
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient synthesis of 4-sulfanylcoumarins from 3-bromo-coumarins via a highly selective DABCO-mediated one-pot thia-Michael addition/elimination process†
Vitor B. Mostardeiroa,
Marina C. Dilelioa,
Teodoro S. Kaufman *b and
Claudio C. Silveira*a
*b and
Claudio C. Silveira*a
aDepartamento de Química, Universidade Federal de Santa Maria, Santa Maria, RS 97105-900, Brazil. E-mail: silveira@quimica.ufsm.br
bInstituto de Química Rosario (IQUIR, CONICET-UNR), Suipacha 531, 2000 Rosario, Argentina
First published on 2nd January 2020
Abstract
A facile and efficient protocol for the highly selective direct sulfanylation of 3-bromocoumarins under DABCO promotion, was developed. The transformation took place with aromatic and aliphatic thiols as well as with α,ω-dithiols, affording the expected products in very good to excellent yields. Simple and convenient ways to access 4-((ω-mercaptoalkyl) thio)coumarins and the dimeric 4,4′-(alkane-1,4-diylbis(sulfanediyl))bis(coumarins) were also devised with the use of α,ω-alkanedithiols in different ratios with regards to the starting 3-bromocoumarin. The transformation seems to proceed through the DABCO-mediated thia-Michael stereoselective addition of the thiolate anion to the α,β-unsaturated carbonyl system of the coumarin, followed by a DABCO-assisted stereoselective dehydrobromination of the resulting α-bromo carbonyl intermediate.
Introduction
The coumarin core is a privileged scaffold and a ubiquitous heterocyclic structure among natural products, bioactive synthetic compounds and technologically interesting materials. The introduction of sulfur as a heteroatom in many molecules has been demonstrated to be an effective method for changing their characteristics, by endowing them with new properties useful for materials science or conveying significant biological activity.1The 4-sulfanylcoumarin core is found in many relevant natural products, such as in the cytotoxic pheofungins (Ia–d), the tricyclic lactone iotrochotazine A (II), a chemical probe to study Parkinson's disease and other heterocycles, like IIIa–d.2 They are also interesting structural motifs in the area of functional materials, as exemplified by the selective fluorescent probes IV and V.3 It is also known that upon reaction with glutathione, compound IV affords the 4-thiocoumarin adduct VI.
In addition, 4-thiocoumarins have ample applications in medicinal chemistry, exhibiting a wide range of biological and pharmacological activities. These include inhibition of vitamin K epoxide reductase4a and pyruvate kinase M2,4b to inhibition of the benzodiazepine receptors4c and the incorporation of DIG-11-dUTP to BS-C-1 cells modified/infected with vaccinia virus,5a as well as inhibition of interleukin 2 (ref. 5b) and the TNF-α induced expression of ICAM-1 (VII).5c 4-Sulfanylcoumarins like VIII and IX6a–c have been tested as cytotoxic and antiproliferative agents and some of them proved to be interestingly bioactive.6a,b Others displayed anti-Hepatitis C virus activity (Fig. 1).6d
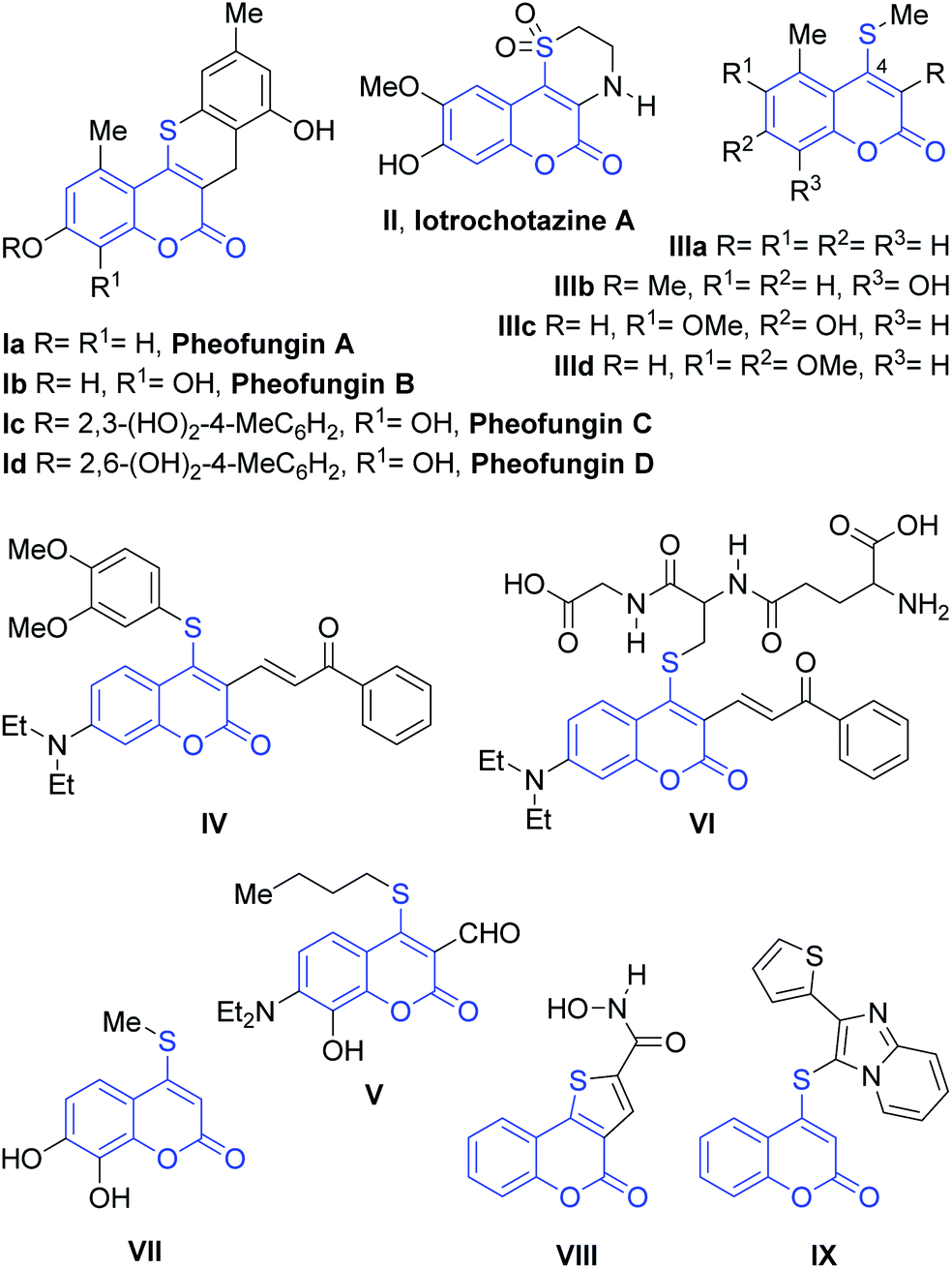 | ||
| Fig. 1 Examples of relevant coumarin derivatives carrying C4–S bonds. | ||
Despite the importance and usefulness of the 4-sulfanyl coumarins, methods for accessing these compounds are scarce, suffer from multiple synthetic steps and harsh reaction conditions, in addition, the available methodologies often use toxic agents and high temperatures, and also display poor substituent tolerance.
