DOI:
10.1039/C9RA09290K
(Paper)
RSC Adv., 2020,
10, 2170-2179
Aggregation-induced emission compounds based on 9,10-diheteroarylanthracene and their applications in cell imaging†
Received
8th November 2019
, Accepted 4th January 2020
First published on 10th January 2020
Abstract
Four centrosymmetric 9,10-diheteroarylanthracene (DHA) derivatives, including 9,10-dithienylanthracene (DTA), 9,10-difurylanthracene (DFA), 9,10-di-(N-t-butyloxycarboryl-2-pyrryl)anthracene (DBPA), and 9,10-dipyrrylanthracene (DPA) have been synthesized and characterized. All of these DHA derivatives displayed distinct aggregation-induced emission (AIE) behaviors except for DBPA, which showed typical aggregation-caused quenching (ACQ) properties. Their crystal structures exhibited nonplanar conformations on account of the intramolecular torsional effects and intramolecular interactions in rigid molecules. The investigation of the effects of the anthracene core and the side heterocyclic units on the AIE properties demonstrated that the heterocycle moiety is the key factor for the AIE features. These DHA AIEgens exhibited excellent bioimaging performance under physiological conditions.
1. Introduction
Luminescent materials have attracted broad interest in the past decade because they offer the opportunity to perform highly sensitive, noninvasive, and both spatially and temporally resolved imaging,1 and have thus become excellent candidates in the fields of sensing, monitoring of biogenic processes, optoelectronic devices, organic light-emitting diodes, and more.2 However, most of them are inherently hydrophobic, which hinders their practical applications in environmental monitoring and in biological systems. Furthermore, the great majority of luminescent materials have strong emission in dilute solutions but are weakly fluorescent or even nonemissive in the solid state or in a concentrated solution.3 These luminegens with high concentration π–conjugated system usually suffer from aggregation caused quenching (ACQ) due to π–π stacking, which limits their applications in cellular imaging and biosensing.4 In contrast, aggregation-induced emission luminogens (AIEgens) and aggregation-induced emission enhancement (AIEE) materials have been studied by Tang and Park, respectively, and they offer an excellent strategy to overcome the challenges of ACQ.5 Due to their unique features, AIEgens and AIEE materials have attracted considerable attention. Under the guidance of the classical restriction of intramolecular rotation (RIR) mechanisms for the AIE behavior, numbers of AIEgens such as tetraphenylethene, distyrylanthracene, triphenylethene, and cyano-substituted diarylethene derivatives have been reported.6
Anthracene is an important π–conjugated compound with outstanding light-emitting properties and has been widely applied in building organic luminescent molecules.7 During the last decade, lots of materials with AIE or AIEE behaviors based on anthracene have been reported. For example, 9,10-bis[4′-(4′′-aminostyryl)-styryl]anthracene derivatives, which exhibit weak emission in solution, show strong fluorescence in solid state or in a nano-aggregated dispersion,8 and the RIR effect between the vinylene moiety and 9,10-anthrylene core is critical for their AIE phenomenon.9 Zhou et al. reported a series of asymmetrical Schiff base modified styrylanthracene AIE derivatives, found that substituent groups and steric hindrance on the terminal benzene ring could effectively modulate their AIE properties.10 However, the synthesis of 9,10-distyrylanthracene derivatives could produce toxic phosphite esters.11 In addition, 9,10-diphenylanthrylene is usually used as a fluorescence quantum yield standard, should be an AIE molecule according to the classical RIR mechanism, but the results were opposite.12 Therefore, it is meaningful to develop new anthracene-AIEgens through introducing different heterocyclic rings to 9,10-position of anthracene ring, which not only could offer a unique chance for chemists to expand the library of anthracene-AIEgens, but also beneficial to seek the ones with ideal AIE properties. Notably, 9,10-diheteroarylanthracene AIE-active compounds with excellent bioimaging performance has not been reported up to present.
In this work, we synthesized four compounds, including 9,10-dithienylanthracene (DTA), 9,10-difurylanthracene (DFA), 9,10-di-(N-t-butyloxycarboryl-2-pyrryl)anthracene (DBPA), and 9,10-dipyrrylanthracene (DPA), and explored their AIE properties and biological applications. Although the DTA and DFA have been reported in previous work,13 their AIE characteristics, crystal structures, and bio-imaging capabilities have not been examined. Through comparing the crystal structures of these 9,10-diheteroarylanthracene (DHA) molecules, we found that the anthracene moiety and side heteroaromatic ring have a nonplanar conformation due to the supramolecular interactions. The RIR effect between the side heterocyclic moieties and the anthracene core plays a crucial role in their AIE properties. Furthermore, these new AIEgens could be used as cellular luminescent dyes, which have good cell staining properties with low cytotoxicity. Scheme 1 illustrates the synthesis routes of these DHA compounds.
 |
| | Scheme 1 Synthesis routes of DHA derivatives. | |
2. Experiment section
2.1 General methods
All solvents were purified by distillation before use. 1H NMR and 13C NMR spectra were recorded on a Bruker AV400 (400 MHz) spectrometer with CDCl3 as the solvent and tetramethylsilane as an internal standard. Infrared spectra (IR) were recorded on a Bruker Vertex-70 spectrometer. Melting point was measured on a WRS-1B melting point apparatus. Absorption spectra were measured using an Agilent 8454 UV/VIS spectrophotometer. Fluorescence spectra were measured using a Hitachi F-4500 spectrophotometer, and the breadths of excitation and emission slit were both selected 5 nm. Luminescent decay experiments were measured bon Edinburgh FLS980 spectrometer. Elemental analysis was carried out with a PE CHN 2400 analyzer. Mass spectra were obtained on a Bruker AmaZon SL spectrometer. Absolute luminescence quantum yields were measured by Hamamatsu Absolute PL Quantum Yield Spectrometer C11347. Their aggregate behaviors were investigated by scanning electron microscopy (SEM, Zeiss, Sigma). Fluorescence images were obtained on an Olympus FV1000 confocal laser scanning microscope. The structures of them were solved by direct methods and refined by full-matrix least-squares procedures on F2 by full-matrix least-squares techniques using SHELXTL-97 program. Further details on the crystal structure investigation have been deposited with The Cambridge Crystallographic Data Centre as supplementary publication CCDC 1874463 for DTA, 1874464 for DFA, 1887433 for DPA, and 1874465 for DBPA.
2.2 Synthesis of 9,10-dithienylanthracene (DTA)
According to the reported methods,13b compound DTA was prepared by 2-thiophenylboric acid (2.6 g, 20.0 mmol) with 9,10-dibromoanthracene (3.8 g, 11.3 mmol) in the presence of Pd(PPh3)4 (0.4 g) and Na2CO3 (10 mL, 2.0 mol L−1) in tetrahydrofuran (THF) (80 mL containing 10% water) for 15 h at reflux temperature. The crude product was distillated in vacuum and purified by column chromatography on silica gel using hexane as the eluent. DTA (2.1 g, 6.0 mmol) was obtained as a green solid in 60% yield. Single crystal DTA was obtained by vaporizing mixed solvents of chloroform and n-hexane (v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v = 4:1), and its melting point was determined to be 164–165 °C. Anal. calcd for C22H14S2 (%): C, 77.15; H, 4.12; S, 18.73. Found C, 77.13; H, 4.12; S, 18.71; 1H NMR (400 MHz, CDCl3, ppm): δ 7.15 (t, 2H, J = 4.0 Hz), 7.24 (t, 2H, J = 4.0 Hz), 7.32–7.34 (d, 4H, J = 8.0 Hz), 7.55 (d, 2H, J = 8.0 Hz), 7.78–7.81 (q, 4H); 13C NMR (100 MHz, CDCl3, TMS) δ = 124.69, 125.71, 125.83, 126.21, 128.57, 129.28, 130.53, 138.0; IR (KBr, v, cm−1): 699, 770, 819, 849, 906, 1026, 1222, 1329, 1373, 1436, 3025, 3058, 3102; HRMS−ESI (m/z): [M + H+]+ of (C22H15S2+): 343.0615 (calcd), 343.0596 (found).
:v = 4:1), and its melting point was determined to be 164–165 °C. Anal. calcd for C22H14S2 (%): C, 77.15; H, 4.12; S, 18.73. Found C, 77.13; H, 4.12; S, 18.71; 1H NMR (400 MHz, CDCl3, ppm): δ 7.15 (t, 2H, J = 4.0 Hz), 7.24 (t, 2H, J = 4.0 Hz), 7.32–7.34 (d, 4H, J = 8.0 Hz), 7.55 (d, 2H, J = 8.0 Hz), 7.78–7.81 (q, 4H); 13C NMR (100 MHz, CDCl3, TMS) δ = 124.69, 125.71, 125.83, 126.21, 128.57, 129.28, 130.53, 138.0; IR (KBr, v, cm−1): 699, 770, 819, 849, 906, 1026, 1222, 1329, 1373, 1436, 3025, 3058, 3102; HRMS−ESI (m/z): [M + H+]+ of (C22H15S2+): 343.0615 (calcd), 343.0596 (found).
