
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidating the ionic liquid distribution in monolithic SILP hydroformylation catalysts by magnetic resonance imaging†
Jakob Maximilian Marinkovic a,
Stefan Bendersb,
Eduardo J. Garcia-Suarez‡§
a,
Alexander Weißc,
Carsten Gundlachd,
Marco Haumannc,
Markus Küppersb,
Bernhard Blümichb,
Rasmus Fehrmanna and
Anders Riisager*a
a,
Stefan Bendersb,
Eduardo J. Garcia-Suarez‡§
a,
Alexander Weißc,
Carsten Gundlachd,
Marco Haumannc,
Markus Küppersb,
Bernhard Blümichb,
Rasmus Fehrmanna and
Anders Riisager*a
aTechnical University of Denmark, Centre for Catalysis and Sustainable Chemistry, Department of Chemistry, Kemitorvet, Building 207, 2800 Kgs. Lyngby, Denmark. E-mail: ar@kemi.dtu.dk
bRWTH Aachen University, Institut für Technische und Makromolekulare Chemie, Worringerweg 2, 52064 Aachen, Germany
cFriedrich-Alexander-Universität Erlangen-Nürnberg (FAU), Lehrstuhl für Chemische Reaktionstechnik (CRT), Egerlandstr. 3, 91058 Erlangen, Germany
dTechnical University of Denmark, Department of Physics, Fysikvej, Building 309, 2800 Kgs. Lyngby, Denmark
First published on 15th May 2020
Abstract
Monolithic silicon carbide supported ionic liquid-phase (SILP) Rh-catalysts have very recently been introduced for gas-phase hydroformylation as an important step toward industrial upscaling. This study investigates the monolithic catalyst system in combination with different impregnation procedures with non-invasive magnetic resonance imaging (MRI). The findings were supported by X-ray microtomography (micro-CT) data of the monolithic pore structure and a catalytic performance test of the catalyst system for 1-butene gas-phase hydroformylation. MRI confirmed a homogeneous impregnation of the liquid phase throughout the full cross-section of the cylindrical monoliths. Consistent impregnations from one side to the other of the monoliths were achieved with a stabilizer in the system that helped preventing inhomogeneous rim formation. External influences relevant for industrial application, such as long-term storage and temperature exposure, did not affect the homogeneous liquid-phase distribution of the catalyst. The work elucidates important parameters to improve liquid-phase catalyst impregnation to obtain efficient monolithic catalysts for industrial exploitation in gas-phase hydroformylation as well as other important industrial processes.
Introduction
Supported ionic liquid-phase (SILP) materials have received much attention in recent years with special interest in the heterogenization of homogeneous catalyst systems.1–3 SILP materials are typically prepared by impregnation of a porous, high-surface oxide support (Al2O3, SiO2, etc.) with an ionic liquid (IL) and a dissolved active catalyst system.4,5 By such impregnation, a thin film layer of the IL catalyst system is distributed in the porous support with a large surface area and short mass transport pathways. Thus, the catalyst system acts as a homogeneous catalyst on the micro-scale, while the SILP material on the macro-scale performs as a heterogeneous catalyst allowing its use in simple, continuous-flow reactions with fixed-bed reactors.2Hydroformylation represents a reaction of major industrial importance to produce aldehydes and derived chemicals used in commodities such as plasticizers, fragrances and detergents.6 Continuous fixed-bed, gas-phase hydroformylation with Rh-SILP catalysts based on micrometer-sized silica supports has been demonstrated successfully on the small scale with C2–C4 alkenes, using both phosphine- and phosphite-modified catalysts with imidazolium-based IL (e.g. [C2C1Im][NTf2]).3 With diphosphite-modified catalysts, stable operation over 900 h time-on-stream with a high selectivity towards the targeted product (n-pentanal) has been reported with 1-butene as reactant,7 but such powdered catalysts are challenging to implement in up-scaled reactors due to concerns of large pressure drop over the catalyst bed and low heat transfer within the bed.8,9 Structured supports, such as monoliths (see Fig. 1 left), made by an inert material like silicon carbide (SiC) with excellent heat conductivity, mechanical strength and chemical inertness10 can overcome these drawbacks and their modular design further eases up-scaling.11 Currently, manufacturing processes of SiC monoliths are being developed, so that the pore structures can be adjusted and hence the gas/liquid flow and the pressure drop can be tailored to the application.12–15
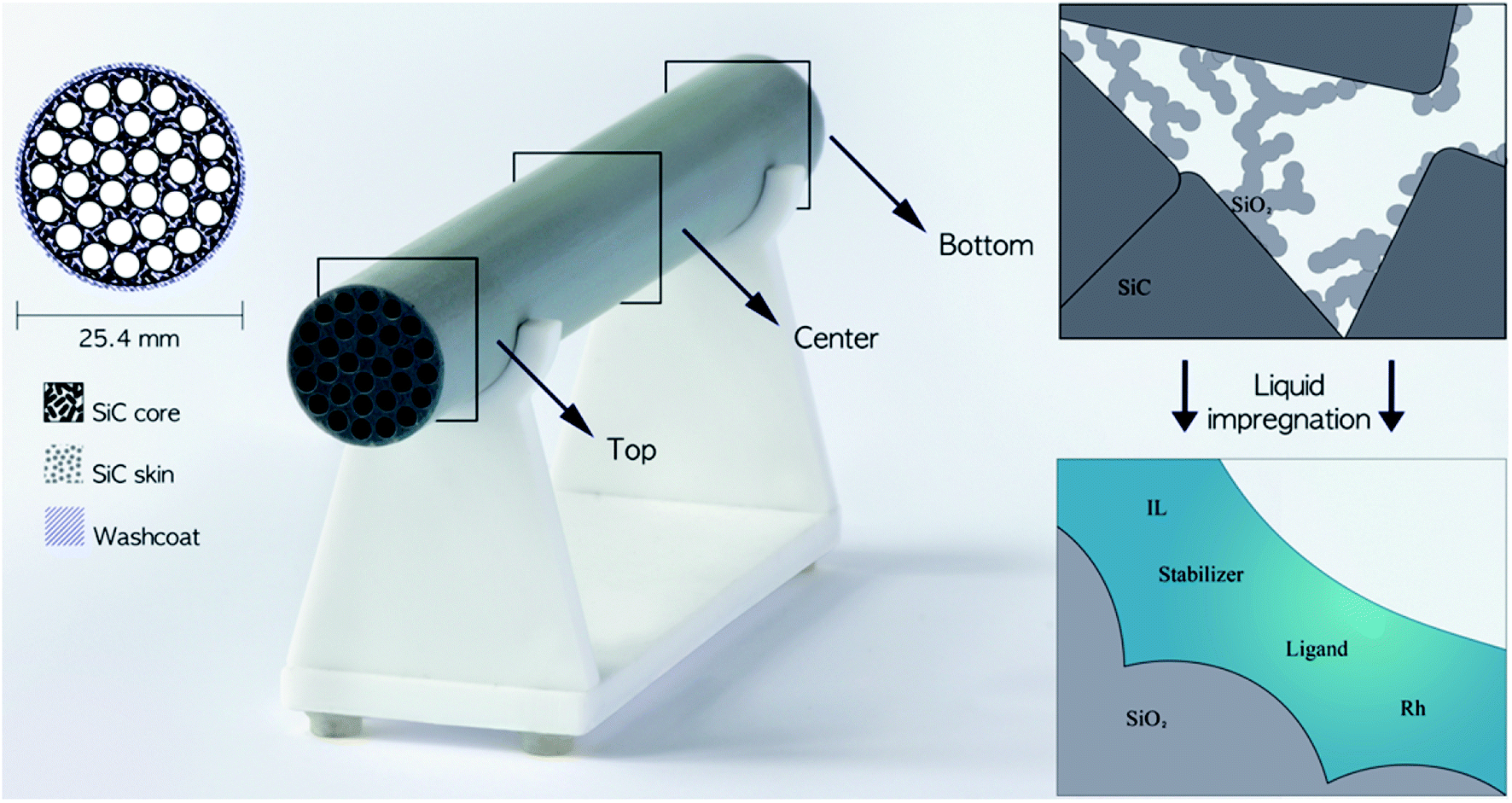 | ||
| Fig. 1 Left: Schematic drawing and photograph of the cylindrical porous monolith consisting of a SiC core, SiC skin and silica wash-coat with the locations indicated where the cross-sections were imaged in the study. Right: Detailed schematic drawing of the pore structure before and after liquid impregnation. | ||
A novel SILP catalyst system based on a cylindrical SiC monolithic support structure has recently been introduced and demonstrated for 1-butene hydroformylation with the IL-catalyst phase immobilized by wet-impregnation.16 In such systems, the impregnation and drying procedures have a strong influence on the final composition and distribution of the catalyst on the monolith.17 The weight uptake can control the overall degree of impregnation, but the distribution of the catalyst system on the monolith is challenging to reveal. For surface-sensitive analytic techniques, such as scanning electron microscopy (SEM) or transmission electron microscopy (TEM), the monoliths need to be divided into small samples, which might influence the catalyst system. To this end, less invasive techniques are therefore preferred. X-ray absorption techniques, such as computed tomography (CT), are sensitive to heavy elements and therefore lack in contrast between IL in the pores and the solid SiC matrix. For high resolution, micro-tomography (micro-CT) is favorable but also requires invasive cutting of the monolith. An alternative, non-invasive technique with lower resolution focusing on the liquid catalyst system is magnetic resonance imaging (MRI) being one of the modes of nuclear magnetic resonance (NMR). MRI applies radiofrequency radiation and provides therefore access to optically opaque materials while unique contrast parameters such as NMR relaxation, can be exploited to increase the information content. Accordingly, MRI has been used for the examination of a wide range of both, soft and hard materials such as polymers, food, plants and wood18–22 as well as in a range of chemical engineering applications23 including the characterization of flow in different geometries24–30 and reactive systems.31–33 NMR relaxation as the primary contrast parameter differs considerably for liquids in bulk and pores,34 and many studies have investigated the distribution and evaporation of liquids in rocks and soil by MRI.35–37 Similarly, MRI has been used to localize gas and liquid flows through monoliths.38,39
