
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-pressure formation of antimony nitrides: a first-principles study†
Lili Liana,
Yan Liub,
Da Li *b and
Shuli Wei*c
*b and
Shuli Wei*c
aThe First Hospital of Jilin University, Jilin University, Changchun, P. R. China
bState Key Laboratory of Superhard Materials, Jilin University, Changchun 130012, China. E-mail: dali@jlu.edu.cn
cSchool of Physics and Optoelectronic Engineering, Shandong University of Technology, Zibo 255049, China. E-mail: weishuli@sdut.edu.cn
First published on 14th January 2020
Abstract
The structural phase transition, electronic properties, and bonding properties of antimony nitrides have been studied by using the first principles projector augmented wave method. The relationship between the formation enthalpy and the composition of the Sb–N system has been explored. The novel Sb2N3 with the Cmcm space group is stable in a narrow pressure range from 100 GPa to 120 GPa. Apart from the Sb2N3, two nitrogen-rich phases SbN2 and SbN4 were predicted. The SbN2 with the C2/m space group is stable at 12 GPa and then transforms to the high-pressure phase at 23 GPa. The nitrogen-rich SbN4 appears at 14 GPa then undergoes C2/m → P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) → P phase transitions, and the calculated pressures of the phase transitions are 31 and 60 GPa, respectively. The nitrogen-rich SbN2 and SbN4 have similar structural features. Both SbN2 and SbN4 can be seen as a sandwich structure composed of the Sb–N layers and N2 dimers. The pressure-induced phase transitions of SbN2 and SbN4 are accompanied by the electron transfer between the Sb–N layers and N2 dimers. Moreover, the nitrogen-rich SbN4 has a higher energy density of 2.42 kJ g−1 and is a potentially high energy density material.
→ P phase transitions, and the calculated pressures of the phase transitions are 31 and 60 GPa, respectively. The nitrogen-rich SbN2 and SbN4 have similar structural features. Both SbN2 and SbN4 can be seen as a sandwich structure composed of the Sb–N layers and N2 dimers. The pressure-induced phase transitions of SbN2 and SbN4 are accompanied by the electron transfer between the Sb–N layers and N2 dimers. Moreover, the nitrogen-rich SbN4 has a higher energy density of 2.42 kJ g−1 and is a potentially high energy density material.
Introduction
The single-bonded cubic gauche cg-N is the most famous polymeric nitrogen phase which was first predicted by a theoretical study, then experimentally synthesized by Eremets et al. by using a high-pressure and high-temperature method (P = 100 GPa and T = 2000 K).1 Previous studies report that the single-bonded cg-N has more than three times higher energy storage capacity than traditional energetic materials. It is well known that the polynitrogens can transform to N2 molecules under ambient conditions, accompanied by large energy release. There is a large energetic gap between a single bond (∼167 kJ mol−1) and a double/triple bond (∼419 kJ mol−1 and ∼954 kJ mol−1). The polynitrogen materials composed of abundant single and double bonds can be seen as high energy density materials (HEDMs). Although the cg-N is a potential high energy density material, it is predicted to be metastable at 40 GPa and it cannot be quenched under ambient conditions. This is a big problem for practical application. To solve this problem, many polynitrogen materials with lower synthesis pressures and good stabilities have attracted great attention in scientific and industrial research. Firstly, the metal azides have drawn considerable attention due to their unique covalent linear N3 units in the crystal structures.2–6 These covalent linear N3 units have the ability to improve the speed of polymerization and lower the synthesis pressures with respect to the pure nitrogen gas. The metal