2.3 Synthesis of 9,10-difurylanthracene (DFA)
Similar to DTA, compound DFA was prepared by 2-furanboronic acid (2.2 g, 20.0 mmol) with 9,10-dibromoanthracene (3.8 g, 11.3 mmol) in the presence of Pd(PPh3)4 (0.4 g) and Na2CO3 (10 mL, 2 mol L−1) in tetrahydrofuran (THF) (80 mL containing 10% water) for 15 h at reflux temperature. The crude product was distillated in vacuum and purified by column chromatography on silica gel using hexane as the eluent. Compound DFA (2.3 g, 7.6 mmol) was obtained as a yellow green solid in 76% yield. Single crystal DFA was obtained by vaporizing mixed solvents of chloroform and n-hexane (v:v = 4:1), and its melting point was determined to be 144–145 °C. Anal. calcd for C22H14O2 (%): C, 85.14; H, 4.55. Found C, 85.15; H, 4.54; 1H NMR (400 MHz, CDCl3, ppm): δ 6.71–6.75 (m, 4H), 7.43–7.46 (q, 4H), 7.78 (s, 2H), 7.91–7.93 (q, 4H); 13C NMR (100 MHz, CDCl3, TMS) δ = 109.92, 124.94, 125.42, 126.72, 130.39, 142.01, 149.41; IR (KBr, v, cm−1): 594, 629, 754, 814, 906, 957, 1014, 1074, 1160, 1191, 1255, 1326, 1439, 1495, 1623, 1730, 2925, 2962, 3042, 3076, 3107, 3145; HRMS calcd for C22H14S2, 310.0994; found 310.1042.13a
2.4 Synthesis of 9,10-di-(N-t-butyloxycarboryl-2-pyrryl)anthracene (DBPA)
Compound DBPA was prepared by N-Boc-2-pyrroleboronicacid (4.2 g, 20.0 mmol) with 9,10-dibromoanthracene (3.8 g, 11.3 mmol) in the presence of Pd(PPh3)4 (0.4 g) and Na2CO3 (10 mL, 2.0 mol L−1) in tetrahydrofuran (THF) (80 mL containing 10% water) for 15 h at reflux temperature. The crude product was distillated in vacuum and purified by column chromatography on silica gel using hexane/dichloromethane (v:v = 1:1) as the eluent. Compound DBPA (4.5 g, 8.8 mmol) was obtained as a yellow solid in 88% yield. Single crystal DBPA was obtained by vaporizing mixed solvents of chloroform and n-hexane (v:v = 4:1), and its melting point was determined to be 170–171 °C. Anal. calcd for C32H32N2O4 (%): C, 75.57; H, 6.34; N, 5.51. Found C, 75.55; H, 6.34; N, 5.52; 1H NMR (400 MHz, CDCl3, ppm): δ 0.60 (s, 18H), 6.29 (d, 2H, J = 4.0 Hz), 6.51 (t, 2H, J = 4.0 Hz), 7.34–7.37 (q, 4H), 7.66–7.69 (q, 6H); 13C NMR (100 MHz, CDCl3, TMS) δ = 25.69, 81.84, 109.88, 115.38, 120.94, 124.27, 125.34, 128.42, 129.37, 130.52, 148.32; IR (KBr, v, cm−1): 675, 734, 778, 814, 849, 876, 924, 1016, 1109, 1158, 1256, 1299, 1335, 1370, 1399, 1473, 1622, 1737, 2932, 2981, 3009, 3096, 3143, 3442; HRMS-ESI (m/z): [M + K+]+ of (C32H32KN2O4+): 547.1999 (calcd), 547.2535 (found).
2.5 Synthesis of 9,10-dipyrrylanthracene (DPA)
Compound DPA was prepared by DBPA. First, to a stirred toluene solution (20 mL) of DBPA (1.5 g, 3.0 mmol), some silica-gel powder were added at 273 K. The reaction mixture was stirred for 2 h at reflux temperature. Then the mixture was filtered and washed by dichloromethane and ethyl acetate. The organic layer was evaporated. The crude product was purified by column chromatography on silica gel using petroleum ether/dichloromethane (v:v = 1:1) as the eluent to afford 0.88 g, 2.9 mmol compound DPA as a yellow green solid in 96%. Single crystal DPA was obtained by vaporizing mixed solvents of chloroform and n-hexane (v:v = 4:1), and its melting point was determined to be 272–273 °C. Anal. calcd for C22H16N2 (%): C, 85.69; H, 5.23; N, 9.08. Found C, 85.69; H, 5.22; N, 9.07; 1H NMR (400 MHz, CDCl3, ppm): δ 6.50–6.56 (m, 4H), 7.10 (t, 2H, J = 4.0 Hz), 7.39–7.41 (q, 4H), 7.92–7.95 (q, 4H), 8.37 (s, 2H); 13C NMR (100 MHz, CDCl3, TMS) δ = 101.00, 108.06, 110.58, 117.18, 124.60, 125.80, 126.29, 128.56, 130.58; IR (KBr, v, cm−1): 652, 731, 768, 895, 1027, 1086, 1111, 1361, 1407, 1439, 1469, 1517, 1619, 2941, 3035, 3058, 3096, 3336, 3368, 3411, 3433; HRMS−ESI (m/z): [M + H+]+ of (C22H17N2+): 309.1392 (calcd), 309.1393 (found).
2.6 TDDFT study
To study the electronic behavior of DHA derivatives, the ground and excited states of molecules were optimized with the Becke-3-Lee-Yang-Parr (B3LYP) by time-dependent density functional theory (TDDFT) calculations at the level of 6-311G (d) with Gaussian 09 program. The b3lyp density functional method[−] was employed in this work to carry out all the calculations. The 6-311g(d) basis set was used for all atoms. Vibrational frequency analyses were performed on the optimized geometries at the same level of theory to characterize the stationary points as local minima (no imaginary frequency) or transition states (one imaginary frequency). In addition, intrinsic reaction coordinate (IRC) calculations were used to verify that the transition state connect with appropriate reactant and product.
2.7 Cell culture and incubation
HeLa cells were cultivated in DMEM medium which supplemented with 10% FBS and 1% penicillin–streptomycin in an atmosphere of 5% CO2 and 95% air at 37 °C. Place the cells in a confocal dish and make them adhere overnight. On the next day before the experiments, the cells were washed with phosphate-buffered saline (PBS) buffer. For imaging in living cells, the cells was cultured using fresh medium and 20 μM DHA compounds were added to each well. After being incubated for 30 min, fluorescence image of the cells were observed on a confocal laser scanning microscope (Olympus FV1000). Excitation energy of 405 nm was used, and the fluorescence emission was measured at 440–540 nm and 480–580 nm.