They mainly comprise formation of the C–S bond by 4-sulfanylation through nucleophilic vinylic substitution of suitable 4-substituted heterocyclic precursors, such as 4-hydroxycoumarins,7a and their sulfonates,7b–d 4-halocoumarins8 and 4-bromomethyl coumarins.9a,b An oxidative cross-coupling on 3-aminocoumarin has been reported as an alternative.9c
In addition, elemental sulfur, inorganic sulfides, KSCN, CS2 and thiourea have also served as sources of the sulfur atom,6c,8b,10 whereas the alkylation of the resulting 4-mercaptocoumarins11 and their arylation through the use of electrochemical means12 have been disclosed as other options.
An exhaustive literature search revealed that there are also few and scattered cases related to the use of 3-bromocoumarins as starting materials;13 however, their use seems discouraging, since the transformations proceed under rather harsh conditions and with low selectivity, resulting in poor to moderate product yields and/or giving mixtures of the isomeric 3- and 4-sulfanyl coumarins. These characteristics seriously limit their general applicability.
We have reported the synthesis of different compounds through functional group transposition,14 such as between C-3 and C-4 among tetrahydroisoquinoline derivatives and have disclosed a vinylic nucleophilic substitution approach toward the synthesis of 4-aryl- and 4-benzyl-selanylcoumarins.15
To the best of our knowledge, despite some scattered precedents,13 there have been no systematic studies of the functional transposition of 3-bromocoumarins toward their corresponding 4-sulfanyl congeners. Therefore, due to our continued interest in the preparation of new coumarin derivatives,15,16 herein we report a facile and metal-free, selective direct 4-sulfanylation of 3-bromocoumarins (1) with aromatic and aliphatic thiols (2), under DABCO promotion. The transformations resulted in the synthesis of diverse novel 4-organylsulfanyl-coumarins (3), as shown in Scheme 1.
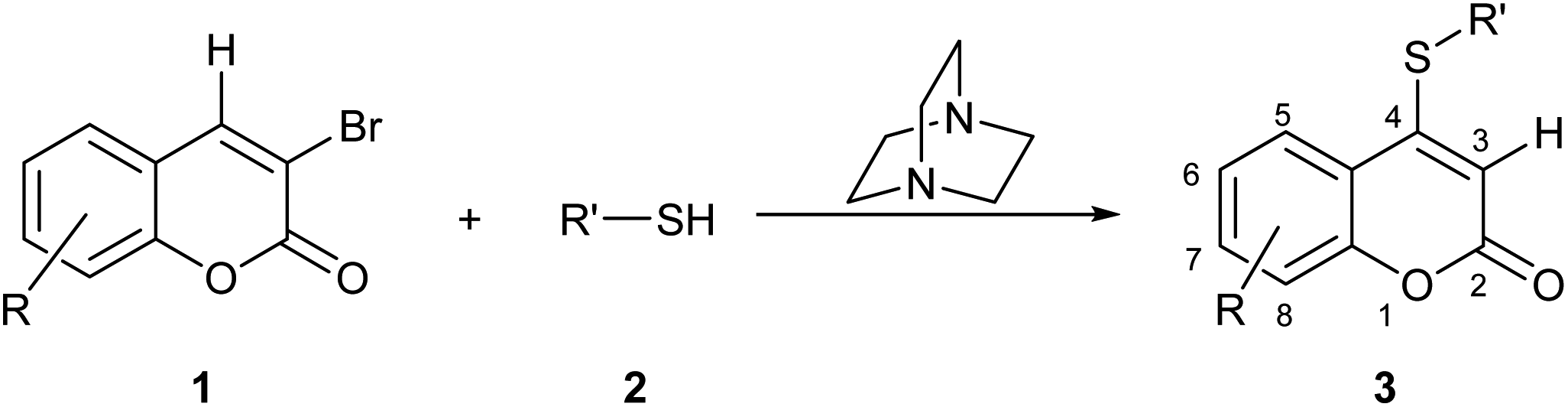 | ||
| Scheme 1 Proposed DABCO-promoted approach for the sulfanylation of 1 toward 4-organyl sulfanyl-coumarin derivatives (3). | ||
Results and discussion
The required 3-bromocoumarin17 starting materials 1a (R = H) and 1c (R = 8-Me) were conveniently obtained by bromination of the corresponding coumarins with the HBr/oxone reagent system,18 whereas the derivative 1b (R = 6-Cl) was prepared by bromo-decarboxylation of 6-chlorocoumarin-3-carboxylic acid with NBS.19At the outset of our debromosulfanylation studies, we employed the reaction between 3-bromocoumarin (1a) and 4-chlorobenzenethiol (2b) as model system.
Therefore, with the intention of developing a more efficient and selective protocol, we tested a small set of bases as promoters, including inorganic (KOH,20a K2CO3 (ref. 20b and c)) and organic (pyridine,13b,c DMAP, Et3N,13a,20d DBU20e and DABCO20f) examples, under different reaction conditions (Table 1).
|
||||
|---|---|---|---|---|
| Entry no. | Base | Temp. (°C) | Yieldb (%) | (3b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4b)c :4b)c |
| a Reaction conditions: 3-bromocoumarin (1a, 1 mmol), p-chlorothiophenol (2b, 1.5 mmol), THF (5 mL), base (1.5 mmol), 2 h.b Isolated yields by column chromatography.c Determined by GC/MS.d The yield was not determined; only the isomer ratio was estimated by GC.e No precipitate was formed after 3 h at room temperature.f No precipitate was formed after 3 h. The GC-MS analysis of the crude mixture revealed the formation of the disulfide, coumarin and 1a. | ||||
| 1 | K2CO3 | r.t | 82 | 46:54 |
| 2 | K2CO3 | 70 | —d | 28:72d |
| 3 | KOH | r.t | 68 | 35:65 |
| 4 | Pyridine | r.t. | —e | — |
| 5 | Pyridine | 70 | —f | — |
| 6 | DMAP | r.t. | 44 | 96:4 |
| 7 | DMAP | 70 | 74 | 92:8 |
| 8 | Et3N | r.t. | 71 | 64:36 |
| 9 | Et3N | 60 | —d | 63:37d |
| 10 | DBU | r.t | 82 | 66:34 |
| 11 | DBU | 70 | —d | 66:34d |
| 12 | DABCO | r.t | 72 | 97:3 |
| 13 | DABCO | 70 | 86 | 100:0 |
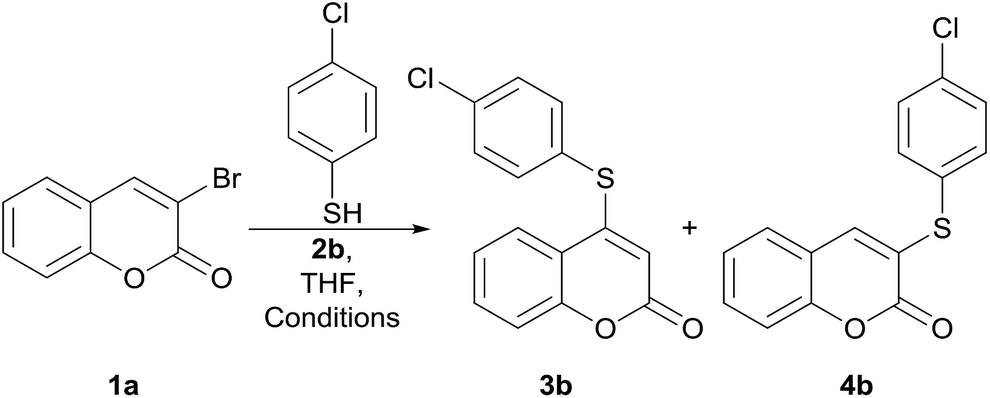
At first, the transformations were performed at room temperature and monitored by GC/MS. Under these conditions, a white precipitate began to form after ca. 10 min and the obtention of mixtures of C-3 and C-4 isomers was consistently observed in the presence of the tested promoters, except pyridine.