azides are good starting materials in the high-pressure synthesization of the polynitrogens. Over the years, many metal azides have been studied as HEDMs such as LiN3, NaN3, KN3, AlN3 and so on.2,7–13 Furthermore, some nitrogen-rich metal nitrides with higher energy density such as LiN5, CsN5, BeN4, MgN4, CaN4, and CaN5 have also been proposed in experimental and theoretical research.9,14–21 Recently, in addition to metal nitrides, many non-metal nitrides have also been studied as HEDMs. In the N–H system, a novel N2H compound with higher energy density (∼4.4 kJ g−1) and higher nitrogen content (96.6%) has been predicted by Yin et al. using the CALYPSO method.22 Batyrev predicted three stable crystalline structures of N–H solids N9H, N4H, and N3H at the pressure range of 10 and 50 GPa.23 Using the USPEX method, Steele theoretically found two novel compounds ammonium pentazolate (NH4)(N5) and pentazole (N5H) at 30 and 50 GPa, respectively.24 In the B–N system, it is well known that only one stable compound BN is reported in the B–N system. However, an unusual B3N5 with high hardness (∼44 GPa) and high energy density (∼3.44 kJ g−1) had been predicted by Li et al.25 Besides, novel high-pressure compounds in the C–N system and S–N system are also considered in the exploration of high-energy materials.26,27 Up to now, several binary systems have been studied. And some new polynitrogen forms including tetrazadiene (N4), pentazole (N5), hexazine (N6) and extended chains (N∞) have been reported in these binary systems. However, there are little reports about the pnictogen nitrides, especially in high-pressure studies. Only one report about the P–N system in the high-pressure studies of pnictogen nitrides was published.28 In the P–N system, two novel compounds PN2 and PN3 were predicted at high pressures. The metallic PN3 is a superconductor at 10 GPa with a Tc value of 18 K. The insulating PN2 is stable at pressures in excess of 200 GPa. The PN3 has the highest nitrogen content in the P–N compounds. However, only N2 dimers appear and no polynitrogen forms are observed in the structure of PN3. Previous reported N4, N5, and N6 units have not been observed in the P–N system. For pnictogen nitrides, the pnictogen elements (As, Sb, and Bi) have the same valence electrons number as that of nitrogen but with a big atomic radius. Therefore, pnictogen elements can enhance the chemical precompression in the pnictogen nitrides while do not change the charge of compounds. Therefore, it can be expected that there are high energy density materials in the pnictogen nitrides. The high-pressure studies of pnictogen nitrides would help to theoretically investigate more promising high-energy materials. These stimulate our interest to prompt in-depth investigations of pnictogen nitrides.In the present work, we did systemic theoretical study of Sb–N system at high pressure. The formation enthalpy and structural stability of SbxNy have been well investigated. Three novel stable compound Sb2N3, SbN2 and SbN4 have been found. The Sb2N3 with the Cmcm space group is stable in a narrow pressure range from 100 GPa to 120 GPa. The structural phase transition of SbN2 under high pressure has been uncovered. The monoclinic SbN2 with C2/m space group is stable at 12 GPa and then transform to high-pressure phase with the same space group at 23 GPa. A nitrogen-rich stoichiometric SbN4 with the C2/m space group appears at 14 GPa then undergo two phase transitions to low symmetry P space group phases at 31 GPa and 60 GPa, respectively. The pressure-induced phase transitions of SbN2 and SbN4 are accompanied by the electrons transfer between the Sb–N layers and N2 dimers. Furthermore, we found that the nitrogen-rich SbN4 phase has a higher energy density of 2.42 kJ g−1.
Computation details