3. Results and discussion
3.1 Photophysical properties
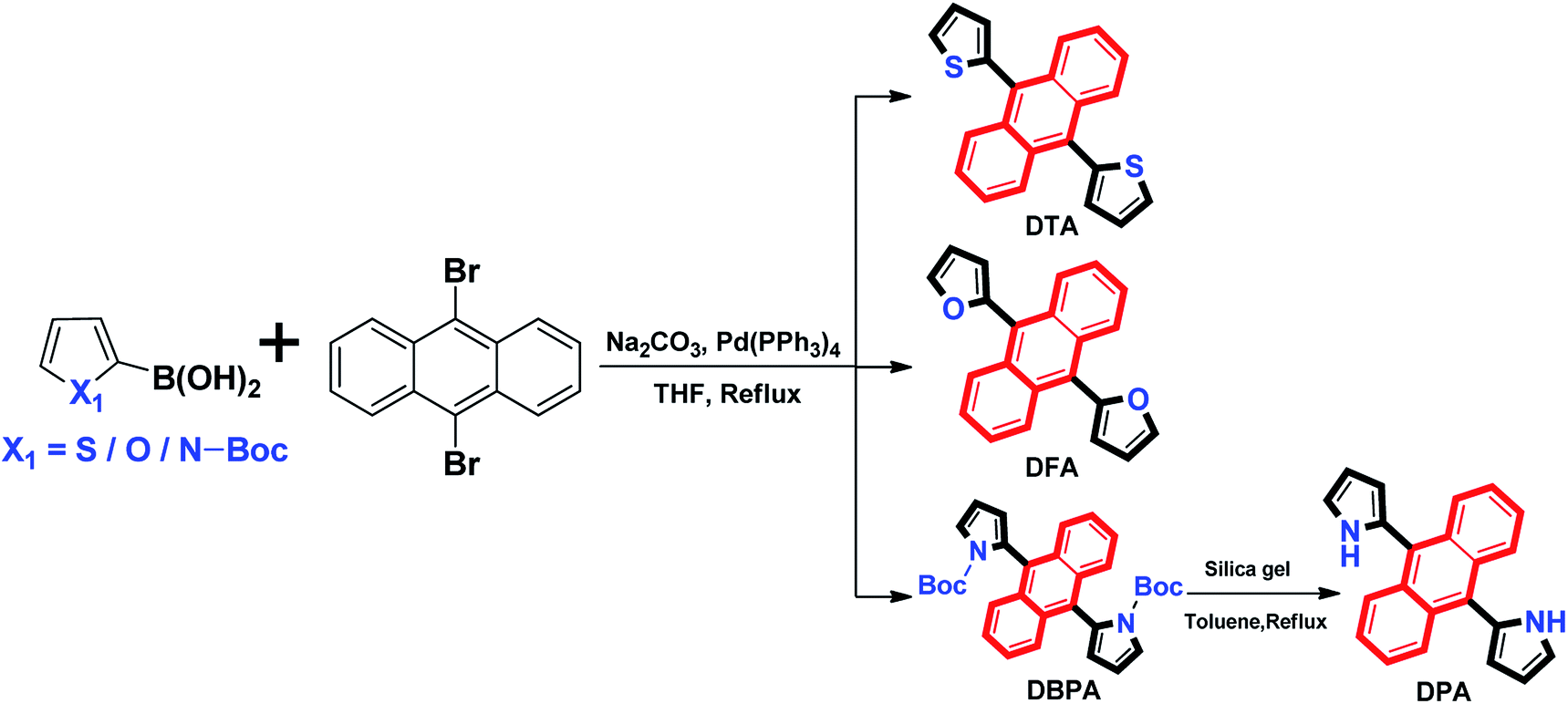
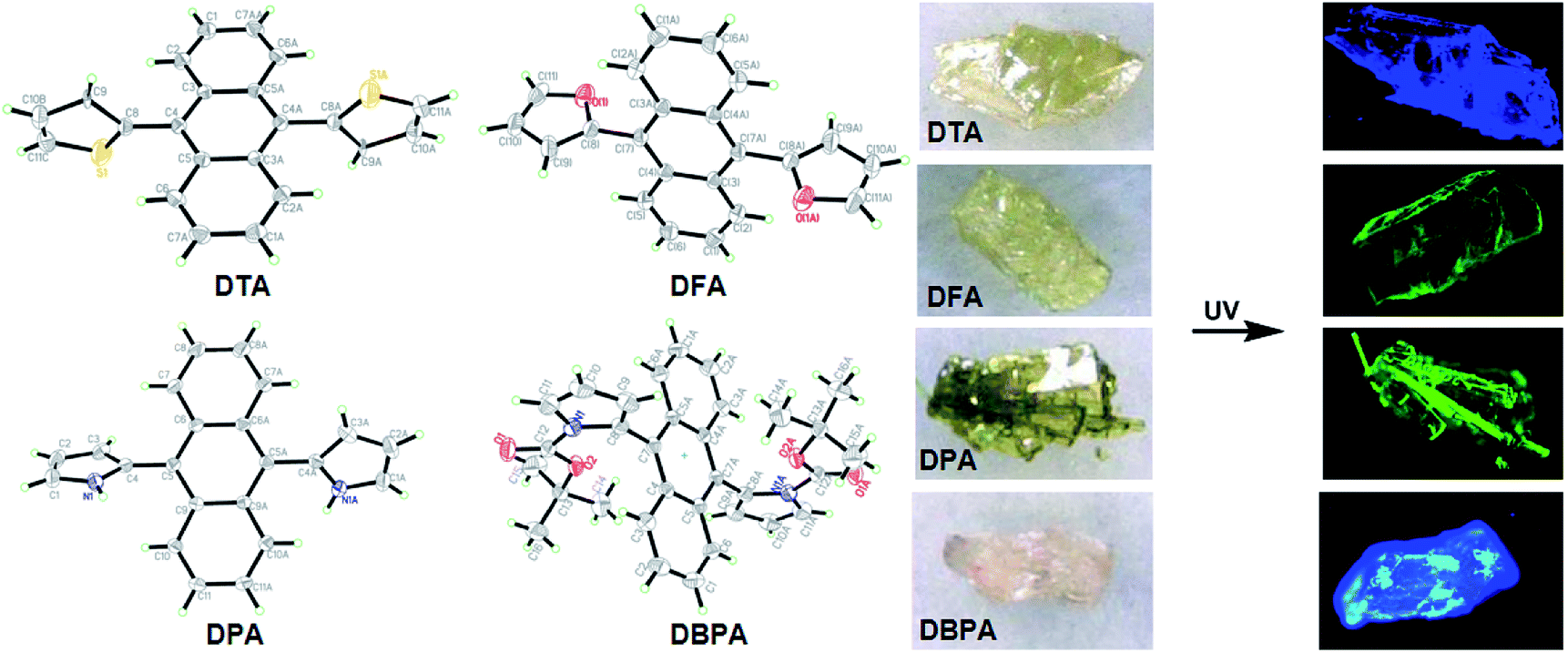
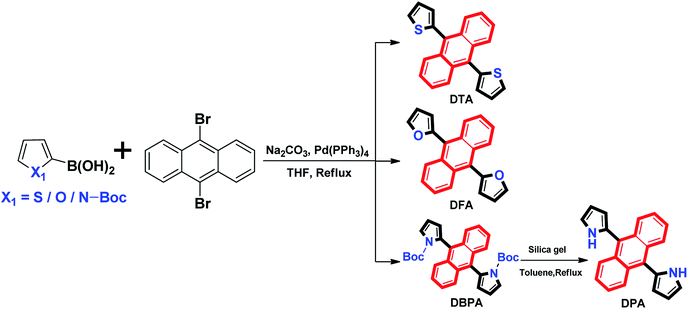
The absorption spectra of 9,10-diheteroarylanthracene (DHA) derivatives (20 μM) in dimethylformamide (DMF) solutions are shown in Fig. 1A. There are three absorption peaks in 9,10-dithienylanthracene (DTA, 360 nm, 379 nm and 403 nm) and 9,10-di-(N-t-butyloxycarboryl-2-pyrryl)anthracene (DBPA, 358 nm, 376 nm and 396 nm), which corresponded to the absorption of the anthracene core. When we introduced furan (DFA) or pyrrole (DPA) into the 9,10-position of anthracene, strong absorption peaks at 398 nm and 405 nm were observed due to the π → π* transition. It should be noted that there are weak absorbance tails at a range longer than 450 nm of DFA and DBPA, the solvation effects caused by strong electronegative oxygen atoms in their molecules may be the main reason for these observations. The fluorescence spectra of these compounds in DMF solutions is shown in Fig. 1B. All of the compounds showed weak fluorescence emission upon photoexcitation, except for DBPA. Compared to DTA (λem = 476 nm), the fluorescence spectra of the DFA (λem = 488 nm) and DPA (λem = 516 nm) have longer wavelengths. Thus red-shifts in the fluorescence spectra are probably due to the weaker electronegativity of the sulphur atom compared to the oxygen and nitrogen atoms, which could enhance the conjugation of DFA and DPA. The absolute fluorescence quantum yield of DTA, DFA and DPA in DMF were determined to be 0.009, 0.010 and 0.024, respectively, by using the absolute fluorescence quantum yields spectrometer QYC11347-11. The results are consistent with their fluorescence spectra. Compared to 9,10-diphenylanthracene (Φf, acetone = 0.99),12 the fluorescence quantum yields of DTA, DFA and DPA are much lower, indicating that introduce five-membered heterocyclic rings to the 9,10-positions of anthracene could efficiently reduce the luminescence in dilute solution. For DBPA, the fluorescence intensity and fluorescence quantum yield (Φf = 0.86) were remarkably enhanced, the existence of intra- and intermolecular interactions may be weakened the steric hindrance of t-butyloxycarboryl (Boc-group) on the pyrrole ring, which could enhance the coplanarity of DBPA and induce its strong fluorescence emission the dilute solution. The spectroscopic features of these four compounds in DMF are summarized in Table 1.