The 1H NMR and GC/MS analyses of the products revealed that the C-3 isomer (4b) prevailed in case of the inorganic bases K2CO3 (46:54, entry 1) and KOH (35:65, entry 3), whereas the opposite was detected for the organic promoters Et3N (64:36, entry 8) and DBU (66:34, entry 10); however, when pyridine (1.5 equiv.) was employed as base, the expected reaction product was not observed13b,c even under the heating condition (entries 4 and 5). Instead, the disulfide related to 2b, coumarin and 1a was the detected products when the reaction was executed at 70 °C. It is worth noting that a similar process was reported to proceed at 140 °C in refluxing pyridine, employed both, as solvent and base.13b–d
Gratifyingly, however, although not fully suppressing the formation of the undesired C-3 isomer, the use of DMAP (96:4, entry 8) and DABCO (97:3, entry 12) resulted in a comparatively larger preference for the expected C-4 isomer 3b, with the latter affording higher product yields.
Therefore, the performances of the reactions with these bases were examined at higher temperatures, where formation of a white precipitate was observed almost immediately; however, these also furnished mixtures of isomers (entries 2, 9 and 11). After some trial and error experiments, it was observed that heating a mixture of 1a, the thiol 2b and DMAP gave a 92:8 mixture of 3b and 4b in a satisfactory 74% overall yield (entry 7).
Luckily, however, the use of DABCO in THF at 70 °C, led to the exclusive formation of 3b, the product substituted at C-4, in an improved 86% yield (entry 13). Interestingly, to the best of our knowledge, despite the use of DABCO as a promoter of a similar transformation has been recorded,20f this is the first time the reaction is explored under DMAP assistance. The differences between DABCO and DMAP as organocatalysts have been examined.21a
The 1H NMR signals of the vinylic protons of the starting material and product where compared with those of the unsubstituted coumarin (δH-3 6.43 ppm, doublet; δH-4 7.72 ppm, doublet), and taken as the diagnostic signals of this outcome.
The H-4 resonance of 1a was observed as a highly deshielded singlet resonating at δ 8.11 ppm, whereas the product exhibited a considerably more shielded singlet at δ 5.62 ppm, consistent with structure 3b. The reason for this upfield shift could only be assigned to H-3 in the presence of an adjacent sulfur-containing group, due to its electron-donating character.
Since only mixtures of C-3 and C-4 isomers were observed with the different bases other than DABCO, it was concluded that use of the latter in THF at 70 °C was the best choice for the proposed transformation. Once suitably optimized conditions were obtained, the scope of the reaction was examined with four different 4-substituted thiophenols (2a–d) and three coumarins (1a–c).
As shown in Table 2, the use of DABCO as promoter systematically resulted in the expected 4-substituted heterocycles (3a–l) in consistently high yields (71–97%).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry no. | Coumarin no. | R1 | R2 | Thiophenol no. | R3 | Product no. | Yieldb (%) |
| a Reaction conditions: 3-bromocoumarin (0.5 mmol), arenethiol (2, 0.75 mmol), THF (5 mL), DABCO (1.0 equiv.), 2 h.b Isolated yields by column chromatography. | |||||||
| 1 | 1a | H | H | 2a | H | 3a | 90 |
| 2 | 1a | H | H | 2b | Cl | 3b | 86 |
| 3 | 1a | H | H | 2c | Me | 3c | 84 |
| 4 | 1a | H | H | 2d | OMe | 3d | 90 |
| 5 | 1b | Cl | H | 2a | H | 3e | 78 |
| 6 | 1b | Cl | H | 2b | Cl | 3f | 97 |
| 7 | 1b | Cl | H | 2c | Me | 3g | 84 |
| 8 | 1b | Cl | H | 2d | OMe | 3h | 79 |
| 9 | 1c | H | Me | 2a | H | 3i | 80 |
| 10 | 1c | H | Me | 2b | Cl | 3j | 78 |
| 11 | 1c | H | Me | 2c | Me | 3k | 71 |
| 12 | 1c | H | Me | 2d | OMe | 3l | 87 |
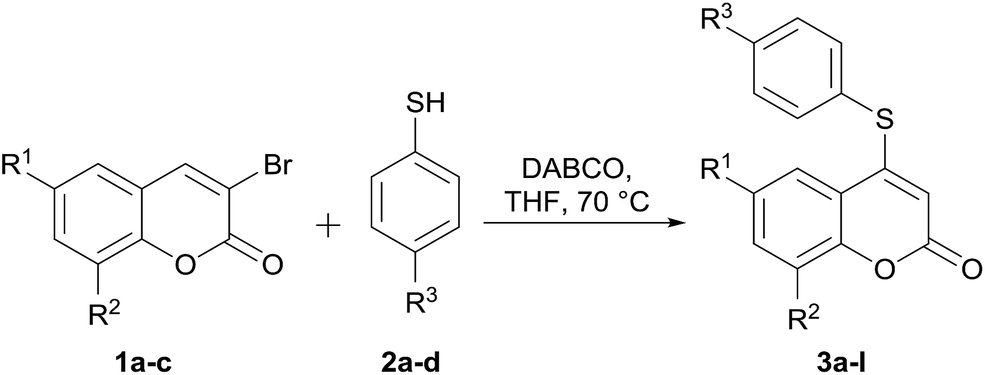
From the analysis of these results, no clear trend emerged with regards to the effect of the substituents of both reaction components on the performance of the transformation. However, it was observed that electron-donating and electron-withdrawing substituents on the thiol side were well tolerated.
Furthermore, the data revealed that the more acidic 4-chloro thiophenol (2b, pKa = 5.46)21b performed better with 6-chlorocoumarin (1b) than with its congeners, the heterocycles 1a and 1c (entry 6 vs. entries 2 and 10). Analogously, the less acidic 4-methoxythiophenol (2d, pKa = 6.21) gave better results in the presence of the coumarins 1a and 1c than with 1b (entries 4 and 12 vs. entry 8).