Hear, the theoretical calculations of Sb–N system are performed within the density functional theory (DFT),29–31 carried out within the Vienna ab initio simulation code VASP, with the projector augmented wave method.32,33 The Sb 5s25p3 electrons and N 2s22p3 electrons are treated as valence electrons, respectively. The Perdew–Burke–Ernzerhof generalized gradient approximation (GGA) exchange and correlation functional are used in our calculations.34 Convergence tests give a kinetic energy cutoff of 800 eV, with a grid of spacing 2π × 0.03 Å−1 for the electronic Brillouin zone integration in all the candidate structures. The geometries of the candidate structures are optimized when the remanent Hellmann–Feynman forces on the ions are less than 0.01 eV Å−1. The phonon frequencies are calculated by using the supercell approach as implemented in the Phonopy code35 and VASP code. The heat of formation (ΔHf) for various SbxNy compositions are calculated by the equation of ΔHf = E(SbxNy) − (xE(Sb) + yE(N)), in which the respective solid phase of Sb and N at different pressures are adopted.Results and discussions
The variable-cell high-pressure structure predictions of the Sb–N system were performed at 0, 20, 40, 60, 80, 100, 120 GPa with the simulation cell containing one to four formula units by using the CALYPSO code.36,37 The ground-state crystal structures of the antimony nitrides with various stoichiometry were uncovered in the pressure range of 0 to 120 GPa. The convex hull plots of the Sb–N system at the selected pressures were constructed by evaluating the averaged formation enthalpy of the candidate structures relative to the elementary Sb and N solids (Fig. 1). According to the hull data, a compound with a formation enthalpy lying on the convex hull is thermodynamically stable with respect to decomposition into other nitrides or elementary Sb and N solids. We can expect that this compound is experimentally synthesizable at their stable pressures.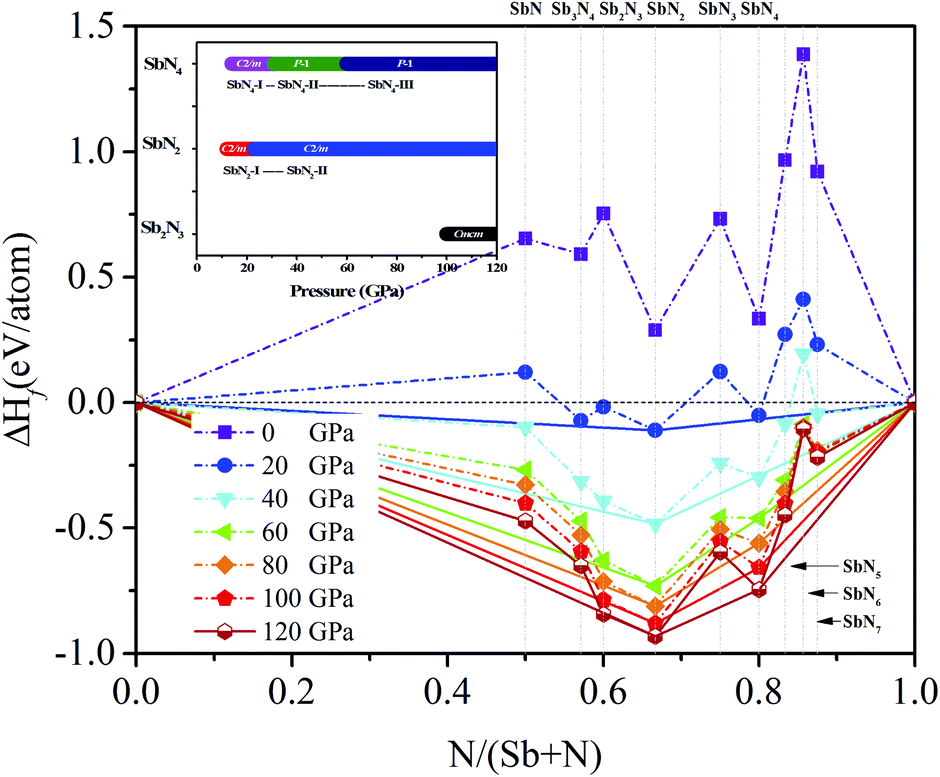 | ||
| Fig. 1 The convex hull plot of Sb–N system at different pressures. Dotted lines are used to connect the stable phases. The inset is the phase transition sequence of the Sb–N compounds. | ||
It is found that three stable stoichiometry (Sb2N3, SbN2, and SbN4) appear in our convex hull plots. The SbN2 is the most stable stoichiometry in the Sb–N system. The Sb2N3 with the Cmcm space group is stable in a narrow pressure range of from 100 GPa to 120 GPa. The monoclinic SbN2 with the C2/m space group (SbN2-I) is stable at 12 GPa and then transform to high-pressure SbN2 (SbN2-II) with the same space group at 23 GPa. A nitrogen-rich stoichiometric SbN4 with the C2/m space group (SbN4-I) appears at 14 GPa then undergo two-phase transitions to the low symmetry P space group at 31 GPa (SbN4-II) and 60 GPa (SbN4-III), respectively. From our calculated convex hull data, it is found that the nitrogen-rich SbNx (x = 5, 6, 7) are also not stable because their formation enthalpy values lie above the convex hull solid lines. Furthermore, there are no stable antimony-rich nitrides in the Sb–N system.
In addition to the thermodynamic stability, we also study the dynamic stabilities of these structures. The phonon spectra are calculated under different pressure conditions. No imaginary frequencies emerge in the whole Brillouin zone of all the predicted phases, indicating the dynamical stability as shown in Fig. 2. At 20 GPa, SbN2-I and SbN4-I have some extremely flat phonon bands at higher frequencies of 60–70 THz, indicating the presence of molecular modes of N2 dimer. In the following part, we mainly study the effect of redundant nitrogen atoms in the Sb–N system. The phase stability, high-pressure phase transition, bonding properties, and potential energy storage applications of antimony nitrides will be discussed.