In this study, the distribution of an IL-catalyst system inside the pore structure of cylindrical SiC monoliths has been examined non-invasively by MRI and micro-CT. For the MRI measurements, the high susceptibility difference between the porous matrix and the fluid gave rise to short relaxation times. In consequence, acquisition times need to be short and as unaffected by relaxation as possible. Thus, single-point imaging (SPI) was chosen as method, which employs a purely phase-encoded pulse sequence with short dephasing times from spatial encoding.40 Various wet-impregnation procedures of the monoliths with the Rh-diphosphite/IL catalyst system were evaluated in the course of the study, and the resulting monoliths characterized. In addition, the active catalyst system was performance tested in continuous, gas-phase 1-butene hydroformylation to demonstrate its potential.
Experimental
General
Cylindrical SiC monoliths with dimensions of 200 mm in length and 25.4 mm outer diameter with 30 channels of inner diameter 3 mm (LiqTech International A/S) were used as supports (Fig. 1 left). The monoliths were manufactured by sintering α-SiC powder (particle diameter 17.3 μm), applying a SiC skin of sub-micro sized α-SiC particles by a patented method41 followed by wash-coating with 7 nm silica particles infiltrated into the inner monolith structure. Before use, the monoliths were washed three times by submerging in de-ionized water followed by drying at 100 °C for 18 h with subsequent vacuum drying for a minimum of 4 h.Catalyst impregnation were performed by Schlenk techniques under argon atmosphere (99.999%), and all chemicals were used as received. The catalyst system comprised the precursor [Rh(acac)(CO)2] (Sigma-Aldrich, 98%), diphosphite ligand biphephos (Evonik Oxeno GmbH, 98%), stabilizer bis(2,2,6,6-tetramethyl-4-piperidyl)sebacate, (Sigma-Aldrich) and IL 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([C2C1Im][NTf2], Sigma-Aldrich, ≥98%), and the catalytic performance test was performed with 1-butene (99.5%), carbon monoxide (99.7%), hydrogen (99.999%), and helium (99.996%) purchased from the Linde Group. The MRI images were recorded on monoliths containing the IL and the stabilizer only.
Preparation of catalyst stock solutions
A batch of 180 ml catalyst stock solution was prepared by dissolving 11.3 g of biphephos (14.4 mmol) in 40 ml anhydrous dichloromethane (DCM, Sigma-Aldrich, ≥99.8%) and [Rh(acac)(CO)2] (3.6 mmol) separately in 40 ml of anhydrous DCM under stirring for 15 min. Stirring was then continued for another 1 h after mixing the two solutions. In parallel, 27.7 g of bis(2,2,6,6-tetramethyl-4-piperidyl)sebacate (57.6 mmol) was dissolved in 60 ml anhydrous DCM and 18 ml of [C2C1Im][NTf2] separately dissolved in 20 ml anhydrous DCM. After stirring for another 1 h both solutions were combined with the Rh–biphephos solution with an additional 2 ml of anhydrous DCM.The amounts of IL and DCM were adjusted to obtain different IL contents resulting in the catalyst system compositions listed in Table S1, ESI.† The simplified impregnations contained only IL and stabilizer as mentioned above.
Impregnation of monoliths with catalyst stock solutions
The monoliths were wet-impregnated by catalyst stock solutions by submerging the monoliths for 5–6 min before removal of excess solution in vertical orientation unless indicated otherwise. The impregnated monoliths were then dried in a downwards argon flow for 24 h followed by drying under vacuum for another 24 h, both at room temperature (see photograph of the drying set-up in Fig. S1, ESI†). The weight increase obtained during impregnation was measured and the Rh content determined by mass fraction of the catalytic system, assuming full evaporation of the solvent.Characterization of the monoliths
The pore volumes of the monoliths were determined by liquid absorption,42 where the monoliths were submerged in de-ionized water until bubbles no longer formed. After careful removal of excess water, the mass of the water-loaded monoliths were determined and the amount of water (mH2O) found as the difference between the dry and the water-loaded monoliths. This allowed calculating the pore volume according to eqn (1) with a density of water of 998.21 kg m−3 at 20 °C.42 The average (![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) ) values determined are arithmetic means, and the relative standard deviation (RSD) was calculated according to eqn (S1), ESI.†
) values determined are arithmetic means, and the relative standard deviation (RSD) was calculated according to eqn (S1), ESI.†
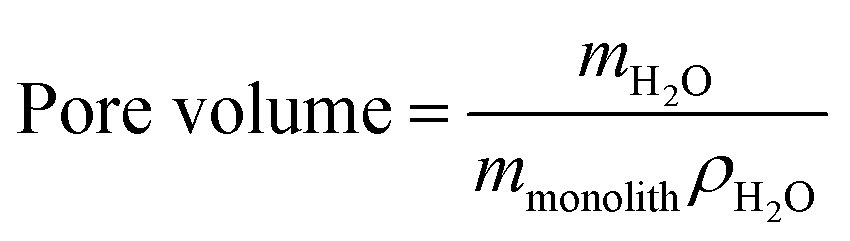 | (1) |
X-ray photoelectron spectra (XPS) were measured with a Thermo Scientific K-Alpha system using a monochromized beam (Al K-α: 1486.6 eV) at a take-off angle of 90° and a spot size of 400 μm. The XPS data was acquired by multiple point-measurements and the data processed with the Avantage software. For analysis, the monoliths were cut transversally into 4 mm thick slices with a precision cutting machine equipped with a diamond blade (Minitom, Struers), and attached to the sample holder without further treatment.