 |
| | Fig. 1 (A) Normalized absorption spectra and (B) fluorescence spectra of the DHA compounds in DMF solutions (20 μM). | |
Table 1 Photophysical properties of DHA derivatives in solution and powder states
| Compds |
DMF |
Solid |
DMF/H2O |
Φfd |
| λmaxa (nm) |
λemb (nm) |
λmaxa (nm) |
λemb (nm) |
Φfd |
λem, max (nm) |
H2O% |
τc (ns) |
fw = 0 |
fw, max |
| Absorption wavelength. The wavelength of emission maximum. The fluorescence lifetime. Fuorescence quantum yield. |
| DTA |
360, 379, 403 |
476 |
405 |
494 |
0.028 |
488 |
80 |
4.32 |
0.009 |
0.244 |
| DFA |
398 |
488 |
417 |
504 |
0.083 |
528 |
90 |
4.41 |
0.010 |
0.286 |
| DPA |
405 |
516 |
412 |
526 |
0.043 |
515 |
90 |
4.22 |
0.024 |
0.354 |
| DBPA |
358, 376, 396 |
473 |
398 |
486 |
0.069 |
474 |
90 |
N/A |
0.860 |
0.227 |
The absorption and fluorescence spectra in different solvents, such as n-hexane, dichloromethane (DCM), tetrahydrofuran (THF), acetonitrile, dimethyl formamide (DMF), and dimethyl sulfoxide (DMSO) of these DHA compounds are shown in Fig. S9 and S10 (ESI†). It could be seen that the absorption peaks exhibited negligible changes in various solvents, whereas the fluorescence peaks of these compounds were red-shifted from λmax, n-hexane = 467 to λmax, DMF = 476 nm for DTA, λmax, n-hexane = 448 to λmax, DMF = 488 nm for DFA, and λmax, n-hexane = 512 to λmax, DMF = 525 nm for DPA. The larger red-shifts of DFA may be attributed to the formation of J-aggregation in polar solvent due to the strong electronegativity of oxygen atoms in furan ring, and conducive to generate hydrogen bonds in polar solvents. The emission intensities were increased with increasing the solvent polarity apart for DBPA. For example, the fluorescence peak of DFA was at 448 nm in nonpolar n-hexane, and was red-shifted to 488 nm in polar DMF. The decrease of π → π* transitions energy in polar solvents maybe the main reason for this phenomenon. The spectroscopic properties of these compounds in various solvents are summarized in Table S1 (ESI†). It should be noted that the fluorescence spectra of DBPA exhibited negligible changes with the increase of solvent polarity, the reason maybe ascribe to the large steric hindrance of Boc-group, which could inhibit the formation of J-aggregation (Fig. S10C, ESI†).
3.2 Aggregation-induced emission properties
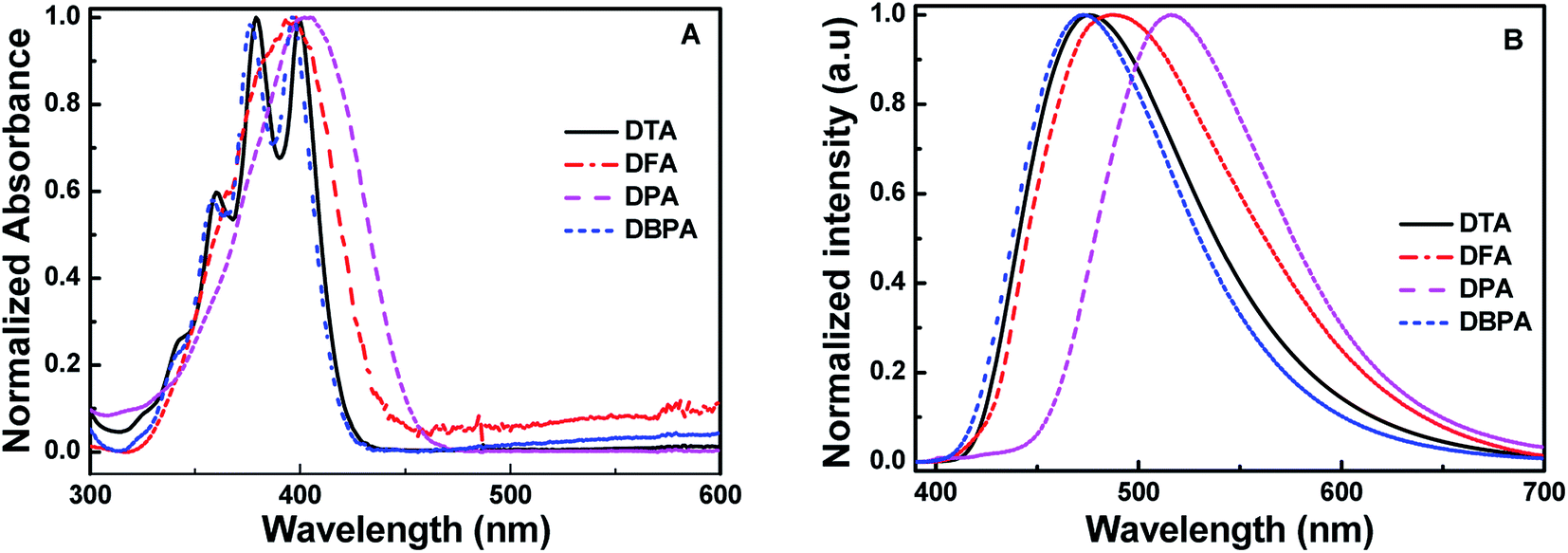
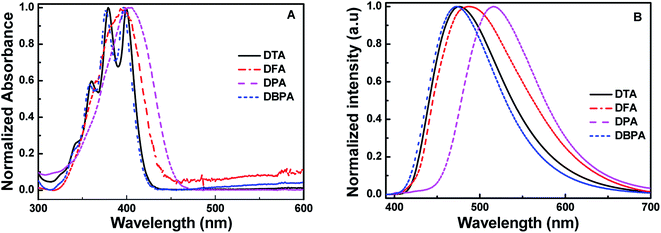
To investigate the AIE properties of the four DHA compounds, their photoluminescence (PL) behaviors in DMF/H2O mixtures are investigated. Importantly, water is a poor solvent for DHA compounds. Fig. 2A illustrates the PL changes for DFA in DMF/H2O mixtures with various water fractions. It could be easily seen that DFA exhibits typical AIE properties. When the water fraction (fw) was in the range of 0–70%, the emission of DFA is very weak under 365 nm excitation, whereas strong cyan emission of DFA was observed with the increase of fw to 80%. When fw increasing to 90%, the emission intensity of DFA enhanced nearly 19-folds, and the PL quantum efficiency increased from 0.01 (fw = 0%) to 0.29 (fw = 90%) (Fig. 2B), which is attributed to the formation of aggregate and the trigging of the RIR effect.14 For DTA, the PL is dramatically enhanced at 476 nm when the fw increased from 60% to 80% (Φf = 0.244) and the emission intensity increased by up to about 19-fold in comparison with pure DMF (Fig. S10A, ESI†). It should be noted that the fluorescence intensity of DTA decreased with further increasing fw to 90%. The rapidly aggregated solute molecules and the shielding of internal molecules may be the main reasons for the decrease in fluorescence intensity of the mixture.15 Similar to DFA, DPA also exhibited typical AIE optical characteristics. The emission intensity of DPA is very weak when the content of water is < 80%, as shown in Fig. S10B (ESI†). With the further addition of water, the fluorescence intensity of DPA dramatically increased, and the highest values was observed when fw reached 90% (Φf = 0.354). In contrast, DBPA exhibited typical ACQ features and displayed strong blue emission in pure DMF. As the fw increased from 0 to 90%, its PL intensity dramatically quenched to 30%, and the absolute fluorescence quantum yield was deceased from 0.86 to 0.23. In addition, the emission lifetimes of DTA, DFA and DPA in 90% water fraction were determined to be 4.32 ns, 4.41 ns or 4.22 ns, respectively.
 |
| | Fig. 2 (A) Fluorescence spectra of DFA in the different DMF/H2O fraction solutions; (B) the changes in absolute fluorescence quantum yields of DTA, DFA and DPA versus water fractions in DMF (inset: photos of DFA in DMF/H2O under 365 nm excitation). | |
To explore the AIE effects of DTA, DFA and DPA in aqueous medium, scanning electron microscopy (SEM) images of their aggregates were studied (Fig. S11, ESI†). It could be seen that DTA and DPA formed globular-shaped nano-particles in the DMF/H2O mixture with fw = 90%, suggesting that self-assembly effects occurred in their aggregate states. Different from that of DTA and DPA, DFA formed in the mixtures were amorphous-like aggregates (Fig. S11B, ESI†). Subsequently, the nano-aggregates obtained were characterized by dynamic light scattering (DLS) experiments (Fig. S12, ESI†). The results indicated that their particle diameter size distribution was approximately in the range of 299–582 nm. Therefore, the strong bright fluorescence emission of DTA, DFA and DPA were attributed to aggregate formation, indicating these compounds belong to AIE-active luminous molecules.