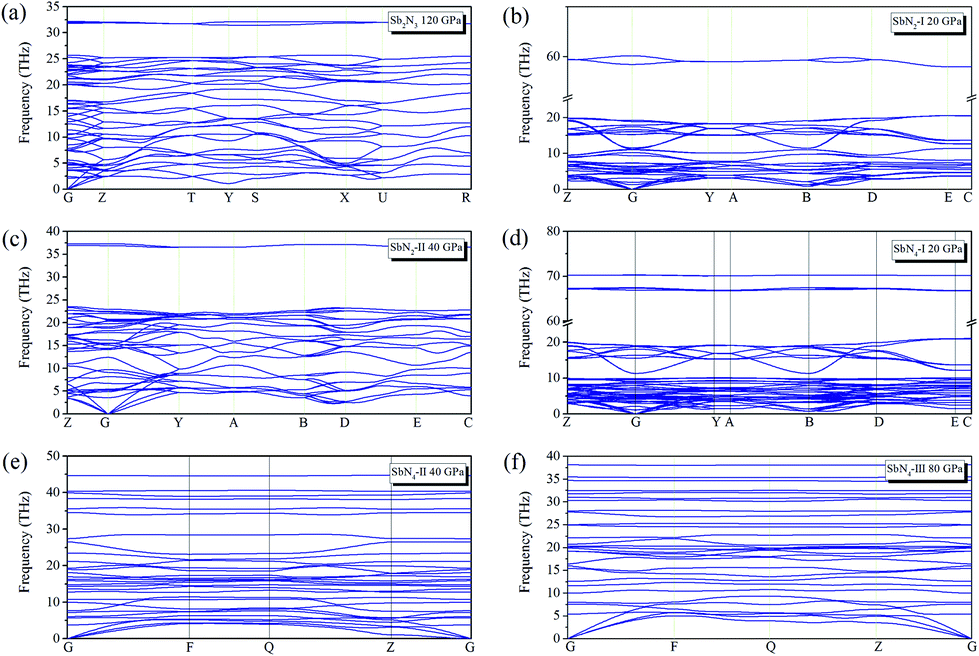 | ||
| Fig. 2 Phonon band structures of (a) Sb2N3 at 120 GPa (b) SbN2-I at 20 GPa (c) SbN2-II at 40 GPa (d) SbN4-I at 20 GPa (e) SbN4-II at 40 GPa (f) SbN4-III at 80 GPa. | ||
The crystal structures of Sb2N3, SbN2, and SbN4 are presented in Fig. 3. The crystallographic parameters of the Sb–N compounds are provided in Table S1.† The high-pressure Sb2N3 adopts an orthorhombic Cmcm-symmetric structure at 120 GPa as shown in Fig. 3a. The equilibrium lattice parameters of Sb2N3 are a = 2.859 Å, b = 8.929 Å, c = 7.002 Å at 120 GPa. The coordination number of Sb atoms is six. While the coordination numbers of N atoms are four and five. There are two type of bonding mode tetrahedron and director plane. SbN2-I crystallizes with the monoclinic symmetry C2/m space group at 12 GPa. SbN2-I can be regarded as a sandwich-type structure, composed of Sb–N layer (A layer) and N2 dimer layer (B layer) with the stacking order of ABABA… along the crystallographic a axis of the monoclinic lattice. The distance between A layers is about 3 Å at 20 GPa. The distances between the Sb and N atoms in A layer are 2.105 Å and 2.183 Å at 20 GPa. The bond length of the N2 dimer is 1.145 Å. Under compression, the distance between the A layer and B layer decreases greatly. New interaction between A layer and B layer appears with the distance decreasing. The N2 dimer gets more electron from the Sb atoms of A layer. New ionic bonds between Sb atoms of A layer and N atoms of B layer appears. Then the SbN2-I phase transforms into the high-pressure SbN2-II phase at 23 GPa. In SbN2-II, due to the presence of new ionic bonds between the Sb atoms of A layer and N atoms of B layer, the structure of A layer occurs distortion to make the crystal structure reach a new mechanical balance. The nitrogen-rich phase SbN4-I adopts the monoclinic C2/m symmetric structure at 14 GPa. SbN4-I has a similar crystal structure with that of SbN2-I. The similar sandwich structure feature appears in the SbN4-I. SbN4-I is composed of Sb–N layer (A layer) and N2 dimer layer (B layer) with the stacking order of ABBBABBB… along the crystallographic a axis of monoclinic lattice. Within this structure, the distance between the A layer and A layer is about 8.5 Å. The distances between the Sb and N atoms in the A layer are 2.105 Å and 2.208 Å. The bond lengths of N2 dimer are 1.113 Å and 1.123 Å. Under compression, the distance between two neighbouring A layers decreases greatly. The N2 dimer gets close to each other. At 31 GPa, the N2 dimers transform to the novel quasi-linear nitrogen armchair chains along the crystallographic [110] direction. The bond lengths of the nitrogen–nitrogen bonds are 1.294 Å, 1.311 Å, and 1.336 Å at 40 GPa. The remanent electrons of Sb atom in the A layers transform into the nitrogen armchair chains. However, the only partial nitrogen atom of the armchair chains form ionic bonds with the Sb atoms of A layers. There are lone pair electrons in the nitrogen armchair chains. The SbN4-I transforms into the P phase SbN4-II at 31 GPa. At higher pressures, the SbN4-II phase transforms into a closed-packed triclinic SbN4-III phase at 60 GPa. The remanent electrons of nitrogen armchair chains forms ionic bonds with the neighboring Sb atoms of A layers in SbN4-III phase.