X-ray micro-tomography (micro-CT) images were recorded with a ZEISS Xradia 410 versa X-ray microscope system equipped with a detector system allowing a combination of geometric and optical magnification at a voltage of 40 kV and a power of 10 W. Due to the dimensional limitation of the system, the monolith was also here cut with the precision cutting machine. The monolith was first analyzed with the large field-of-view objective for an overview (11.3 μm efficient pixel size, 1601 projection images, total acquisition time ca. 3 h), before an area of interest was selected for a high-resolution image (objective 10×, 1.54 μm efficient pixel size, 3201 projection images, total acquisition time ca. 52 h). Each 2D image set was reconstructed using XRMreconstructor software attached to the X-ray microscopy system. The reconstruction software used an FDK algorithm, and the resulting 3D volume for the high-resolution scan was a cylinder with a diameter of approximately 1.5 mm.
1H magnetic resonance images (MRI) were measured with a SPI sequence in ParaVision 5.1 on a Bruker Avance III 300 MHz spectrometer equipped with a MIC2.5 gradient system (max. gradient strength 1.5 T m−1), and a 30 mm quadrature coil using the following parameters: dephasing time 75 μs, spectral width 1 MHz, field-of-view (FOV) 30 × 30 mm2, matrix size 256 × 256, resolution 117.2 × 117.2 μm2, pulse duration 9 us, repetition time 500 ms, number of scans 96, and experiment time: 17.5 h. No slices were selected and the images corresponded to an axial projection of the monolith. The monoliths were stabilized in the resonator with paper and all images were recorded at room temperature with the monoliths positioned at the same location as best as possible for comparison of images. The matching of the radiofrequency circuit did not result in the lowest possible reflection due to the high loading of the coil, which resulted in the variable capacitors reaching their limits and thus possibly leading to deviations in signal intensity between different images. All displayed images have the same absolute value scale and are shown without the lowest 10% of their maximum values to cut off the noise. All pixels of the images were integrated to determine the total spin-density including a small artefact in the center originating from a DC offset, which could not readily be eliminated.
Catalytic testing of the monoliths
Continuous, gas-phase hydroformylation of 1-butene was performed at 110 °C and 10 bar using an in-house designed reactor module described in detail elsewhere.16 Gas flows were regulated by mass flow controllers (Bronkhorst) and 1-butene conveyed with an HPLC pump (Smartline 100, Knauer). The fluids were dosed in a volume ratio of H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CO:1-butene = 2:2:1. The impregnated SiC monolith had a total mass of 103.6 g and contained 31.5 mg of Rh. To ensure inert conditions, the monolith was assembled into the reactor module in a glovebox and the sealed reactor installed in the reactor set-up using helium counter flow. Afterwards, the reaction temperature and pressure were set and the reaction gas flows adjusted accordingly. The downstream gas flow was monitored by on-line GC analysis (Bruker GC 450) equipped with two flame ionization detectors and a thermal conductivity detector.
:CO:1-butene = 2:2:1. The impregnated SiC monolith had a total mass of 103.6 g and contained 31.5 mg of Rh. To ensure inert conditions, the monolith was assembled into the reactor module in a glovebox and the sealed reactor installed in the reactor set-up using helium counter flow. Afterwards, the reaction temperature and pressure were set and the reaction gas flows adjusted accordingly. The downstream gas flow was monitored by on-line GC analysis (Bruker GC 450) equipped with two flame ionization detectors and a thermal conductivity detector.
Results and discussion
Impregnation of monoliths
The SiC monoliths were wet-impregnated with catalyst stock solutions containing different amounts of ligand, stabilizer and the hydrophobic IL [C2C1Im][NTf2] (see Table S1, ESI†), which was mainly chosen to protect the sensitive ligand against water. The pore volume of the pristine monoliths was determined by liquid absorption and resulted in an average of 0.17 ml g−1 with a relative standard deviation of 1.62% (RSD, n = 4, see Table S2, ESI†). The low RSD indicated a high accuracy of the method as well as a consistent support material.The pristine monolith used for the catalytic performance test (vide infra) gained a weight increase of 5.7% after drying (from 98.0 g to 103.6 g) corresponding to a Rh loading of 31.5 mg (0.03 wt%) and ligand loading of 961.6 mg (0.98 wt%), respectively. The immobilization of the catalyst system was assumed to be mainly caused by capillary forces as indicated in literature.3 As the combined mass fraction of the IL and the stabilizer was larger than 80 wt%, the Rh precursor and the ligand were left out in most of the following characterizations.