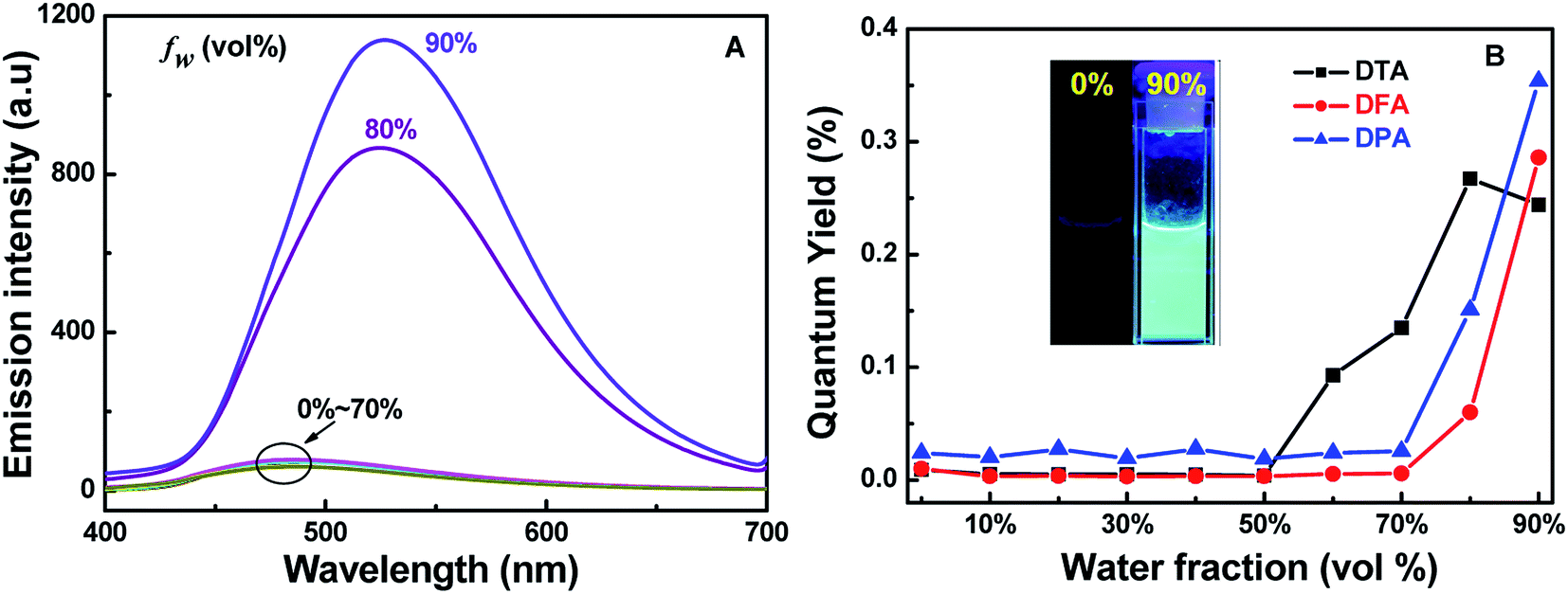
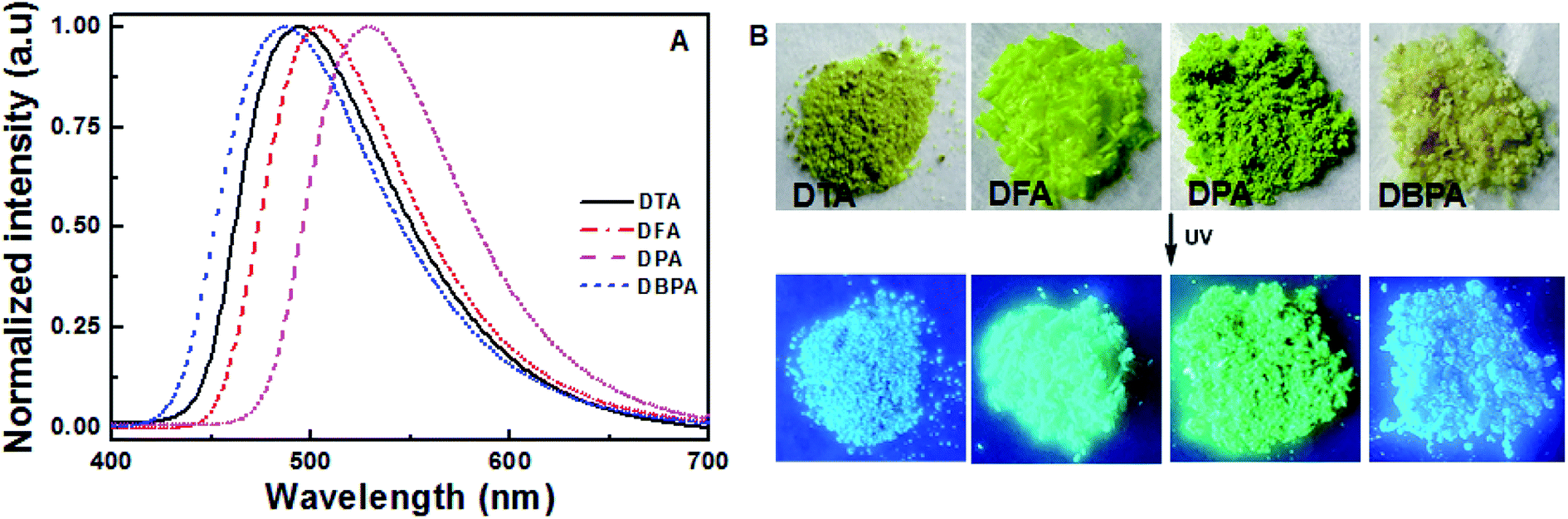
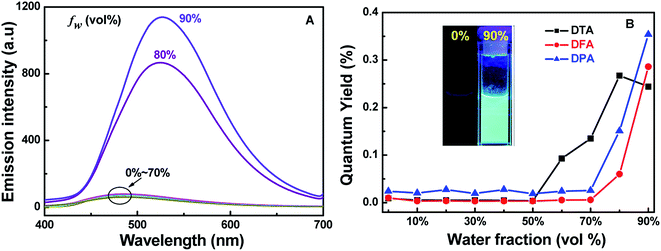
To further evaluate the solid-state fluorescence behaviors of these DHA luminogens, their solid-state PL were studied. As shown in Fig. 3A, the emission spectrum of DTA showed strong emission bands with a peak at 494 nm, corresponding to a bright blue emission under 365 nm UV light (Fig. 3B). Similarly, DFA and DPA displayed strong cyan/green emission with peaks at 504 nm and 526 nm, respectively. However, compared with the DTA, DFA and DPA, the PL of the solid sample DBPA exhibited distinct blue-shifted (λem = 486 nm) with emitted strong bright blue light upon irradiating with 365 nm, in which the steric-hinderance effect of the Boc-group may be responsible for this phenomenon. Furthermore, the emission lifetimes of these compounds in solid-state were tested at the room temperature, and yielded values of 0.07 ns for DTA, 0.16 ns for DFA, 0.19 ns for DPA, and 0.19 ns for DBPA (Fig. S13, ESI†). The low luminescence lifetimes of these molecules maybe due to the large intermolecular distortion induced a nonplanar conformation between anthracene core and side heteroaromatic ring.16
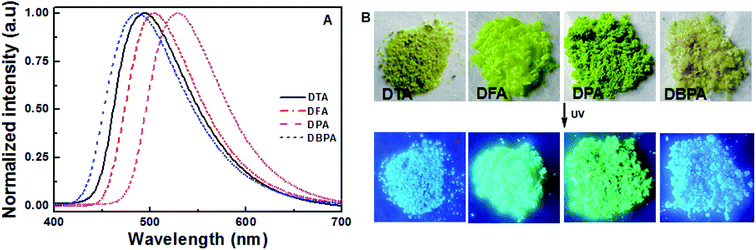 |
| | Fig. 3 (A) Solid state PL spectra of DHA compounds; (B) photographic images of the four compounds under 365 nm UV illumination. | |
3.3 Nonplanar crystal structures
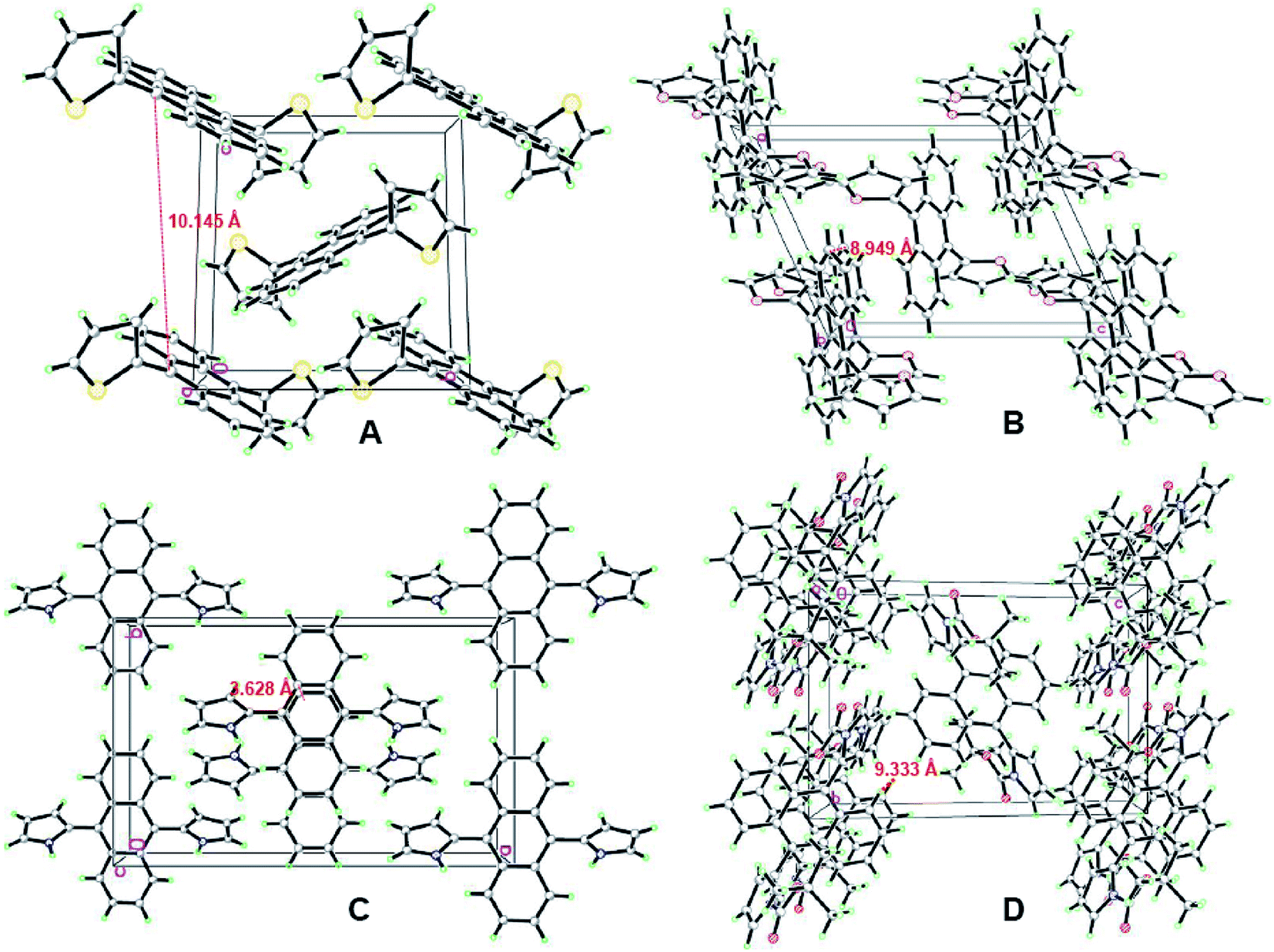
To further examine the luminescence behaviors of these compounds, it was necessary to investigate the structural relationship between the side heteroaromatic rings and the anthracene core. Fortunately, single crystals were obtained for all of them by slowly vaporizing mixed solvents of DCM and n-hexane at room temperature. X-ray diffraction data were collected on a Bruker SMART APEX II CCD diffractometer by using a MULTI scan technique at 296(2) K and Mo Kα radiation. The crystal structures are shown in Fig. 4 and S14–S17 (ESI†). Their crystal data and structure refinements are summarized in Table S2 (ESI†). For DTA and DFA molecules, the crystal systems and space groups were monoclinic with P21/c and P2(1)/n. The crystal density of DTA and DFA were 1.364 g cm−3 and 1.304 g cm−3, respectively. The dihedral angles between the anthracene core and the two heteroaromatic rings were equal to 82.1° (DTA) and 97.7° (DFA), showing a central symmetric structure. The results indicated that DTA and DFA displayed nonplanar conformation in the crystalline states. The different of the dihedral angles indicated the intramolecular heterocyclic effect and different molecular stacking on these molecular conformations. As shown in Fig. 5A, the crystal of DTA was packed into molecular columns along the α-axis slides to their neighbors along the long axis within one column with a ‘zigzag motif’ mode and formed J-stacking.17 The distance between two parallel molecules of anthracene ring within one column was measured