalton Trans., 2013, 42, 2735–2744 RSC.
-
(a) A. C. McQuilken, H. Matsumura, M. Dürr, A. M. Confer, J. P. Sheckelton, M. A. Siegler, T. M. McQueen, I. Ivanović-Burmazović, P. Moënne-Loccoz and D. P. Goldberg, J. Am. Chem. Soc., 2016, 138, 3107–3117 CrossRef CAS;
(b) A. Abhervé, M. J. Recio-Carretero, M. López-Jordà, J. M. Clemente-Juan, J. Canet-Ferrer, A. Cantarero, M. Clemente-León and E. Coronado, Inorg. Chem., 2016, 55, 9361–9367 CrossRef;
(c) G. S. Matouzenko, E. Jeanneau, A. Y. Verata and A. Bousseksou, Dalton Trans., 2011, 40, 9608–9618 RSC;
(d) J. R. Thompson, R. J. Archer, C. S. Hawes, A. Ferguson, A. Wattiaux, C. Mathonière, R. Clérac and P. E. Kruger, Dalton Trans., 2012, 41, 12720–12725 RSC.
- J. Cirera and F. Paesani, Inorg. Chem., 2012, 51, 8194–8201 CrossRef CAS PubMed.
-
(a) S. J. Lippard and C. J. O'Connor, Prog. Inorg. Chem., 1982, 29, 203 Search PubMed;
(b) N. Paradis, G. Chastanet, F. Varret and J.-F. Létard, Eur. J. Inorg. Chem., 2013, 968 CrossRef CAS.
-
(a) J. M. Zadrozny, S. M. Greer, S. Hill and D. E. Freedman, Chem. Sci., 2016, 7, 416–423 RSC;
(b) J. Yuan, M.-J. Liu, C.-M. Liu and H.-Z. Kou, Dalton Trans., 2017, 46, 16562–16569 RSC;
(c) G. Novitchi, S. Jiang, S. Shova, F. Rida, I. Hlavička, M. Orlita, W. Wernsdorfer, R. Hamze, C. Martins, N. Suaud, N. Guihéry, A.-L. Barra and C. Train, Inorg. Chem., 2017, 56, 14809–14822 CrossRef CAS.
- O. Kahn, Molecular Magnetism, VCH, New York, 1993, p. 26 Search PubMed.
-
(a) H. E. Mason, M. L. Hamilton, J. A. K. Howard and H. A. Sparkes, New J. Chem., 2018, 42, 18028–18037 RSC;
(b) Z. Arcís-Castillo, L. Piñeiro-López, M. C. Muñoz, R. Ballesteros, B. Abarca and J. A. Real, CrystEngComm, 2013, 15, 3455–3462 RSC;
(c) B. Fei, X. Q. Chen, Y. D. Cai, J.-K. Fang, M. L. Tong, J. Tucek and X. Bao, Inorg. Chem. Front., 2018, 5, 1671–1676 RSC;
(d) X.-P. Sun, T. Liu, Z.-S. Yao and J. Tao, Dalton Trans., 2019, 48, 9243–9249 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1946253–1946255. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra09782a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c
*c