Characterization by micro-CT
Micro-CT images were recorded of a pristine SiC monolith to visualize the pore structure (Fig. 2). The overview image (Fig. 2a) shows the SiC core (large particles), the SiC skin (small particles) and a fraction of one of the circular bore channels on the left part of the image. An enlarged image of the area of the SiC skin and core (Fig. 2b) revealed the pores of the core created by interparticle voids to be in the micrometer range, while the skin with a thickness around 50 μm (determined via pixel analysis) had pores in the sub-micrometer range. A clear separation between the regions indicated no intrusion of fine skin particles into the core. However, the high-resolution micro-CT image revealed a few defects in the skin with minor cracks and small circular holes.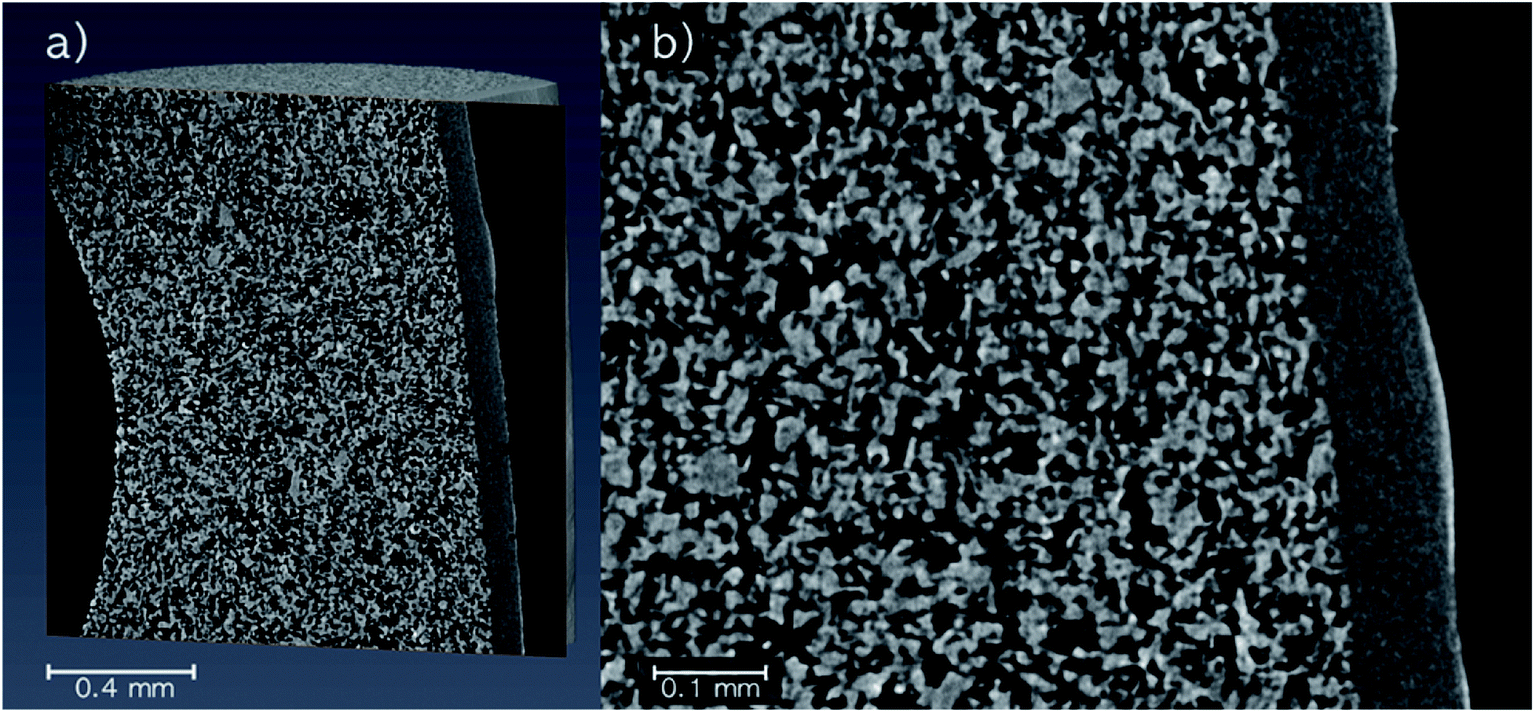 | ||
| Fig. 2 Micro-CT images of a pristine SiC monolith. (a) Overview with the core and skin. (b) Enlarged image with the focus on the SiC skin. High-resolution image: 1.54 μm efficient pixel size, 51.5 h total acquisition time. | ||
Characterization by MRI
MRI was used as a non-invasive method to investigate the distribution of the IL-catalyst system in the monolithic SiC support. Besides background noise, the pristine SiC monolith (Fig. 3a) produced no significant signal, thus verifying that the method was suitable for imaging fluids in the monolith (integrals of all MRI images are given in Fig. S2, ESI†). Moreover, it confirmed that possible water accumulation on the surface of the SiC support was of no importance. | ||
| Fig. 3 (a) MRI images of a pristine monolith. (b) Optical image of a monolith (top view). (c) MRI images of an impregnated monolith (IL, stabilizer). | ||
A MRI image of a SiC monolith with impregnated IL and stabilizer is depicted in Fig. 3c. The comparison with a photograph showing the top of the monolith (Fig. 3b), demonstrates that the impregnation follows the pattern of the support structure. Therefore, it is reasonable to assume that the impregnation solution penetrated the entire porous support structure of the monolith, and that the MRI analysis showed solely the impregnated species on the solid support structure, not the structure itself. Notably, as the thickness of the SiC skin (ca. 50 μm) is less than half of the edge length of a voxel (117.2 μm) of the MRI, it is reasoned to have negligible influence. As the melting point of the stabilizer is above room temperature (82–85 °C) where the MRI images were recorded, an additional measurement was carried out with a stabilizer-only impregnated monolith. Here, also mainly noise was detected (see Fig. S3, ESI†) confirming that the signal detected from the stabilizer originated only from the stabilizer dissolved in the IL and not from its solid form.
MRI images recorded of monoliths impregnated with stock solutions of different compositions of IL and stabilizer are shown in Fig. 4. When the monoliths were impregnated with only IL (Fig. 4a: 2.74 wt% IL; Fig. 4c: 5.80 wt% IL), non-uniform distributions of the IL seemed to form with regions of relatively low concentrations in the center and higher concentrations following the outline of the cylindrical monoliths. In comparison, co-impregnations with stabilizer (Fig. 4b: 2.17 wt% IL and 2.20 wt% stabilizer; Fig. 4d: 3.73 wt% IL and 3.77 wt% stabilizer) clearly resulted in more even distributions of the components. The drying procedure of wet-impregnated monoliths can influence the distribution of the liquid phase, since the fluid may move from more wet to more dry regions due to capillary forces.17 Therefore, the argon flow used for drying may preferentially have dried the monolith from the outside causing the IL to move outwards from the inside (see Fig. S1, ESI† for argon flow direction). In the presence of stabilizer, the IL movement seemed hampered, which possibly could be related to the higher viscosity of the liquid phase in the presence of the stabilizer.
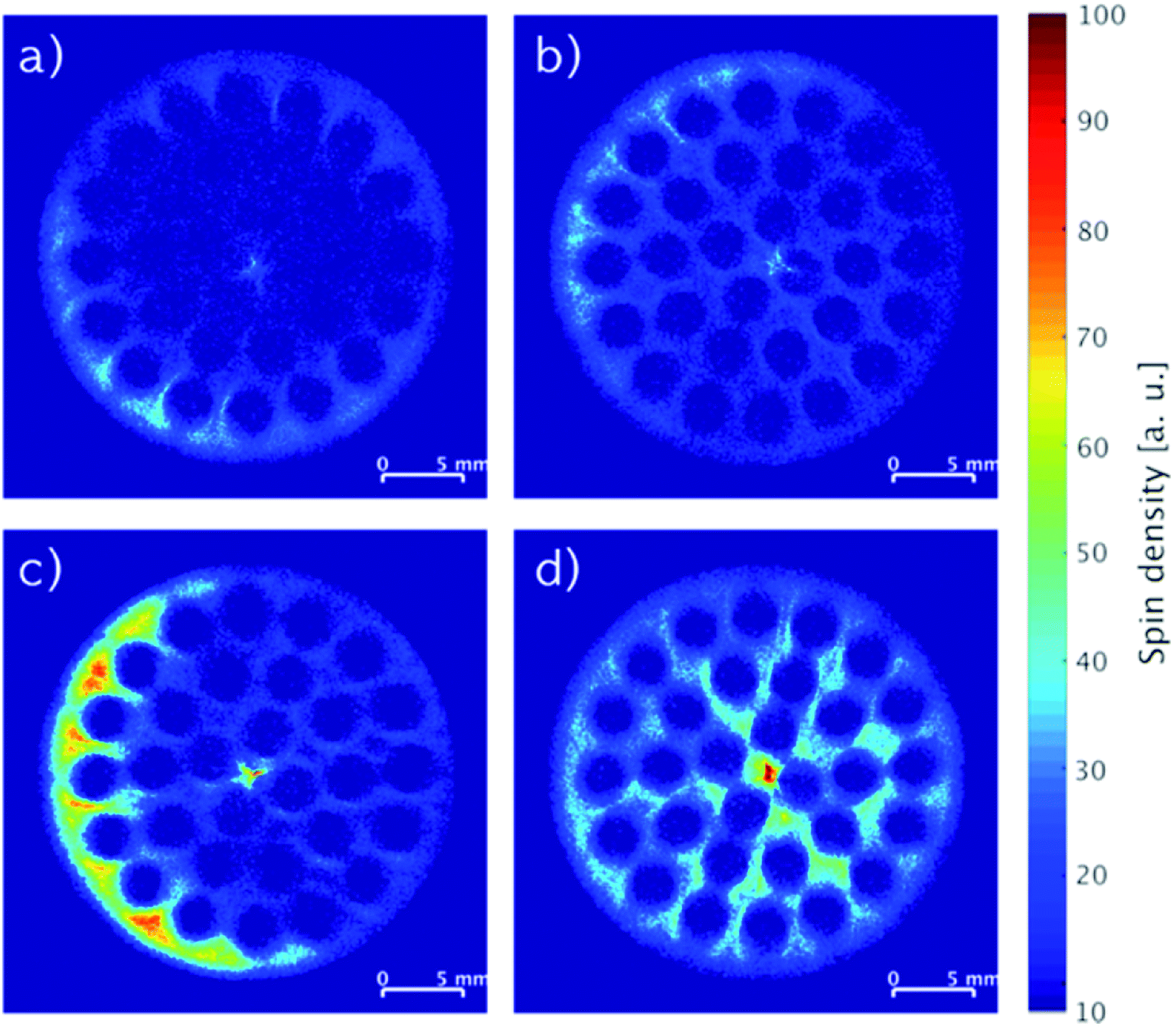 | ||
| Fig. 4 MRI images of SiC monoliths with (a) 2.74 wt% IL, (b) 2.17 wt% IL and 2.20 wt% stabilizer, (c) 5.80 wt% IL, (d) 3.73 wt% IL and 3.77 wt% stabilizer. | ||
The integrated spin density of signal from an image represents the total number of protons detected on the cross-section of the monolith. This density is proportional to the overall concentration of protons in the slice, which is related directly to the protons of the immobilized compounds represented by the mass gained by impregnation. Comparing the monoliths imaged in Fig. 4a and c, the ratio of their integrated spin densities were 1.9, which was close to their expected IL weight-ratio of 2.1. The same ratios for the monoliths with higher IL content and stabilizer (Fig. 4b and d) were both 1.7, indicating that most of the stabilizer was dissolved in the IL in these systems. Notably, the systems with stabilizer displayed also a more even distribution longitudinally over the full monolith compared to the systems containing only IL (see Fig. S4, ESI†).