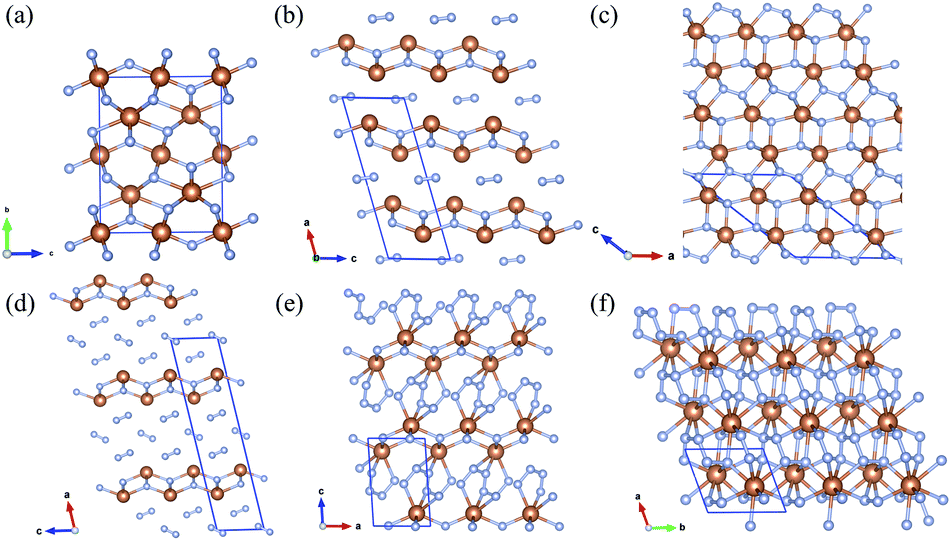 | ||
| Fig. 3 The crystal structures of antimony nitrides (a) Sb2N3 (b) SbN2-I (c) SbN2-II (d) SbN4-I (e) SbN4-II (f) SbN4-III. The golden yellow circles represent Sb atoms; the blue circles represent N atoms. | ||
To investigate the nature of the polymerization and formation mechanism of materials, we calculated the electronic structures and Bader charge of the Sb2N3, SbN2, and SbN4. The projected density of states of these compounds at different pressures is shown in Fig. 4. It is shown that the Sb2N3 exhibits a clear metallic feature by the evidence of the finite electronic DOS at the Fermi level. The Sb_s states mainly contribute to the metallic properties of Sb2N3. We also calculated the contributions of the Sb–N layer and N2 dimers or nitrogen armchair chain for the DOS as shown in Fig. S1.† We can find that the N2 dimer and nitrogen armchair chain have a bigger contribution to the DOS of these compounds. Furthermore, with the pressure increasing more Sb_d states appear in the energy range of −5 and 0 eV in the PDOS of SbN2-I, SbN2-II, SbN4-I, SbN4-II, and SbN4-III. This indicates that there is more charge transfer between Sb atoms and N2 dimers in these nitrogen-rich phases. In addition, in order to better understand the bonding properties between the Sb–N layer and N2 dimers, we investigated the charge density difference of the nitrogen-rich Sb–N compounds. Fig. 5 shown the charge density difference between the Sb–N layer and N2 dimers for the SbN2-I, SbN2-II, SbN4-I, and SbN4-II. For SbN2, upon the compression, it is found that the electrons localized in the nitrogen–nitrogen bonds of the N2 dimers transfer to form ionic bonds with the Sb–N layers. For SbN4, the electrons of the Sb atoms in the Sb–N layers transfer to the N2 dimers. The Bader charge analysis also confirms this view. Each nitrogen atom of the N2 dimer gets 0.235 electrons from the Sb–N layer in SbN2-I. For SbN2-II, each nitrogen atom of the N2 dimer gets 0.94 electrons from the Sb–N layer. For SbN4-I, each nitrogen atom of the N2 dimer gets 0.04 electrons from the Sb–N layer. For SbN4-II, each nitrogen atom of the armchair chain gets 0.288 electrons from the Sb–N layer. These suggest that these phases have ionic characteristics. The Sb–N layers behave as electron-donors whose strongly affect the N–N bonding of the polymerization.