to be 10.145 Å and 9.228 Å, which were too large to form the π–π interaction. Therefore, the AIE effects of DTA was remarkably enhanced due to the restriction of intramolecular π–π stacking. Additionally, large torsional angles and central symmetric configuration further promoted AIE activity of DTA, inducing the crystal to generate a bright blue light. The crystal cell of DFA exhibited similar stacking as DTA (Fig. 5B). It should be noted that the dihedral angles between the anthracene core and furan ring (DFA, 97.7°) is much larger than in DTA (82.1°), demonstrating there are much larger supramolecular interactions than in DTA. The results may be due to stronger electronegativity of the oxygen atom than the sulfur atom, which prompted the rigidity of DFA molecules in the lattice.
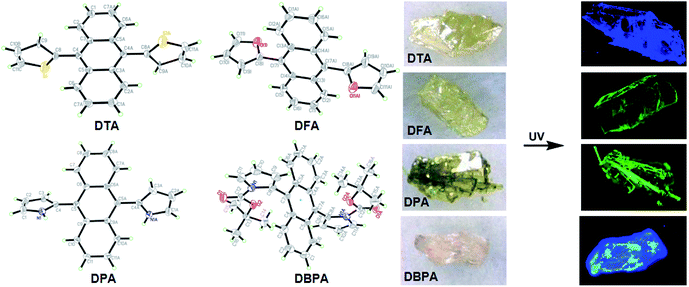 |
| | Fig. 4 ORTEP drawings of DHA crystals and their crystal pictures at room temperature under ambient light and UV light. | |
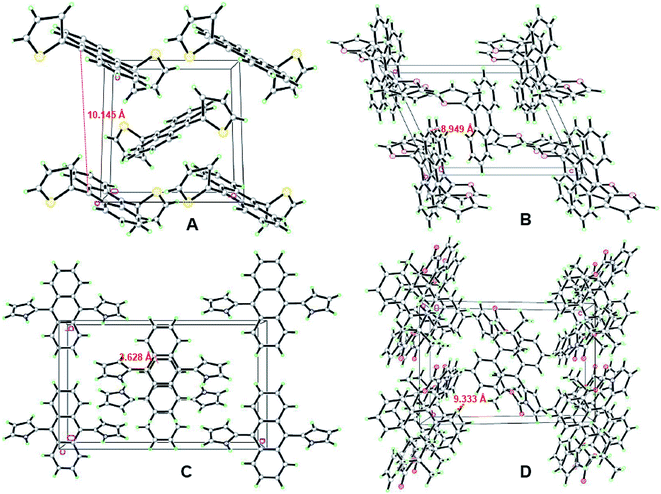 |
| | Fig. 5 Fragment of the crystal packing of the DHA compounds: (A) stacking images of DTA, viewed along the a-axis; (B) stacking images of DFA, viewed along the b-axis; (C) stacking images of DPA, viewed along the c-axis; (D) stacking images of DPA, viewed along the c-axis. | |
Unlike DTA and DFA, the unit cell of DPA was orthorhombic with space group Pbcn. The dihedral angles of DPA between the central benzene ring of anthracene (C5–C6–C6A–C5A–C9A–C9) and the pyrrole ring were 126.9° for N1/C1–C2–C3–C4 and 53.1° for N1A/C1A–C2A–C3A–C4A. Compared with DTA and DFA, the dihedral angles of DPA was much smaller than former. Notably, the plane of the anthracene ring showed distortion to a certain extent, i.e., the dihedral angles between the central benzene ring of anthracene and adjacent benzene ring were 3.2° (C6–C7–C8–C8A–C7A–C6A) and 5.0° (C9–C10–C11–C11A–C10A–C9A), respectively. The crystal cell stacking diagram of DPA was systematically investigated to determine which forces caused the distortion of the anthracene ring plane. As shown in Fig. 5C, there were six molecules in a DPA single crystal cell and these molecules pack into the simple brick wall motif,18 and the anthracene rings of the two molecules at the center of the crystal cell displayed partial overlap in the unit cell. The distance between the overlap surfaces of the anthracene moieties were in the range of 3.604–3.844 Å, which formed the offset face-to-face π–π stacking interactions and induced distortion of the anthracene ring. Additionally, the intramolecular NH/π hydrogen bond of DPA was also crucial for anthracene ring distortion. The stacking arrangement of DPA is attributed to two NH/π hydrogen bonds between two pyrrole rings, where the NH of the pyrrole acts as an H donor and the pyrrole ring of the adjacent molecule acts as an H acceptor.19 The interaction distance and the angle of the N–H–π center for DPA are 2.500 Å and 86.49°. Consequently, the restriction of intramolecular