Fig. 5 depicts MRI images of representative cross-sections from top to bottom at equal distances of a single monolith impregnated with 5.80 wt% IL. Except for the top part (Fig. 5a), all images showed similar patterns and yielded comparable spin density integrals (see Fig. S2, ESI†) throughout the monolith indicating an equal longitudinal distribution of the impregnation. The difference in distribution of the IL found in the top cross-section of the monolith may be a result of the drying procedure as the argon flow entered this part of the monolith first.
 | ||
| Fig. 5 Slice-selective MRI images of an impregnated SiC monolith with 5.80 wt% IL from top (a) to bottom (d) recorded at equal slice distances. | ||
The influence of gravity on the mobility of the liquid phase in the monoliths over time was investigated by acquiring cross-sectional images of horizontally stored monoliths containing 2.74 wt% IL (Fig. 6a and b) and 2.17 wt% IL with 2.20 wt% stabilizer (Fig. 6c and d). The first monolith was stored at room temperature for three weeks and the second at 100 °C for two weeks, and both analyzed by MRI before and after storage (Fig. 6). For the IL-only impregnated monolith, the patterns of the images before and after storage (Fig. 6a and b) were alike confirming a little influence by gravity for long-term storage at room temperature. On the other hand, the concentration apparently increased in the upper left part of the image of the second monolith containing the liquid IL-stabilizer phase after storage at 100 °C for two weeks compared to the monolith before storage (Fig. 6c and d). A slightly different positioning of the monolith in the MRI resonator before and after storage could account for this difference due to varying signal differences derived from the spin density integrals (see Fig. S2, ESI†). The influence of gravity was therefore also low despite the viscosity of the IL changing considerably with increasing temperature7 and melting of the stabilizer.
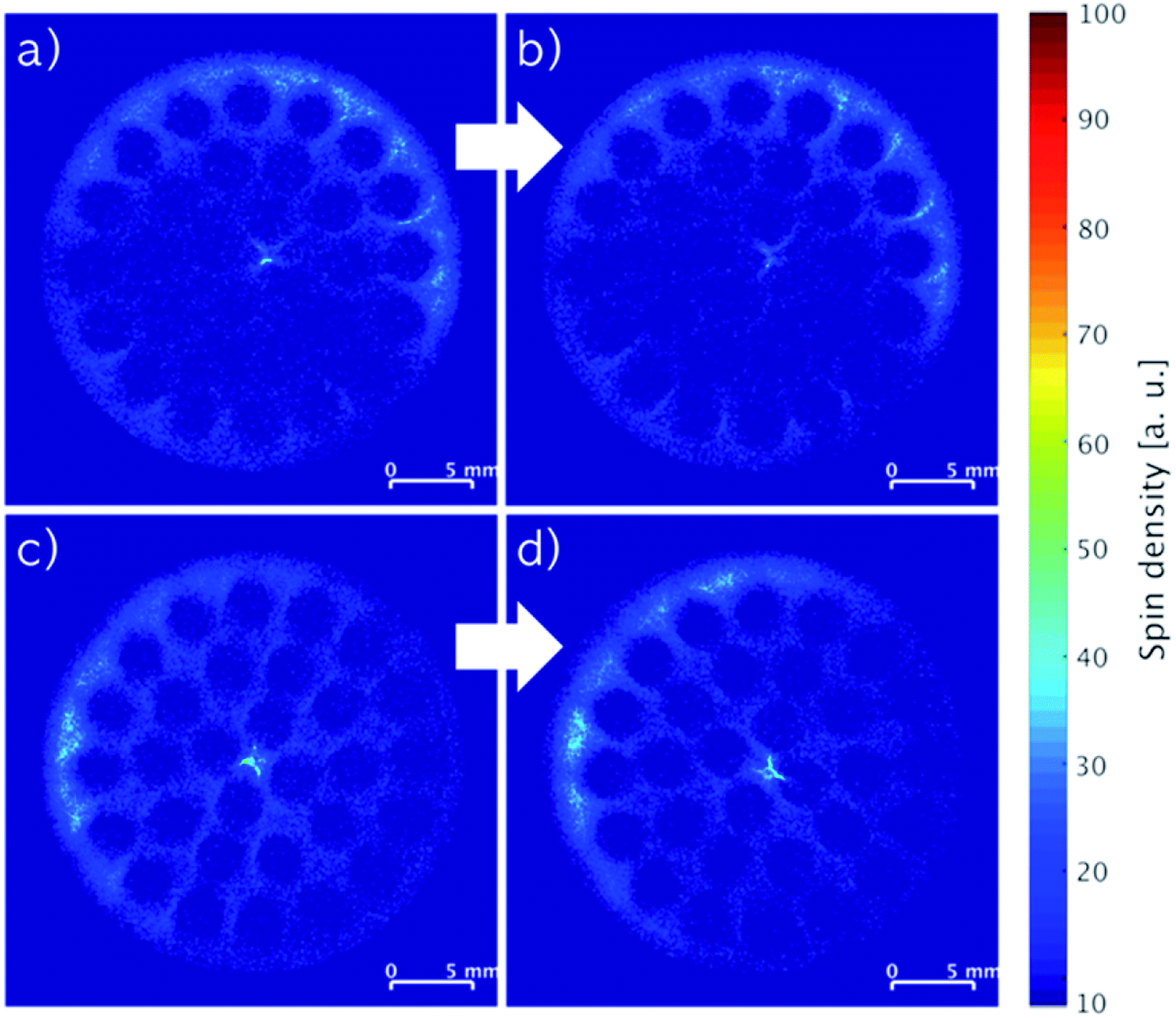 | ||
| Fig. 6 MRI images of SiC monolith with 2.74 wt% IL before (a) and after (b) horizontal storage for three weeks at room temperature, SiC monoliths with 2.17 wt% IL and 2.20 wt% stabilizer before (c) and after (d) horizontal storage for two weeks at 100 °C. | ||
The liquid phase mobility in the monoliths with 3.16 wt% IL and 3.20 wt% stabilizer (Fig. 7a) as well as 3.23 wt% IL and 3.28 wt% stabilizer (Fig. 7b) were examined by impregnating the monoliths horizontally by complete or partial (ca. 50% coverage) submersion in stock solution followed by drying. Notably, the positions of the monoliths were maintained during the impregnation and the MRI analysis, ensuring that the bottom of the images were facing towards the center of gravity. During both horizontal impregnation procedures, the monoliths were found to readily soak up the stock solution due to good wettability of polar solvents (like DCM) on SiC43 resulting in 6.4 and 6.5 wt% uptake, respectively. In line with this, images of both monoliths revealed complete impregnation of the cross-sections independent of gravity with only minor accumulation of species in the lower right part (Fig. 7a) and the upper rim (Fig. 7b), respectively. This firmly demonstrated that the orientation of the monolith during impregnation was not a key factor for obtaining an even liquid distribution with the IL-stabilizer system.