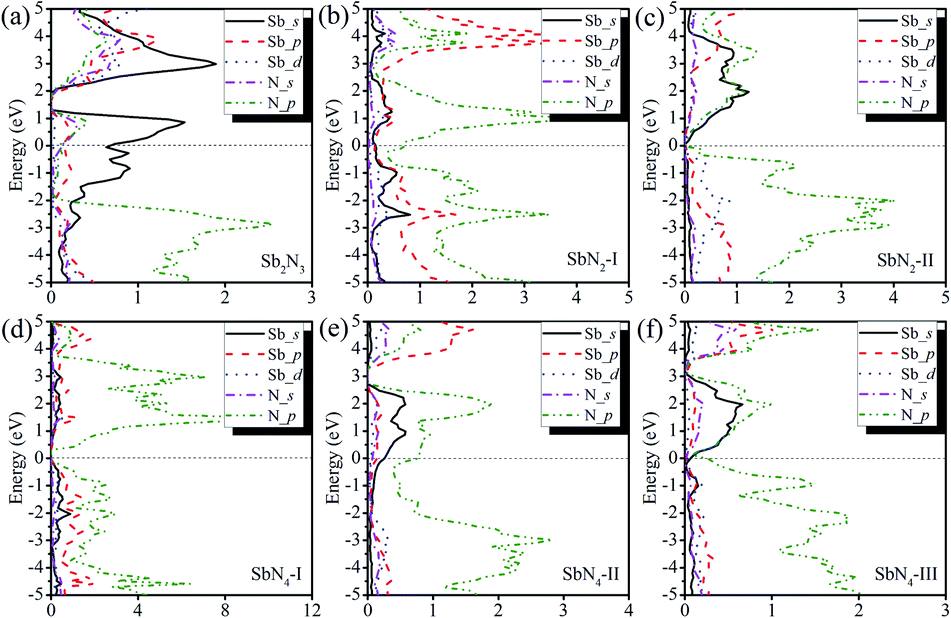 | ||
| Fig. 4 The projected density of state of (a) Sb2N3 at 120 GPa (b) SbN2-I at 20 GPa (c) SbN2-II at 40 GPa (d) SbN4-I at 20 GPa (e) SbN4-II at 40 GPa (f) SbN4-III at 80 GPa. | ||
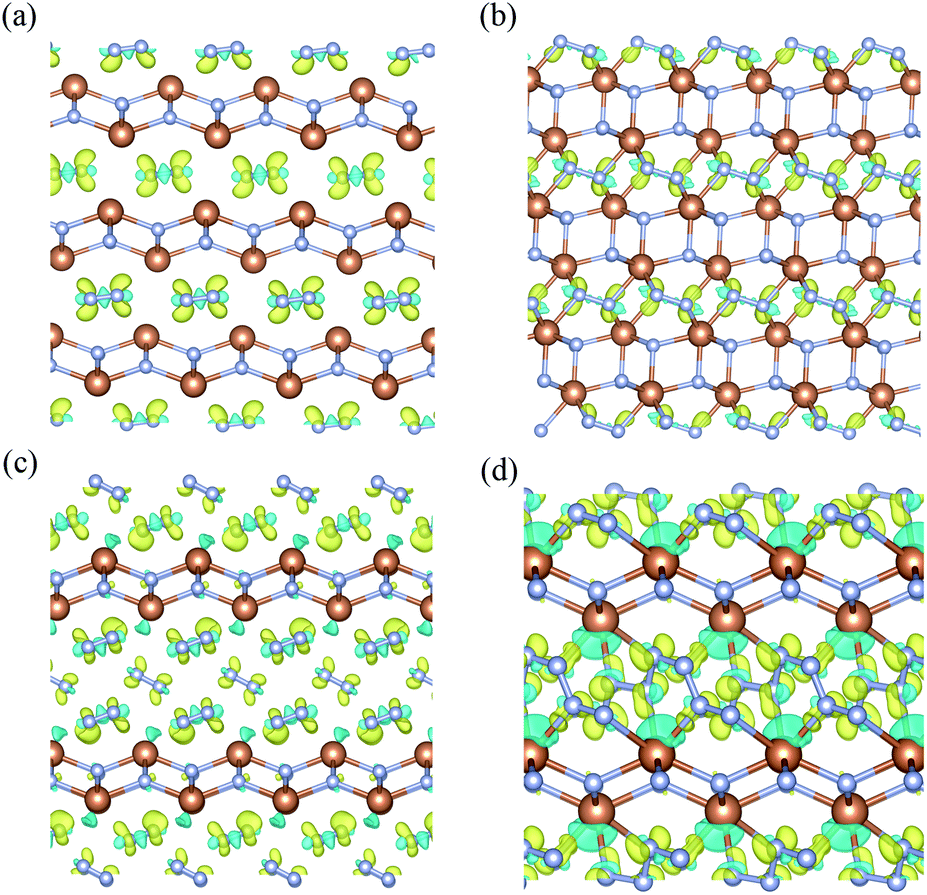 | ||
| Fig. 5 The charge density difference between the Sb–N layer and N2 dimers for (a) SbN2-I with the isosurface level of 0.004 e bohr−3 (b) SbN2-II with the isosurface level of 0.023 e bohr−3 (c) SbN4-I with the isosurface level of 0.003 e bohr−3 (d) SbN4-II with the isosurface level of 0.014 e bohr−3. The yellow represents the positive value and the green represents the negative value. | ||
Moreover, it is well known that the nitrogen-rich materials are considered to be promising candidates for HEDMs because their decomposition reactions will release large energy. Different from the phosphorus nitrides, SbN4 has the highest nitrogen contents among the Sb–N compounds and the polymeric nitrogen chains. The calculated energy density of SbN4, relative to products N2 and Sb solids, is 2.42 kJ g−1 which is larger than that of typical covalent high energy density material CNO (2.2 kJ g−1)38 and is comparable with other traditional HEDMs (TATB, RDX, and HMX with energy densities of 1 to 3 kJ g−1).39 So SbN4 can be expected as a possible candidate of HEDMs.
Conclusions
In conclusion, the high-pressure ground-state phases of the stoichiometric binary antimony nitrides are extensively explored. The formation enthalpy and structural stability of the candidate structures have been well investigated. Three novel stable compound Sb2N3, SbN2, and SbN4 have been predicted. The Sb2N3 with the Cmcm space group is stable in a narrow