rotation (RIR) in the structure of DPA were realized through hydrogen bond effects. The strong supramolecular interactions not only strengthened the rigidity and stability in the crystal lattice, but also induced tight intermolecular packing, which could result in distinct AIE behavior. The crystal system and space group of DBPA were monoclinic and P2(1)/n. The dihedral angles of DBPA between the anthracene core and the two pyrrole rings are equal to 85.5°, which were much larger than that of DPA due to strong steric hindrance. As shown in Fig. 5D, there were nine molecules in the DBPA single crystal cell with a large volume. The distance between two parallel molecules within one column was measured to be 9.333 Å, indicating that π–π interaction cannot be formed in the molecule. Meanwhile, the large steric hindrance of Boc-group on the pyrrole ring caused severe twisting, which inhibited the conformational planarization of DBPA both in solution and in aggregative states. The result induced DBPA to have strong emission in solution due to the restriction of molecular rotation, blocked the non-radiative path, and activated radiative decay.20 Therefore, the ACQ effect of DBPA in the aggregate states could be attributed to the agglomeration of nanoparticles.21
3.4 Theoretical investigations
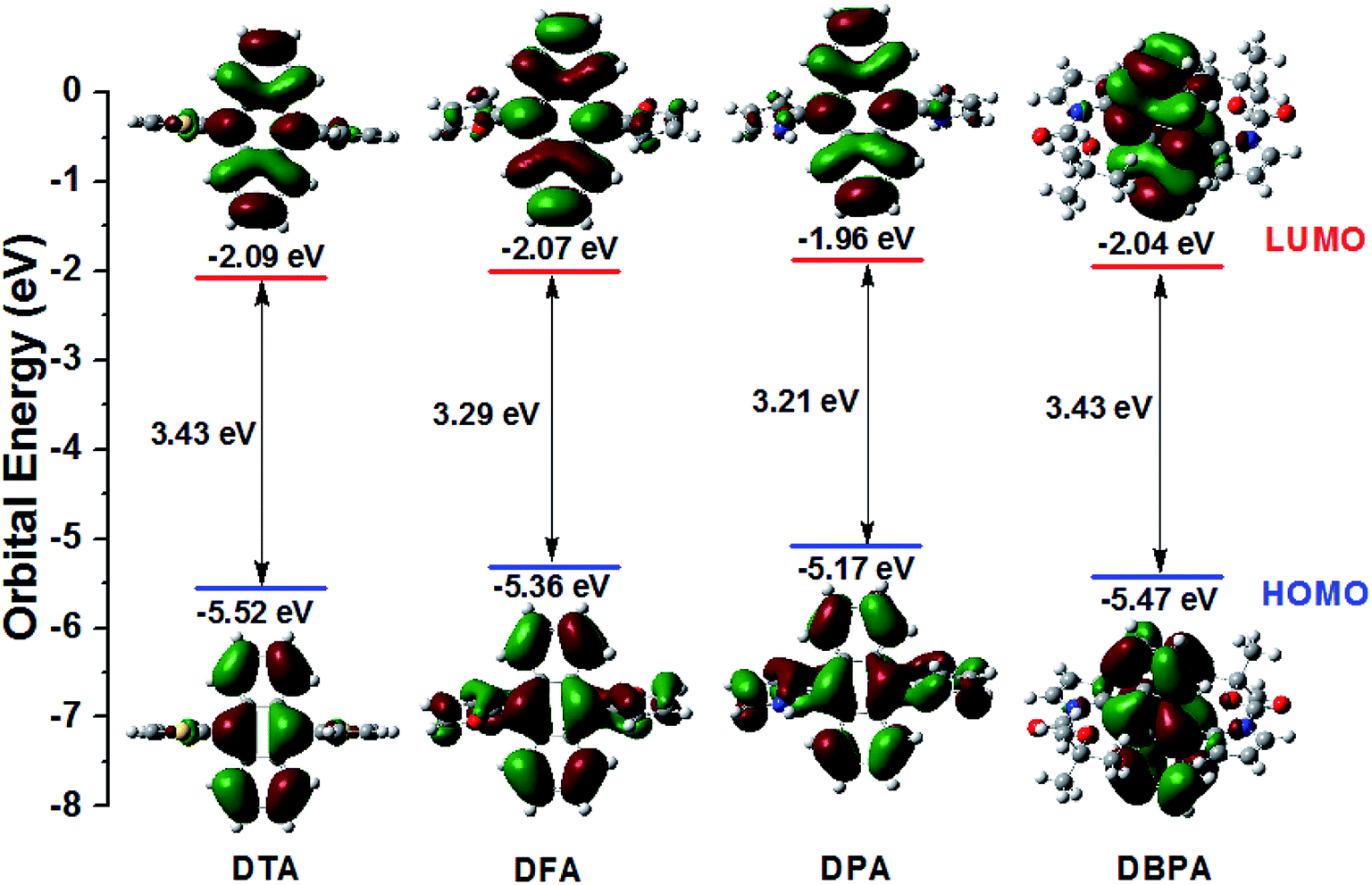
To further exploration the electronic structures of DTA, DFA, DPA and DBPA, density functional theory (DFT) calculation using Gaussian 09. The Symbolic Z-Matrix and computed total energies are given in Tables S3–S10 (ESI†). It was noteworthy that all of the compounds adopted twisted spatial conformations from the optimized structure. As shown in Fig. 6, the electron clouds in the HOMOs of DPA (−5.17 eV) and DFA (−5.36 eV) were spread on anthracene, pyrrole, and furan subunits. Whereas their LUMOs were mainly located on the anthracene core. Accordingly, the HOMOs and LUMOs of DPA and DFA supplied an obvious spatial separation, leading to calculated energy gaps between HOMOs and LUMOs of 3.21 eV for DPA and 3.29 eV for DFA. Compared to DPA/DFA, the electron cloud of DTA in the HOMO was located on both the anthracene core and part of the thiophene ring, whereas the electron cloud of the LUMO showed concentrated distribution on the anthracene core, and the calculated HOMO–LUMO gap for DTA was 3.43 eV. These observations indicated that the electron clouds followed the flow direction from heteroaromatic rings. It is worth noting that the electron clouds of DBPA both in HOMO and LUMO levels were mainly located on the anthracene core, suggesting that the electron clouds followed the flow direction from the Boc group to the intermediate anthracene in the excited state, which is consistent with the strong fluorescence intensity of DBPA in DMF solution and ACQ features in the aggregate states. In addition, the energy gaps of these DHA derivatives were consistent with the position of their fluorescence emission peaks.