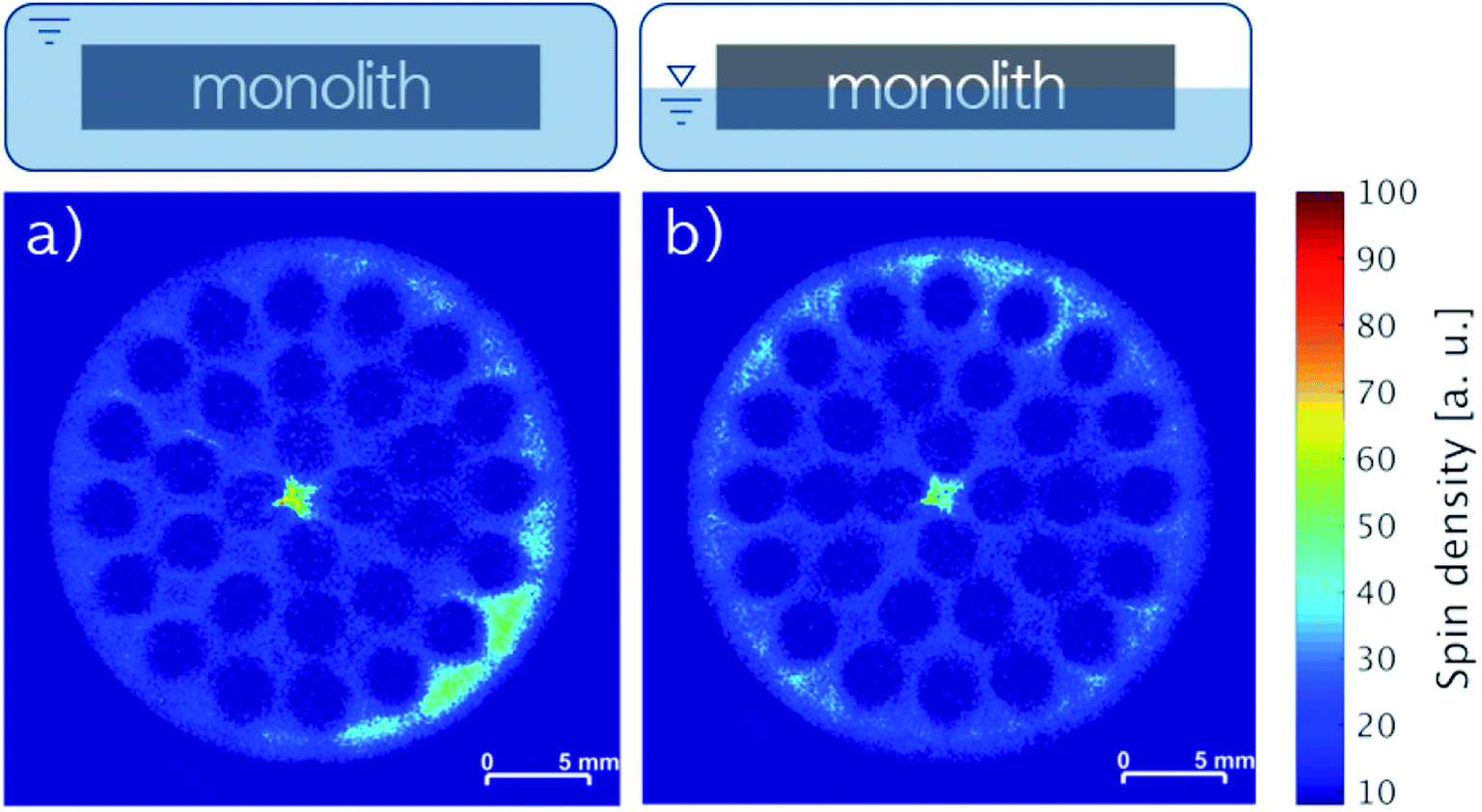 | ||
| Fig. 7 MRI images of SiC monoliths with (a) 3.16 wt% IL and 3.20 wt% stabilizer and (b) 3.23 wt% IL and 3.28 wt% stabilizer impregnated horizontally by (a) full submersion or (b) partial submersion into stock solution. | ||
Characterization by XPS
Monoliths impregnated with 2.57 wt% IL and 2.61 wt% stabilizer revealed an even distribution of impregnated species throughout the full cross-section, but some accumulation occurred as shown by MRI (Fig. 8a). To examine if the pore structure of the silica wash-coat accumulated the impregnated species by improved immobilization of the liquid phase, the monolith was laterally opened and a slice (Fig. 8b) scanned by five point-measurements for oxygen, as a representative element of the wash-coat, by XPS at the positions shown in Fig. 8c (see Fig. S5, ESI† for survey spectra). Oxygen was found at all five points with a signal above the arithmetic mean of two measurements. The recorded MRI was expected to cover the two points of the XPS measurement, which could indicate that the higher concentration of impregnated species followed an inhomogeneous distribution of the wash-coat. The atomic% of sulfur, nitrogen and fluorine were less than 5% in all measurements and the influence of the liquid phase was therefore considered negligible. It is important to mention, that the roughness of the core support material can be disadvantageous for XPS and uncertainties of the measurements were therefore present.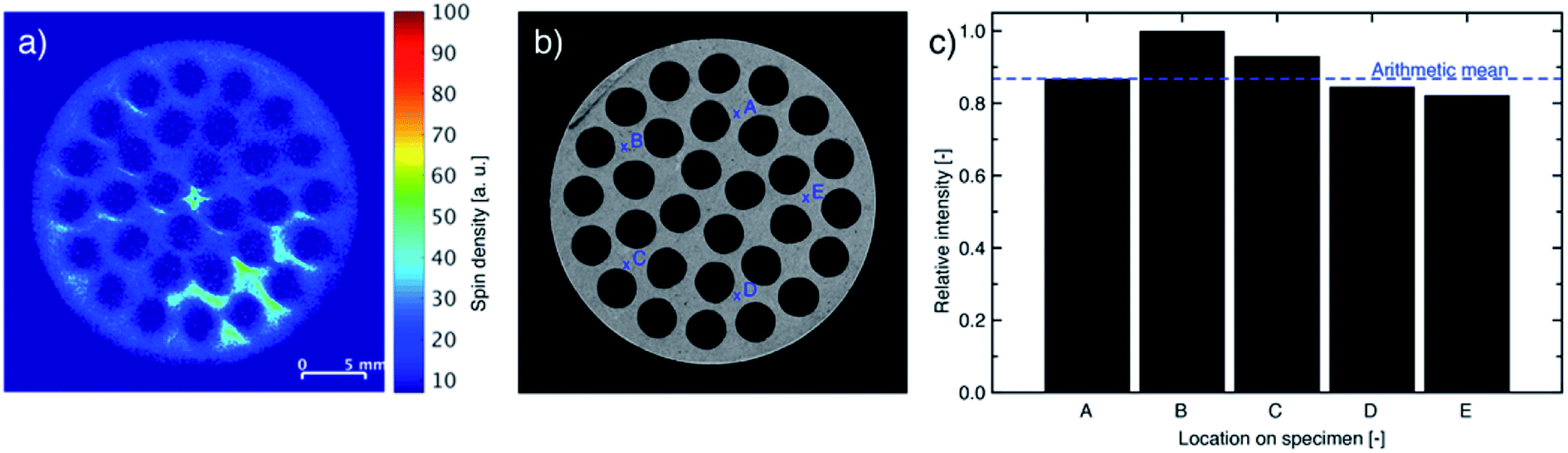 | ||
| Fig. 8 (a) MR images, (b) photograph and (c) XPS analysis for oxygen (wash-coat: silica) of slice of SiC monolith with 2.57 wt% IL and 2.61 wt% stabilizer loading using locations as indicated in (b). | ||
Catalytic performance
The performance of the monolithic SILP catalyst system impregnated with Rh–biphephos and stabilizer was tested for continuous, gas-phase hydroformylation of 1-butene. The 1-butene conversion, aldehyde and aldol yields as well as the n-pentanal selectivity (desired product) over 20 h time-on-stream are depicted in Fig. 9. A steady-state conversion of 1-butene close to 80% was obtained after an activation period of about 5 h. Importantly, the system showed high selectivity of 98% to n-pentanal product indicating formation of active Rh–biphephos complexes, while the detected aldol-condensation side products remained low around 2–3%. Noteworthy, the monolith was impregnated with a biphephos-to-Rh molar ratio of only 4:1, while the ratio reported for SILP hydroformylation catalysis in literature is usually 10:1.3 The lower ratio reduces the costs of the catalyst system as less amounts of the expensive ligand are needed, which will be important for the possible implementation in larger scale.
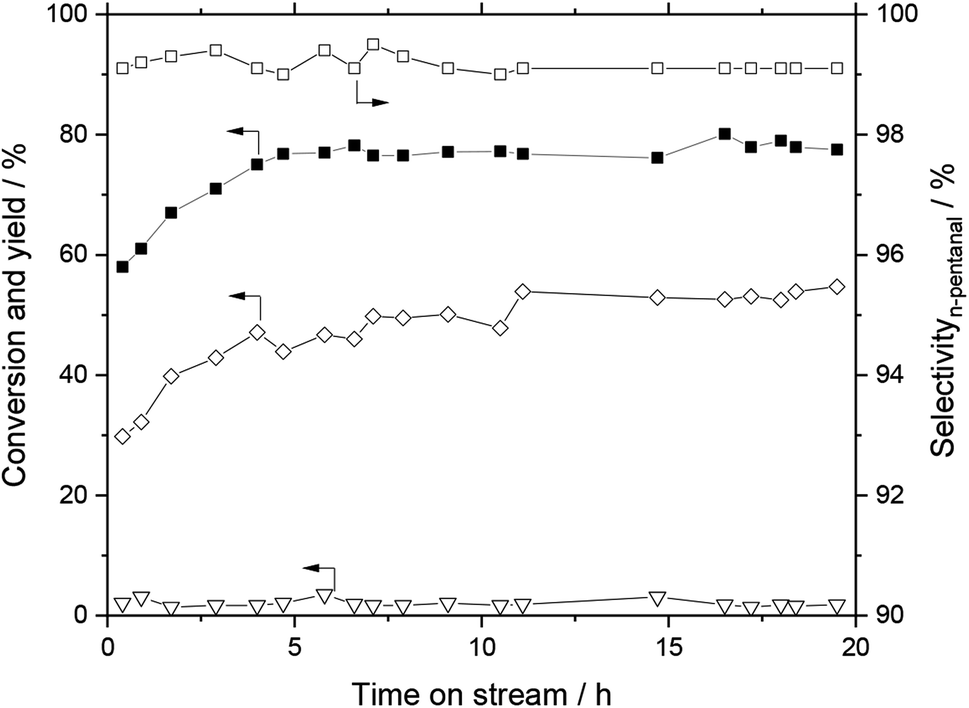 | ||