Taking advantage of the good performance of the transformation when arenethiols were employed, and in order to expand its scope, the reaction was also carried out with α,ω-dithiols (5a,d). Rewardingly, it was observed (Table 3) that in the presence of the α,ω-dithiols in excess, the reaction furnished the expected 4-((ω-mercaptoalkyl)thio)coumarins 6a,d.
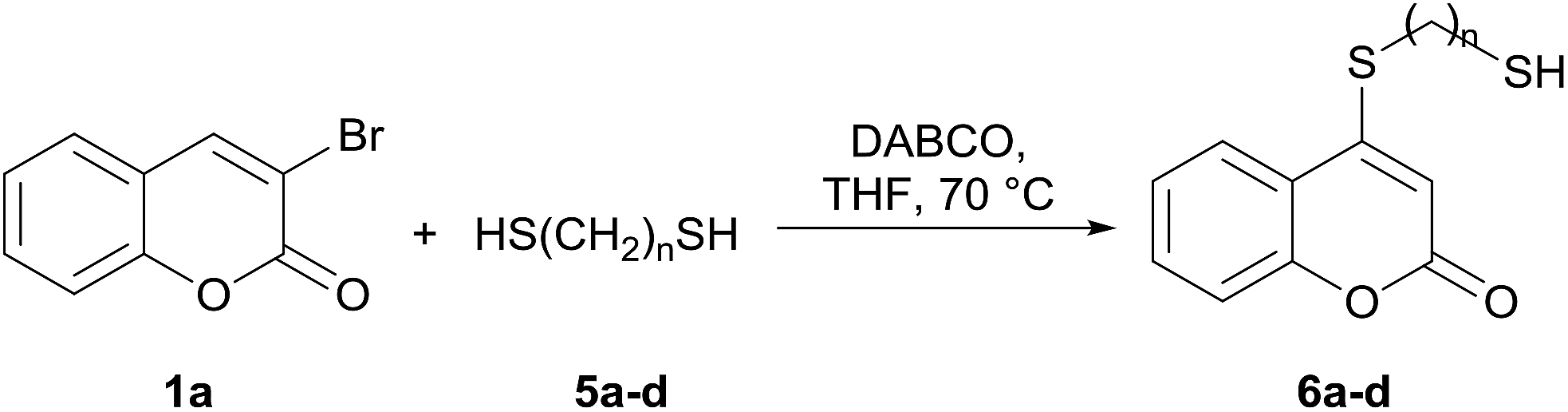
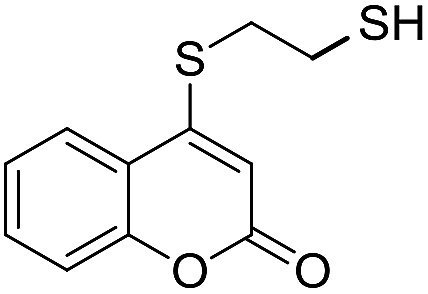
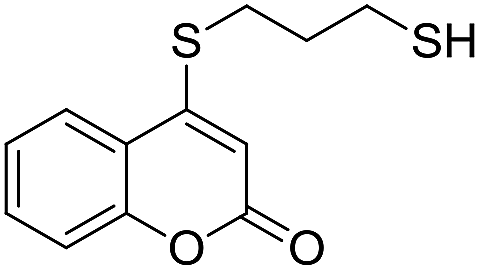
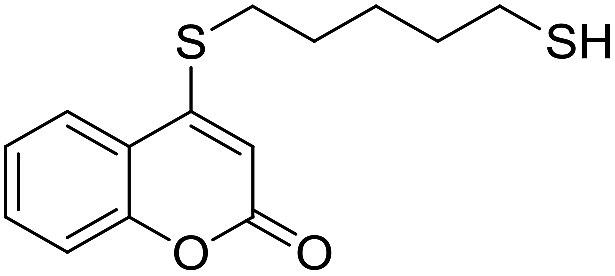
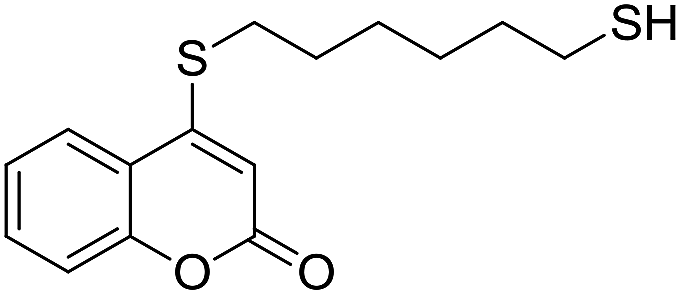
Thus, in the presence of 3.0 equiv. of the dithiols with three (5b) and six (5d) carbon atoms and 1.5 equiv. of the dithiol with five carbon atoms (5c), the coumarin 1a afforded good yields of the expected coumarin thiols 6b–d (71–79%, entries 2–4), with slightly decreased with the increase of the chain length. Contrastingly, however, only a meagre 39% yield of 6a was isolated (entry 1), despite the use of 3.0 equiv. of the more reactive 1,2-ethanedithiol (5a).
Encouraged by these results, the scope of this selective sulfanylation was also explored in the presence of excess of the coumarin 1a, aiming to obtain the more challenging 4,4′-(alkane-α,ω-diylbis(sulfanediyl))bis(coumarins). As shown in Table 4, under our optimized conditions, the corresponding reactions of 1a with 5c and 5d gave good yields of the expected products 7a and 7b, respectively. Sadly, however, when analogous transformations were attempted with the more reactive 1,2-ethanedithiol (5a) and 1,3-propanedithiol (5b), they met with failure, giving insoluble white solids, which could not be analyzed by NMR.
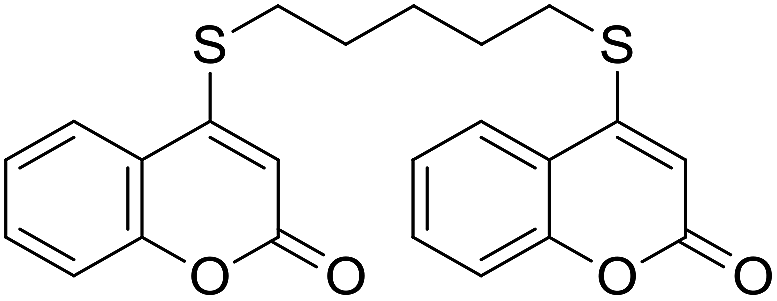
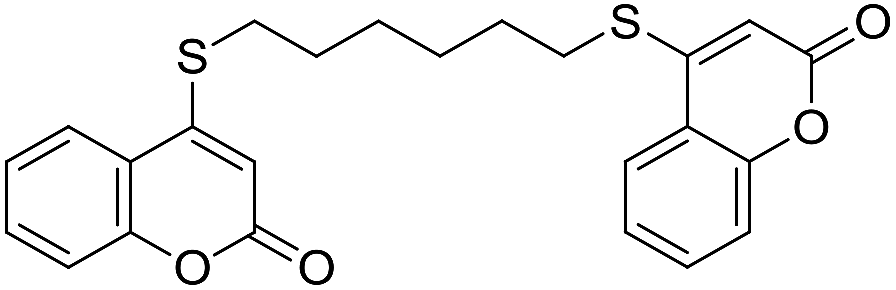
Noteworthy, compounds such as 7a and 7b are very rare. A related 4,4′-sulfanediyl-biscoumarin was prepared as a side product of the reaction of 4-mercaptocoumarin with diphenylketene.22
On the other hand, their synthesis complements earlier work by Drozd and coworkers who prepared analogous heterocycles in a more cumbersome way, by vinylic substitution on 3-nitro-4-halocoumarins.23
Further, these authors also found out that the presence of a good leaving group on C-3 enables short chain dithiols to displace the leaving group, resulting in intramolecular cyclization, to afford tricyclic products which carry 1, n + 2 dithiane motifs (n = number of carbon atoms of the α,ω-dithiol), and bear sulfur atoms attached to both positions, C-3 and C-4. In addition, they discovered that 4-((ω-mercaptoalkyl)thio) coumarins can be preferentially obtained (<40% yield) by running the transformation at low temperature.