pressure range from 100 GPa to 120 GPa. The nitrogen-rich SbN2 and SbN4 have similar structural features. Both SbN2 and SbN4 can be seen as a sandwich structure composed of the Sb–N layers and N2 dimers. Furthermore, the structural phase transition of SbN2 under high pressure has been uncovered. The SbN2-I is stable at 12 GPa and then transforms to a high-pressure SbN2-II phase at 23 GPa. The nitrogen-rich SbN4-I appears at 14 GPa then undergoes two phase transitions to low symmetry P space group SbN4-II and SbN4-III at 31 GPa and 60 GPa, respectively. The pressure-induced phase transitions of SbN2 and SbN4 are accompanied by the electrons transfer between the Sb–N layers and N2 dimers. Moreover, the nitrogen-rich SbN4 can be expected to be a potential high energy density material (energy density: 2.42 kJ g−1). The established high-pressure phase diagram of the Sb–N system is of fundamental interest and important for future experiments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 91745203, 11404134), Jilin Provincial Science and Technology Development Project of China (20160520016JH).References
- M. I. Eremets, A. G. Gavriliuk, I. A. Trojan, D. A. Dzivenko and R. Boehler, Nat. Mater., 2004, 3, 558–563 CrossRef CAS PubMed.
- X. Wang, J. Li, H. Zhu, L. Chen and H. Lin, J. Chem. Phys., 2014, 141, 044717 CrossRef PubMed.
- J. Jiang, P. Zhu, D. Li, Y. Chen, M. Li, X. Wang, B. Liu, Q. Cui and H. Zhu, J. Phys. Chem. B, 2016, 120, 12015–12022 CrossRef CAS PubMed.
- H. Zhu, F. Zhang, C. Ji, D. Hou, J. Wu, T. Hannon and Y. Ma, J. Appl. Phys., 2013, 113, 033511 CrossRef.
- H. Zhu, X. Han, P. Zhu, X. Wu, Y. Chen, M. Li, X. Li and Q. Cui, J. Phys. Chem. C, 2016, 120, 12423–12428 CrossRef CAS.