| Fig. 9 Hydroformylation of 1-butene with SiC SILP monolith. Conversion of 1-butene (■), yield of aldehydes (◇), yield of aldols (▽), n-pentanal selectivity (□). Reaction conditions: T = 110 °C, ptotal = 10 bar (H2:CO:1-butene = 2:2:1), Rh loading = 0.03 wt% (31.5 mg), biphephos loading = 0.98 wt%, [C2C1Im][NTf2] loading = 2.20 wt%, stabilizer loading = 2.40 wt% (biphephos:Rh = 4:1, stabilizer:Rh = 4:1). | ||
Conclusions
A homogeneous SILP Rh-catalyst system for gas-phase hydroformylation has been immobilized by wet-impregnation on porous SiC monoliths. The liquid-phase distribution was successfully measured by non-invasive MRI providing a close-to-reality picture of the catalyst system. The IL-phase alone formed an inhomogeneous rim on the monolith, which was attributed to the gas-flow drying-procedure during impregnation. However, addition of the stabilizer enhanced an even distribution over the full cross-section of the monolith and prevented movement of the liquid. Surprisingly, an excellent liquid-phase distribution was also obtained when the monolith was impregnated horizontally and submerged only partially into the stock solution, thus enabling impregnation by lower amounts of stock solution with the benefit of cost reduction in catalyst preparation. This is contradictory to previous studies, where continuous rotation of the monolith during drying is recommended in order to prevent movement of the liquid by gravity. Temperature studies and storage experiments with the immobilized IL-stabilizer system further revealed no negative impact on the liquid-phase distribution making the monolithic catalyst system industrially applicable.For future studies, non-invasive techniques that allow analysis of specimens on the cm-or-larger scale with high resolution on the nm-scale will be valuable as supplement to MRI with resolution on μm-scale (voxel with edges of 117.2 μm in length). Even though the costs of the introduced catalyst system are drastically reduced by the low biphephos-to-Rh molar ratio of 4:1, long-term catalytic studies and catalyst reuse evaluation will be valuable in the future to facilitate industrial implementation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the European Commission within the Horizon2020-SPIRE project ROMEO (grant agreement number 680395) and donation of modified SiC monoliths with silica wash-coat by LiqTech International A/S. C. G. is grateful for financial support from the Innovation Fund Denmark via grant agreement number 5152-00005B. Prof. Jakob Birkedal Wagner from the 3D Imaging Centre at the Technical University of Denmark is acknowledged for fruitful discussions, and the thematic DAAD Network ACalNet of student and staff exchange (https://www.acalnet.com) for support and Vicky Sun for helping carrying out some of the MRI experiments.References
- J. C. Bailar, Catal. Rev., 1974, 10, 17–36 CrossRef CAS.
- Supported Ionic Liquids - Fundamentals and applications, ed. R. Fehrmann, A. Riisager, and M. Haumann, Wiley-VCH, 2014 Search PubMed.