It was reported that α-bromo Michael acceptors such as ketones and esters, react with thiols to undergo ipso-substitution, furnishing the α-sulfanyl derivatives alone or in mixtures with the corresponding dithiosubstituted compounds.20c,24a,b Further, cyclic α-haloenones react with thiols providing mainly the α-sulfanyl derivatives,24c and α-haloenals derived from crotonaldehyde and cinnamaldehyde gave moderate yield of the ipso-substitution product upon reaction with butanethiol.24d,e
The actual case is different and, although the exact reaction mechanism of the disclosed transformations remains unknown, a good mechanistical picture can be drawn based on literature precedents.25a As shown in Scheme 2, it can be assumed that the addition of DABCO effects deprotonation of the acidic thiol moieties 2 and results in the formation of the thiolate anions (2S−), which are stronger nucleophiles and can react with the α,β-unsaturated carbonyl system of 1a.
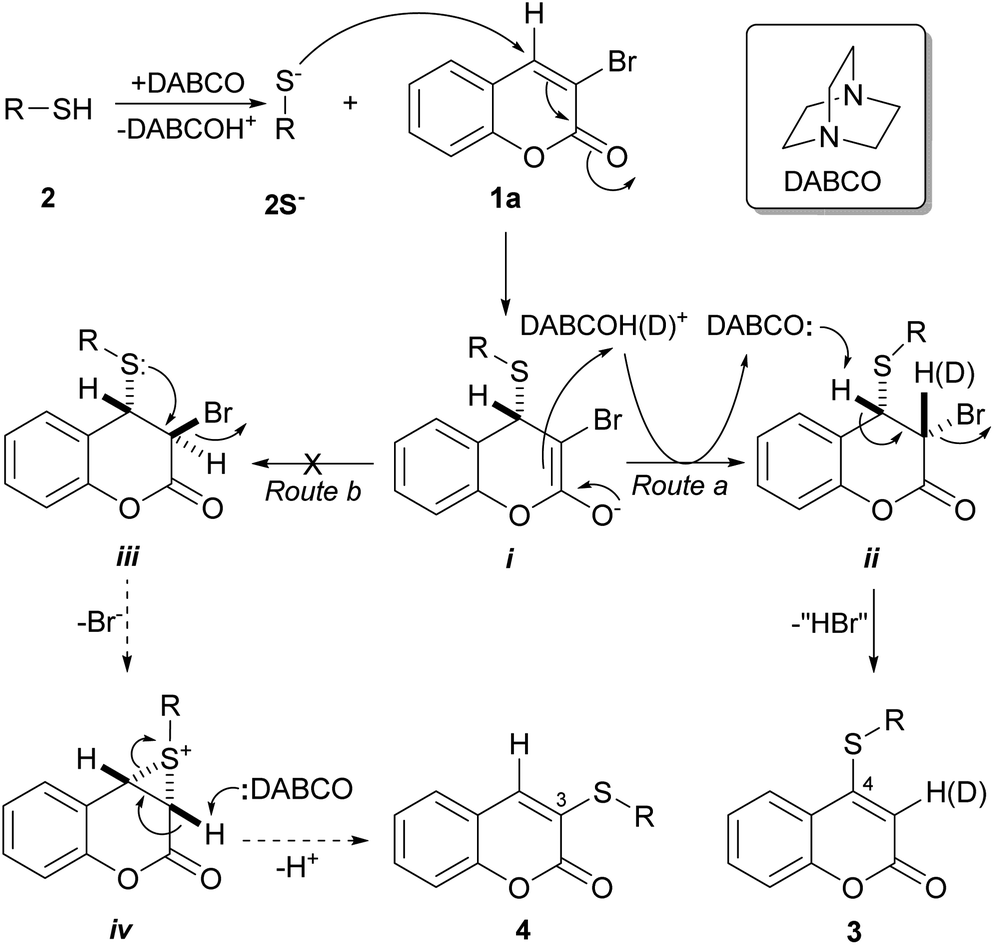 | ||
| Scheme 2 Plausible reaction mechanism for the DABCO-promoted selective sulfanylation of 3-bromocoumarins to afford 4-sulfanylcoumarins 3. | ||
In general, the α,β-unsaturated carbonyl system of coumarins is prone to undergo a Michael type (C-4) addition, and it has been observed that the latter transformation takes place preferentially in these heterocycles when they are 3,4-disubstituted with good leaving groups, such as halides and sulfonates.7b,25b
This in sharp contrast with the reactivity of the neighbor C-3 position. It is known that in α,β-unsaturated carbonyl systems like those of coumarins, when an halogen atom is placed at the α-position to the carbonyl moiety (C-3), the activation of the α-carbon atom is usually too low to afford a substitution product via an addition–elimination mechanism. Therefore, all the reactivity characteristics of the coumarin unsaturated system strongly favor a C-4 attack.
Accordingly, the thiolate anions might trigger a thia-Michael addition, where the incoming nucleophile becomes attached to the β-end of the double bond (C-4) system, resulting in the enolate intermediate i. In turn, the latter may capture a proton (from DABCOH+), furnishing a 3,4-disubstituted 3,4-dihydro coumarin intermediate and returning the nucleophilic catalyst DABCO to the reaction medium. If the transformation path takes the Route a, the structure of this intermediate is ii, which can then undergo a base-mediated dehydrohalogenation to furnish the expected 4-sulfanylcoumarin 3.
Some literature precedents suggest that, at least in the case of the coumarins, both stages, the formation intermediate ii as well as its dehydrohalogenation, should be stereospecific processes, with the latter stage being an 1,2-anti-type elimination.25c In view of the observed results, it is most likely that the bulk of DABCO, a sterically hindered tertiary amine, may play some key role in inducing the delivery of the proton to intermediate i in an anti-fashion to the incoming sulfur reactant, to selectively afford the intermediate ii.