- D. Li, X. Wu, J. Jiang, X. Wang, J. Zhang, Q. Cui and H. Zhu, Appl. Phys. Lett., 2014, 105, 071903 CrossRef.
- N. Holtgrewe, S. S. Lobanov, M. F. Mahmood and A. F. Goncharov, J. Phys. Chem. C, 2016, 120, 28176–28185 CrossRef CAS.
- M. Zhang, H. Yan, Q. Wei and H. Liu, RSC Adv., 2015, 5, 11825–11830 RSC.
- J. Zhang, Z. Zeng, H.-Q. Lin and Y.-L. Li, Sci. Rep., 2014, 4, 4358 CrossRef PubMed.
- F.-C. Liu, Y.-F. Zeng, J.-P. Zhao, B.-W. Hu, E. C. Sañudo, J. Ribas and X.-H. Bu, Inorg. Chem., 2007, 46, 7698–7700 CrossRef CAS.
- W. Zhu and H. Xiao, J. Phys. Chem. B, 2006, 110, 18196–18203 CrossRef CAS.
- M. I. Eremets, M. Y. Popov, I. A. Trojan, V. N. Denisov, R. Boehler and R. J. Hemley, J. Chem. Phys., 2004, 120, 10618–10623 CrossRef CAS.
- Z. Liu, D. Li, S. Wei, W. Wang, F. Tian, K. Bao, D. Duan, H. Yu, B. Liu and T. Cui, Inorg. Chem., 2017, 56, 7494–7500 CrossRef CAS.
- S. Zhang, Z. Zhao, L. Liu and G. Yang, J. Power Sources, 2017, 365, 155–161 CrossRef CAS.
- S. Zhu, F. Peng, H. Liu, A. Majumdar, T. Gao and Y. Yao, Inorg. Chem., 2016, 55, 7550–7555 CrossRef CAS PubMed.
- Y. Shen, A. R. Oganov, G. Qian, J. Zhang, H. Dong, Q. Zhu and Z. Zhou, Sci. Rep., 2015, 5, 14204 CrossRef.
- F. Peng, Y. Yao, H. Liu and Y. Ma, J. Phys. Chem. Lett., 2015, 2363–2366 CrossRef CAS PubMed.
- F. Peng, Y. Han, H. Liu and Y. Yao, Sci. Rep., 2015, 5, 16902 CrossRef CAS.
- S. Wei, D. Li, Z. Liu, X. Li, F. Tian, D. Duan, B. Liu and T. Cui, Phys. Chem. Chem. Phys., 2017, 19, 9246–9252 RSC.
- S. Yu, B. Huang, Q. Zeng, A. R. Oganov, L. Zhang and G. Frapper, J. Phys. Chem. C, 2017, 121, 11037–11046 CrossRef CAS.
- S. Wei, D. Li, Z. Liu, W. Wang, F. Tian, K. Bao, D. Duan, B. Liu and T. Cui, J. Phys. Chem. C, 2017, 121, 9766–9772 CrossRef CAS.
- K. Yin, Y. Wang, H. Liu, F. Peng and L. Zhang, J. Mater. Chem. A, 2015, 3, 4188–4194 RSC.
- I. G. Batyrev, J. Phys. Chem. A, 2017, 121, 638–647 CrossRef CAS PubMed.
- B. A. Steele and I. I. Oleynik, J. Phys. Chem. A, 2017, 121, 1808–1813 CrossRef CAS PubMed.
- Y. Li, J. Hao, H. Liu, S. Lu and J. S. Tse, Phys. Rev. Lett., 2015, 115, 105502 CrossRef.
- D. Li, F. Tian, Y. Lv, S. Wei, D. Duan, B. Liu and T. Cui, J. Phys. Chem. C, 2017, 121, 1515–1520 CrossRef CAS.
- K. Xia, J. Sun, C. J. Pickard, D. D. Klug and R. J. Needs, Phys. Rev. B, 2017, 95, 144102 CrossRef.
- Z. Raza, I. Errea, A. R. Oganov and A. M. Saitta, Sci. Rep., 2014, 4, 05889 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- J. P. Perdew and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 5048–5079 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- Z. Raza, C. J. Pickard, C. Pinilla and A. M. Saitta, Phys. Rev. Lett., 2013, 111, 235501 CrossRef.
- W. J. Evans, M. J. Lipp, C. S. Yoo, H. Cynn, J. L. Herberg, R. S. Maxwell and M. F. Nicol, Chem. Mater., 2006, 18, 2520–2531 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09438e |
| This journal is © The Royal Society of Chemistry 2020 |