- J. M. Marinkovic, A. Riisager, R. Franke, P. Wasserscheid and M. Haumann, Ind. Eng. Chem. Res., 2019, 58, 2409–2420 CrossRef CAS.
- A. Riisager, P. Waasercheid, R. van Hal and R. Fehrmann, J. Catal., 2003, 219, 452–455 CrossRef CAS.
- M. Jakuttis, A. Schönweiz, S. Werner, R. Franke, K.-D. Wiese, M. Haumann and P. Wasserscheid, Angew. Chem., Int. Ed., 2011, 50, 4492–4495 CrossRef CAS PubMed.
- A. Börner and R. Franke, Hydroformylation: Fundamentals, processes, and applications in organic synthesis, Wiley-VCH, 2016 Search PubMed.
- S. Walter, M. Haumann, P. Wasserscheid, H. Hahn and R. Franke, AIChE J., 2015, 61, 893–897 CrossRef CAS.
- H. F. Rase, Chemical Reactor Design for Process Plants, Wiley New York, 1977 Search PubMed.
- J. M. Herman, P. J. van den Berg and J. J. F. Scholten, Chem. Eng. J., 1987, 34, 133–142 CrossRef CAS.
- M. J. Ledoux and C. Pham-Huu, CATTECH, 2001, 5, 226–246 CrossRef CAS.
- D. A. Reay, C. Ramshaw, and A. P. Harvey, Process Intensification. Engineering for Efficiency, Sustainability and Flexibility, 2nd edn, Oxford: Butterworth-Heinemann, 2013 Search PubMed.
- L. Montanaro, B. Coppola, P. Palmero and J.-M. Tulliani, Ceram. Int., 2019, 45, 9653–9673 CrossRef CAS.
- F. Wang, D. Yao, Y. Xia, K. Zuo, J. Xu and Y. Zeng, Ceram. Int., 2016, 42, 4526–4531 CrossRef CAS.
- R. E. Chinn, K. H. Kate and S. V. Atre, Met. Powder Rep., 2016, 71, 460–464 CrossRef.
- H. Zhong, X. Yao, Y. Zhu, J. Zhang, D. Jiang, J. Chen, Z. Chen, X. Liu and Z. Huang, J. Ceram. Sci. Technol., 2015, 6, 133–140 Search PubMed.
- M. Logemann, J. Marinkovic, M. Schörner, E. J. Garcia-Suarez, C. Hecht, R. Franke, M. Wessling, A. Riisager, R. Fehrmann and M. Haumann, 2020, submitted.
- T. A. Nijhuis, A. E. W. Beers, T. Vergunst, I. Hoek, F. Kapteijn and J. A. Moulijn, Catal. Rev., 2001, 43, 345–380 CrossRef CAS.
- P. Blümler and S. Hafner, Nuclear Magnetic Resonance, Imaging of Polymers in Encyclopedia of Analytical Chemistry, American Cancer Society, 2009 Search PubMed.
- P. Adriaensens, L. Storme, R. Carleer, J. D'Hae, J. Gelan, V. M. Litvinov, R. Marissen and J. Crevecoeur, Macromolecules, 2002, 35, 135–140 CrossRef CAS.
- L. Borisjuk, H. Rolletschek and T. Neuberger, Plant J., 2012, 70, 129–146 CrossRef CAS PubMed.
- V. De Schepper, D. van Dusschoten, P. Copini, S. Jahnke and K. Steppe, J. Exp. Bot., 2012, 63, 2645–2653 CrossRef CAS PubMed.
- H. Van As and J. van Duynhoven, J. Magn. Reson., 2013, 229, 25–34 CrossRef CAS PubMed.
- S. Benders and B. Blümich, Phys. Sci. Rev., 2019, 4, 1–17 Search PubMed.
- D. A. Clarke, A. J. Sederman, L. F. Gladden and D. J. Holland, Ind. Eng. Chem. Res., 2018, 57, 3002–3013 CrossRef CAS.
- A. Adair, I. V Mastikhin and B. Newling, Magn. Reson. Imaging, 2018, 49, 71–77 CrossRef PubMed.
- A. B. Tayler, D. J. Holland, A. J. Sederman and L. F. Gladden, J. Magn. Reson., 2011, 211, 1–10 CrossRef CAS PubMed.
- S.-I. Han, S. Stapf and B. Blümich, Phys. Rev. Lett., 2001, 87, 144501 CrossRef CAS PubMed.
- A. Amar, B. Blümich and F. Casanova, ChemPhysChem, 2010, 11, 2630–2638 CrossRef CAS PubMed.
- M. Wiese, S. Benders, B. Blümich and M. Wessling, Chem. Eng. J., 2018, 343, 54–60 CrossRef CAS.
- L. F. Gladden and A. J. Sederman, J. Magn. Reson., 2012, 229, 2–11 CrossRef PubMed.
- S. Benders, F. Strassl, B. Fenger, B. Blümich, S. Herres-Pawlis and M. Küppers, Magn. Reson. Chem., 2018, 56, 826–830 CrossRef CAS PubMed.
- R. Evans, C. R. Timmel, P. J. Hore and M. M. Britton, Chem. Phys. Lett., 2004, 397, 67–72 CrossRef CAS.
- M. E. Skuntz, D. Perera, J. E. Maneval, J. D. Seymour and R. Anderson, Chem. Eng. Sci., 2018, 190, 164–172 CrossRef CAS.
- K. R. Brownstein and C. E. Tarr, J. Magn. Reson., 1977, 26, 17–24 CAS.
- M. Li, D. Xiao, L. Romero-Zerón, F. Marica, B. MacMillan and B. J. Balcom, J. Magn. Reson., 2016, 269, 13–23 CrossRef CAS PubMed.
- N. Proietti, D. Capitani, S. Cozzolino, M. Valentini, E. Pedemonte, E. Princi, S. Vicini and A. L. Segre, J. Phys. Chem. B, 2006, 110, 23719–23728 CrossRef CAS PubMed.
- S. Merz, B. J. Balcom, R. Enjilela, J. Vanderborght, Y. Rothfuss, H. Vereecken and A. Pohlmeier, Vadose Zone J., 2018, 17, 1–15 CrossRef CAS.
- A. K. Heibel, F. J. Vergeldt, H. van As, F. Kapteijn, J. Moulijn and T. Boger, AIChE J., 2003, 49, 3007–3017 CrossRef CAS.
- A. K. Heibel, T. W. J. Scheenen, J. J. Heiszwolf, H. Van As, F. Kapteijn and J. A. Moulijn, Chem. Eng. Sci., 2001, 56, 5935–5944 CrossRef CAS.
- S. Emid and J. H. N. Creyghton, Phys. B+C, 1985, 128, 81–83 CrossRef CAS.
- P. Stobbe and U. Haok, US Pat. US2006/0236668A1, 2006.
- M. P. McDaniel and T. D. Hottovy, J. Colloid Interface Sci., 1980, 78, 31–36 CrossRef CAS.
- V. Médout-Marère, A. El Ghzaoui, C. Charnay, J. M. Douillard, G. Chauveteau and S. Partyka, J. Colloid Interface Sci., 2000, 15, 205–234 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09515b |
| ‡ Current address: Tecnalia, Energy and Environment Division, Parque Tecnológico de Álava, Leonardo Da Vinci, 11, 01510 Miñano, Spain. |
| § Current address: IKERBASQUE, Basque Foundation for Science, Maria Diaz de Haro 3, 48013 Bilbao, Spain. |
| This journal is © The Royal Society of Chemistry 2020 |