Notably, when the transformation of 1a with thiol 2b was performed in the presence of a small amount of D2O (≈0.1 mL), it was observed that the product 3b exhibited deuteration on C-3 (δ 5.62 ppm),25c,d as stemmed from the reduction of its 1H NMR resonance integral, clearly demonstrating the participation of intermediates i and ii in the reaction. Furthermore, a GC/MS analysis of the reaction products confirmed this observation, and indicated over 90% deuterium incorporation.
Interestingly, Nambara found out that the reaction between thiophenol and 3-bromothiachromone-1,1-dioxide affords 2-phenylsufanylthiachromone-1,1-dioxide. Despite the reaction may take place through an analogous mechanism, it is noteworthy that the transformation was performed in KOH,20a which proved to give unsatisfactory results in the case of 1a. In addition, a similar mechanism may be at the heart of a recently reported modification of Fiesselmann's thiophene synthesis.25e
Alternatively, the reaction could take place through Route b and proceed through the intermediacy of iii, which has the proper geometric array for an intramolecular attack by the sulfur moiety of the sulfanyl group from behind to the bromine atom, to furnish the episulfonium (thiiranium) ion intermediate iv.20e This step entails a 3-exo-tert process,24e which is favorable according to Baldwin's rules. In turn, the intermediate iv could suffer an attack of the base on the most acidic α-carbonyl hydrogen, resulting in opening of the strained episulfonium ion ring, to finally give the 3-sulfanylcoumarin 4.
The same outcome could be achieved by means of either a thiolate-mediated nucleophilic ring opening of intermediate iv, or a nucleophilic bromide substitution on intermediate iii, followed in both cases by elimination of a molecule of thiol,20e,24c,24e which would result in a formal ipso-substitution of the halogen atom. This sequence of events is reminiscent of intracellular reactions of molecules containing the α-haloacryloyl motif with biological thiols in living organisms, which generate episulfonium ion intermediates; the latter are DNA-alkylating agents and play a key role in processes of DNA-damaging and cytotoxicity.26
Contrastingly, however, in the actual case and under DABCO promotion in refluxing THF, no 3-thio-substituted coumarin derivatives 4 were isolated in any of the studied cases. Furthermore, the high yields recorded in most of the examples suggest that the reaction takes place exclusively through Route a or that involvement of Route b, if any, should be minimal.
Experimental
General information
The solvents were purified and dried according to usual procedures.27 The reagents for synthesis were obtained commercially and were used as received, without further purification. The progress of the reactions was monitored by thin layer chromatography on silica gel GF254 plates. For detection of the spots the plates were exposed to UV light (254 and 365 nm), I2 or H2SO4/vanillin solution. The chromatographic purifications were carried out by column chromatography employing silica gel (230–400 mesh, 40–63 μm) and eluting with hexane–EtOAc (10:1, v/v).
Equipment
The melting points were taken on a MQAPF-301 melting point apparatus and are reported uncorrected.The 1H and 13C NMR spectra were recorded on a Bruker 400 MHz NMR spectrometer, with the samples dissolved in CDCl3, unless otherwise noted. The chemical shifts are informed in ppm downfield from the signal of TMS, used as internal standard, and the coupling constants (J) are expressed in Hertz (Hz).
The high-resolution mass spectral data were obtained on a LTQ Orbitrap Discovery mass spectrometer (Thermo Fisher Scientific), using sodium formate as reference.
:1, v/v). After 5 minutes, the stirred reaction was treated with NBS (0.748 g, 4.2 mmol) and allowed to further stir overnight at room temperature. Then, the reaction was diluted with H2O (10 mL) and extracted with EtOAc (3 × 25 mL); the combined organic phases were washed with brine (10 mL), dried over MgSO4 and concentrated under reduced pressure. Chromatographic purification of the residue afforded 1b (0.838 g, 81%), as a white solid, mp 174 °C (lit. 172–174 °C). 1H NMR δ 8.02 (s, 1H), 7.51 (dd, J = 2.4, 8.8, 1H), 7.44 (d, J = 2.4, 1H), 7.29 (d, J = 8.8, 1H).19General procedure for the 4-sulfanylation of 3-bromocoumarins
The coumarin (0.5 mmol) was transferred to a 25 mL two-necked round bottomed flask containing THF (3 mL) and the mixture was heated to 70 °C. Then, the thiol (0.75 mmol) and DABCO (0.5 mmol) were successively added (specific modifications to the amounts of the reagents of this protocol are given in the tables). The reaction was monitored by TLC. Upon complete consumption of the starting material, the reaction was diluted with H2O (10 mL) and the products were extracted with EtOAc; the combined organic extracts were washed with 10% w/v Na2CO3 solution, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by column chromatography. In the labelling experiment, D2O (0.1 mL) were mixed with the thiol and the coumarin, before adding DABCO.Conclusion
In conclusion, we have developed an efficient DABCO-promoted approach for the direct 4-sulfanylation of 3-bromocoumarins, to afford a ready access to different 4-organylsulfanyl-coumarin derivatives in good yield. This methodology entails a one-pot thia-Michael addition/dehydrobromination process and results in a direct C–S bond formation.In the presence of DABCO, the introduction of the sulfanyl moiety is highly stereospecific, in such a way that it favors the following anti-elimination step and precludes thiirane formation, to afford only 4-substituted coumarins. This approach is simple and metal-free; therefore, it may find application in the synthesis of other relevant compounds from the biological, functional or structural points of view.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are thankful to CNPq, CAPES and CONICET for financial support.Notes and references
- (a) Z. J. Witczak, Curr. Med. Chem., 1999, 6, 165–178 CAS; (b) L. Wu, X. Wang, W. Xu, F. Farzaneh and R. Xu, Curr. Med. Chem., 2009, 16, 4236–4260 CrossRef CAS PubMed; (c) M. E. Riveiro, N. De Kimpe, A. Moglioni, R. Vázquez, F. Monczor, C. Shayo and C. Davio, Curr. Med. Chem., 2010, 17, 1325–1338 CrossRef CAS PubMed; (d) A. Lacy and R. O'Kennedy, Curr. Pharm. Des., 2004, 10, 3797–3811 CrossRef CAS PubMed; (e) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed.
- (a) K. C. Majumdar and S. K. Ghosh, Tetrahedron Lett., 2002, 43, 2115–2117 CrossRef CAS; (b) K. Scherlach, H.-W. Nützmann, V. Schroeckh, H.-M. Dahse, A. A. Brakhage and C. Hertweck, Angew. Chem., Int. Ed., 2011, 50, 9843–9847 CrossRef CAS PubMed; (c) T. Grkovic, R. H. Pouwer, M.-L. Vial, L. Gambini, A. Noel, J. N. A. Hooper, S. A. Wood, G. D. Mellick and R. J. Quinn, Angew. Chem., Int. Ed., 2014, 53, 6070–6074 CrossRef CAS PubMed; (d) R. D. Waigh, B. M. Zerihun and D. J. Maitland, Phytochemistry, 1991, 30, 333–335 CrossRef CAS; (e) F. Bohlmann, C. Zdero, R. M. King and H. Robinson, Phytochemistry, 1980, 19, 689–691 CrossRef CAS.
- (a) J. Liu, Y.-Q. Sun, Y. Huo, H. Zhang, L. Wang, P. Zhang, D. Song, Y. Shi and W. Guo, J. Am. Chem. Soc., 2014, 136, 574–577 CrossRef CAS PubMed; (b) Y. Kim, S. V. Mulay, M. Choi, S. B. Yu, S. Jon and D. G. Churchill, Chem. Sci., 2015, 6, 5435–5439 RSC.
- (a) M. Gebauer, Bioorg. Med. Chem., 2007, 15, 2414–2420 CrossRef CAS PubMed; (b) L. C. Cantley, M. G. Vander Heiden and H. R. Christofk, Inhibitors of pyruvate kinase and methods of treating disease, Patent WO 2008/19139, 2008; (c) V. Colotta, D. Catarzi, F. Varano, L. Cecchi, G. Filacchioni, C. Martini, L. Giusti and A. Lucacchini, Farmaco, 1998, 53, 375–381 CrossRef CAS.
- (a) M. Ciustea, J. E. Y. Silverman, A. M. D. Shudofsky and R. P. Ricciardi, J. Med. Chem., 2008, 51, 6563–6570 CrossRef CAS PubMed; (b) N. H. Vo, Q. Che and S. Chen, Fused ring compounds for inflammation and immune-related uses, Patent WO 2008/39520, 2008; (c) S. Kumar, B. K. Singh, N. Kalra, V. Kumar, A. Kumar, A. K. Prasad, H. G. Raj, V. S. Parmar and B. Ghosh, Bioorg. Med. Chem., 2005, 13, 1605–1613 CrossRef CAS PubMed.
- (a) A. Brakhage, H.-M. Dahse, C. Hertweck, H.-W. Nützmann and K. Scherlach, Benzopyranobenzothiazinones and their use as fungicides, antibiotics and antitumor agents, Patent WO 2013/00543, 2013; (b) K. Wittine, I. Ratkaj, K. Benci, T. Suhina, L. Mandić, N. Ilić, S. K. Pavelić, K. Pavelić and M. Mintas, Med. Chem. Res., 2016, 25, 728–737 CrossRef CAS; (c) T. Guo, X.-N. Wei, H.-Y. Wang, Y.-L. Zhu, Y.-H. Zhao and Y.-C. Ma, Org. Biomol. Chem., 2017, 15, 9455–9464 RSC; (d) J. Wu, Z. Yang, R. Fathi and Q. Zhu, Thio-coumarins, US Pat., 7148253, 2006.
- (a) Y.-Y. Peng, Y. Wen, X. Mao and G. Qiu, Tetrahedron Lett., 2009, 50, 2405–2406 CrossRef CAS; (b) W. Wang, Q. Ding, R. Fan and J. Wu, Tetrahedron Lett., 2007, 48, 3647–3649 CrossRef CAS; (c) U. Kazmaier, T. Doroshenko, K. Bauer, I. Filbrich, A. Grueter, G. Jung and A. Ullrich, Synthesis, 2017, 49, 2743–2748 CrossRef CAS; (d) J. Wu, Chem. Lett., 2006, 35, 562–563 CrossRef CAS.
- (a) A. R. Kapdi, A. Karbelkar, M. Naik, S. Pednekar, C. Fischer, C. Schulzke and M. Tromp, RSC Adv., 2013, 3, 20905–20912 RSC; (b) S. Anwar, S. B. Paul, K. C. Majumdar and S. Choudhury, Synth. Commun., 2014, 44, 3304–3313 CrossRef CAS; (c) V. O. Iaroshenko, F. Erben, S. Mkrtchyan, A. Hakobyan, M. Vilches-Herrera, S. Dudkin, A. Bunescu, A. Villinger, V. Y. Sosnovskikh and P. Langer, Tetrahedron, 2011, 67, 7946–7955 CrossRef CAS.
- (a) M. Jeyachandran, P. Ramesh, D. Sriram, P. Senthilkumar and P. Yogeeswari, Bioorg. Med. Chem. Lett., 2012, 22, 4807–4809 CrossRef CAS PubMed; (b) D. Kong, Z. Jiang, S. Xin, Z. Bai, Y. Yuan and Z. Weng, Tetrahedron, 2013, 69, 6046–6050 CrossRef CAS; (c) K. Mahato, N. Arora, P. Ray Bagdi, R. Gattu, S. S. Ghosh and A. T. Khan, Chem. Commun., 2018, 54, 1513–1516 RSC.
- (a) J. R. Merchant, G. Martyres and M. S. Venkatesh, Indian J. Chem., 1981, 20, 493–495 Search PubMed; (b) M. Belal and A. T. Khan, RSC Adv., 2015, 5, 104155–104163 RSC; (c) Z. Yin, Y. Wang, Q. Yang, Y. Wang, J. Xu, Z. Deng and Y. Peng, Synthesis, 2013, 45, 759–766 CrossRef.
- (a) M. S. Shepard and E. M. Carreira, J. Am. Chem. Soc., 1997, 119, 2597–2605 CrossRef CAS; (b) K. C. Majumdar, U. K. Kundu and S. K. Ghosh, Org. Lett., 2002, 4, 2629–2631 CrossRef CAS PubMed; (c) G. Wang, L. Chen, T. Xian, Y. Liang, X. Zhang, Z. Yang and M. Luo, Org. Biomol. Chem., 2014, 12, 8048–8060 RSC; (d) K. C. Majumdar and A. Biswas, Monatsh. Chem., 2004, 135, 1001–1007 CAS.
- (a) D. Nematollahi, J. Azizian, M. Sargordan-Arani, M. Hesari, S. Jameh-Bozorghi, A. Alizadeh, L. Fotouhi and B. Mirza, Chem. Pharm. Bull., 2008, 56, 1562–1566 CrossRef CAS PubMed; (b) D. Nematollahi, J. Azizian, M. Sargordan-Arani, M. Hesari and B. Mirza, J. Heterocycl. Chem., 2009, 46, 1000–1002 CrossRef CAS.
- (a) G. Eisenbrand, M. Otteneder and W. Tang, Toxicology, 2003, 190, 249–258 CrossRef CAS PubMed; (b) J. R. Merchant and N. M. Koshti, Org. Prep. Proced. Int., 1983, 15, 321–327 CrossRef CAS; (c) J. R. Merchant and P. J. Shah, Chem. Ind., 1980, 791–792 CAS; (d) J. R. Merchant, A. S. Gupta, P. J. Shah and S. S. Shirali, Chem. Ind., 1979, 351–352 CAS.
- (a) V. L. Ponzo, D. A. Bianchi and T. S. Kaufman, Tetrahedron Lett., 1998, 39, 3409–3412 CrossRef CAS; (b) A. B. J. Bracca and T. S. Kaufman, Synth. Commun., 2006, 36, 299–310 CrossRef CAS.
- G. Padilha, P. T. Birmann, M. Domingues, T. S. Kaufman, L. Savegnago and C. C. Silveira, Tetrahedron Lett., 2017, 58, 985–990 CrossRef CAS.
- (a) G. Padilha, B. A. Iglesias, D. F. Back, T. S. Kaufman and C. C. Silveira, ChemistrySelect, 2017, 2, 1297–1304 CrossRef CAS; (b) J. L. Pergomet, M. G. Di Liberto, M. G. Derita, A. B. J. Bracca and T. S. Kaufman, Fitoterapia, 2018, 125, 98–105 CrossRef CAS PubMed; (c) J. L. Pergomet, A. B. J. Bracca and T. S. Kaufman, Org. Biomol. Chem., 2017, 15, 7040–7049 RSC.
- (a) V. Bansal, S. Kanodia, P. C. Thapliyal and R. N. Khanna, Synth. Commun., 1996, 26, 887–892 CrossRef CAS; (b) K. Bowden and S. Battah, J. Chem. Soc., Perkin Trans. 2, 1998, 1603–1606 RSC; (c) D. Audisio, S. Messaoudi, J.-D. Brion and M. Alami, Eur. J. Org. Chem., 2010, 1046–1051 CrossRef CAS; (d) J. Su, Y. Zhang, M. Chen, W. Li, X. Qin, Y. Xie, L. Qin, S. Huang and M. Zhang, Synlett, 2019, 30, 630–634 CrossRef CAS.
- K. Kim and I. Park, Synthesis, 2004, 16, 2641–2644 CrossRef.
- C. Cho, J. Park, I. Jung and H. Lee, Tetrahedron Lett., 2001, 42, 1065–1067 CrossRef CAS.
- (a) T. Nambara, Yakugaku Zasshi, 1958, 78, 624–627 CrossRef CAS; (b) F. Farina, M. C. Maestro, M. R. Martin, M. V. Martin, F. Sánchez and M. L. Soria, Tetrahedron, 1986, 42, 3715–3722 CrossRef CAS; (c) V. Rosnati, A. Saba, A. Salimbeni and U. Vettori, Gazz. Chim. Ital., 1981, 111, 249–256 CAS; (d) R. Hunter, J. P. Michael and D. S. Walter, Tetrahedron Lett., 1994, 35, 5481–5484 CrossRef CAS; (e) E. V. Kondrashov, A. R. Romanov, I. A. Ushakov and A. I. Rulev, J. Sulfur Chem., 2017, 38, 18–33 CrossRef CAS; (f) M. Bio, G. Nkepang and Y. You, Chem. Commun., 2012, 48, 6517–6519 RSC.
- (a) M. Baidya, S. Kobayashi, F. Brotzel, U. Schmidhammer, E. Riedle and H. Mayr, Angew. Chem., Int. Ed., 2007, 46, 6176–6179 CrossRef CAS; (b) The pKa values were calculated with MarvinSketch v. 18.4.0.
- (a) F. Eiden and E. Zimmermann, Arch. Pharm., 1976, 309, 619–625 CrossRef CAS; (b) K. Tabakovic and I. Tabakovic, J. Heterocycl. Chem., 1992, 29, 383–385 CrossRef CAS; (c) E. A. Parfenov and L. D. Smirnov, Chem. Heterocycl. Compd., 1991, 27, 825–830 CrossRef.
- (a) V. N. Drozd, N. L. Nam, V. N. Knyazev, D. S. Yufit, Y. T. Struchkov and V. L. Savel'ev, Journal of Organic Chemistry of the USSR, 1992, 28, 1184–1193 (Z. Org. Khim., 1992, 28, 1496–1507) Search PubMed; (b) T. Y. Mozhaeva, V. P. Lezina, V. L. Savel'ev, V. N. Knyazev and V. N. Drozd, Journal of Organic Chemistry of the USSR, 1991, 27, 163–169 (Z. Org. Khim., 1991, 27, 185–191) Search PubMed; (c) V. N. Drozd, V. N. Knyazev, N. L. Nam, V. P. Lezina, T. Y. Mozhaeva and V. L. Savel'ev, Journal of Organic Chemistry of the USSR, 1993, 29, 653–658 (Z. Org. Khim., 1993, 29, 782–788) Search PubMed.
- (a) V. Rosnati, A. Saba and A. Salimbeni, Tetrahedron Lett., 1981, 22, 167–168 CrossRef CAS; (b) J.-L. Boucher and L. Stella, Tetrahedron, 1988, 44, 3595–3605 CrossRef CAS; (c) X. Xu, X. Li, A. Wang, Y. Sun, W. B. Schweizer and R. Prins, Helv. Chim. Acta, 2011, 94, 1754–1763 CrossRef CAS; (d) N. A. Keiko, L. G. Stepanova, I. D. Kalikhman and M. G. Voronkov, Russ. Chem. Bull., 1986, 35, 1524–1526 CrossRef; (e) L. He, H. Guo, Y.-Z. Li, G.-F. Du and B. Dai, Chem. Commun., 2014, 50, 3719–3721 RSC.
- (a) T. Kauffmann, A. Risberg, J. Schulz and R. Weber, Tetrahedron Lett., 1964, 3563–3568 CrossRef CAS; (b) M. S. Newman and S. Schiff, J. Am. Chem. Soc., 1959, 81, 2266–2270 CrossRef CAS; (c) C. Oh, I. Yi and K. P. Park, J. Heterocycl. Chem., 1994, 31, 841–844 CrossRef CAS; (d) S. Kumar, S. S. Chimni and H. Singh, J. Chem. Soc., Perkin Trans. 1, 1991, 1391–1395 RSC; (e) R. A. Irgashev, A. S. Steparuk and G. L. Rusinov, Org. Biomol. Chem., 2018, 16, 4821–4832 RSC.
- M. I. Fekry, N. E. Price, H. Zang, C. Huang, M. Harmata, P. Brown, J. S. Daniels and K. S. Gates, Chem. Res. Toxicol., 2011, 24, 217–228 Search PubMed.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Butterworth–Heinemann, Oxford, UK, 5th edn, 2003 Search PubMed.
- (a) T. Mahajan, L. Kumar, K. Dwived and D. D. Agarwal, Ind. Eng. Chem. Res., 2012, 51, 3881–3886 CrossRef CAS; (b) M. N. Deshmukh, R. Burud, C. Baldino, P. C. M. Chan and J. Liu, Synth. Commun., 2003, 33, 3299–3303 CrossRef CAS; (c) X. Cao, X. Oan, P. Zhou, J. Zou and O. Asekun, Chem. Commun., 2014, 25, 3359–3362 RSC.
- (a) J. Wu, Z. Yang, R. Fathi, Q. Zhu and L. Wang, Thio-coumarins, US Pat., 6703514, 2004; (b) R. Grigg and D. Vipond, Tetrahedron, 1989, 45, 7587–7592 CrossRef CAS; (c) G. Peinhardt and L. Reppel, Pharmazie, 1970, 25, 68–69 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of the final products. See DOI: 10.1039/c9ra09545d |
| This journal is © The Royal Society of Chemistry 2020 |