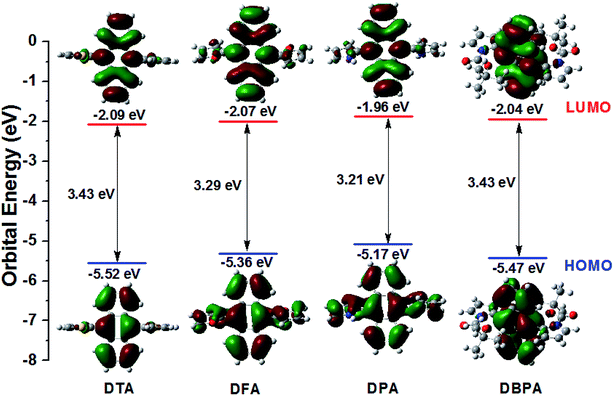 |
| | Fig. 6 Calculated spatial electron distributions of LUMO and HOMO, and the optimized conformation structure of DHA compounds. | |
3.5 Cell imaging applications of DHA derivatives
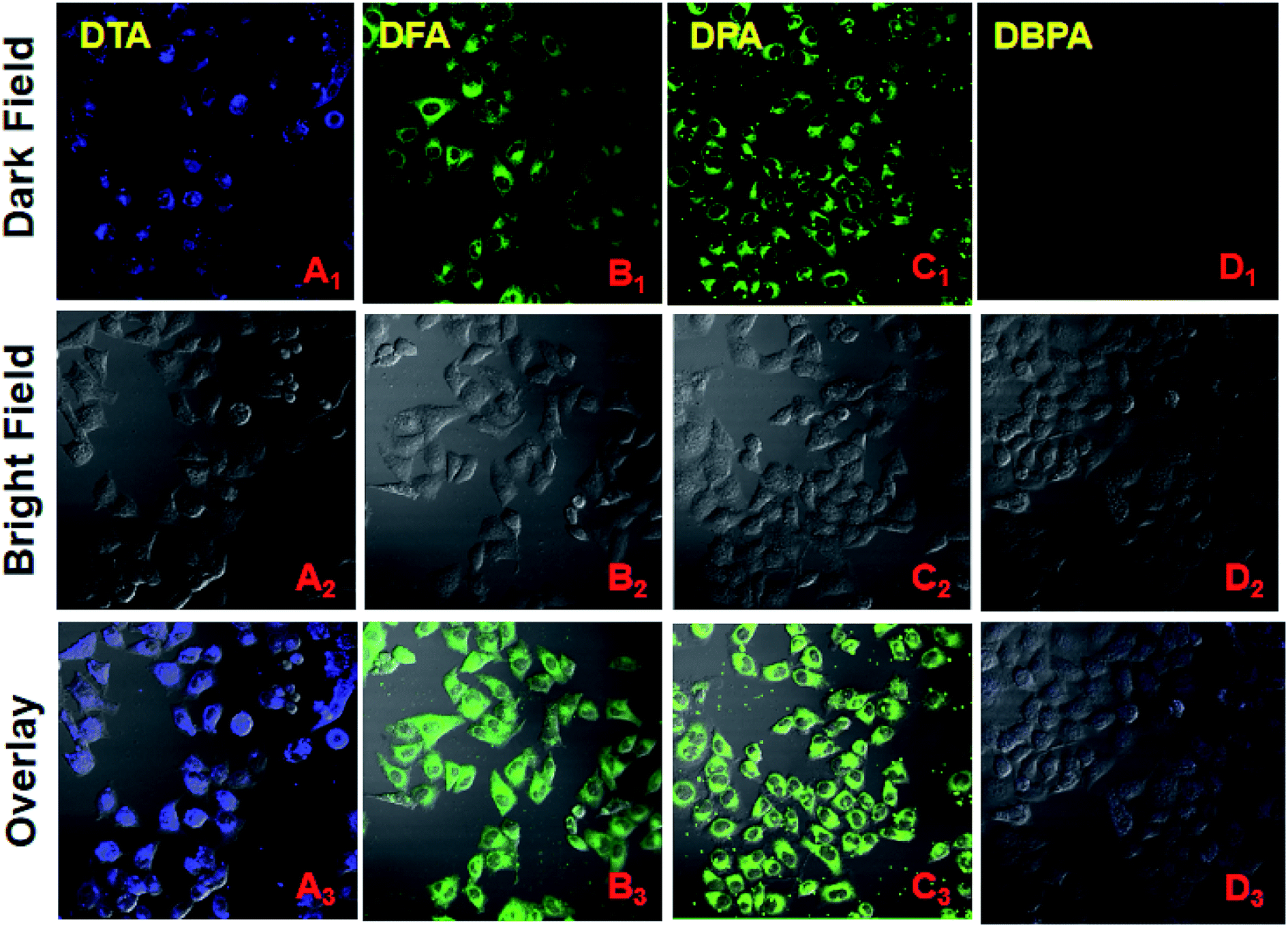
Considering the properties of these DHA compounds such as their unique nanoscale size, high water dispersibility, and intense fluorescence, their biological applications were monitored by fluorescence microscopy. HeLa cell culture and cytotoxicity assays were performed according to the reported method.22 HeLa cells stained with 20 μM of DPA, DFA, DPA and DBPA for 30 min, respectively, and exhibited strong fluorescent signals in the blue channel (DTA, 440–540 nm) and green channel (DFA/DPA, 480–580 nm) under the fluorescence microscopy, indicating the superior cell labeling ability of these compounds (Fig. 7). In contrast, DBPA showed weak fluorescence signal in the blue channel (440–540 nm) due to the notorious ACQ effect (Fig. 7D). Additionally, the MTT assays confirmed that DPA have no significant cytotoxic effects on HeLa cells up to 24 h of treatment (Fig. S18, ESI†). These results suggested that these AIE DHA molecules had good staining ability for HeLa cells, thus making them a promising candidate biological dyes for cell imaging.
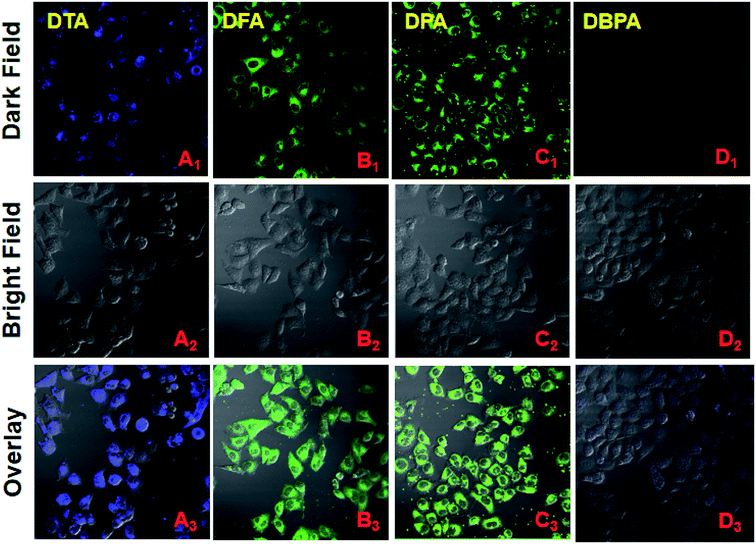 |
| | Fig. 7 Fluorescent confocal microscopy images of HeLa cells stained with 20 μM of DHA compounds for 30 min. (A1 → D1) Dark field images; (A2 → D2) Bright field images, A2 and D2 blue channel images: 440–540 nm, B2 and C2 green channel images: 480–580 nm; (A3 → D3) overlay of dark and bright field images. | |
4. Conclusion
In this study, a class of DHA AIEgens based on anthracene have been rationally designed. These DHA molecules displayed nonplanar conformation in crystals. They showed obvious AIE behavior and emitted strong blue/green fluorescence in aggregate states except for DBPA, which exhibited ACQ property due to large steric hindrance. The large torsional effects and intramolecular interactions between the anthracene core and the side heterocyclic units were the critical factors for their AIE performance. Additionally, their potential as practical biological imaging agents was verified by confocal fluorescence microscopy in HeLa cells. This study provides a new class of AIEgens, which could be useful in understanding AIE properties of 9,10-diheteroarylanthracene derivatives and designing AIE materials with high PL quantum efficiency.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (41867053, 41867052). We were also grateful for the Jiangxi Science & Technology Normal University Doctoral Research Startup fund (2018BSQD021), and the Open Project Program of “2011 Collaborative Innovation center” (KFGJ18001).
References
-
(a) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed;
(b) W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC;
(c) P. Choudhury, S. Sarkar and P. K. Das, Langmuir, 2018, 34, 14328–14341 CrossRef CAS PubMed.
-
(a) K. Das, S. Sarkar and P. K. Das, ACS Appl. Mater. Interfaces, 2016, 8, 25691–25701 CrossRef CAS PubMed;
(b) M. Vendrell, D. Zhai, J. C. Er and Y. T. Chang, Chem. Rev., 2012, 112, 4391–4420 CrossRef CAS PubMed;
(c) H. H. Gorris and O. S. Wolfbeis, Angew. Chem., Int. Ed., 2013, 52, 3584–3600 CrossRef CAS PubMed;
(d) Y. Chen, W. Zhang, Z. Zhao, Y. Cai, J. Gong, R. T. Kwok, J. W. Y. Lam, H. Y. S. Herman, H. Y. Sung, I. D. Williams and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 130, 5105–5109 CrossRef;
(e) M. Zhang, W. Yang, K. Li, W. Zhou, T. Gong and R. Xue, Mater. Chem. Phys., 2018, 204, 37–47 CrossRef CAS.
- M. Chen, R. Chen, Y. Shi, J. Wang, Y. Cheng, Y. Li, X. D. Gao, Y. Yan, J. Z. Sun, A. J. Qin, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1704689–1704699 CrossRef.
-
(a) J. Cornil, D. Beljonne, J. P. Calbert and J. L. Brédas, Adv. Mater., 2001, 13, 1053–1067 CrossRef CAS;
(b) T. Usuki, M. Shimada, Y. Yamanoi, T. Ohto, H. Tada, H. Kasai, N. Eiji and H. Nishihara, ACS Appl. Mater. Interfaces, 2018, 10, 12164–12172 CrossRef CAS PubMed.
-
(a) J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 18, 1740–1741 RSC;
(b) S. J. Lim, B. K. An and S. Y. Park, Macromolecules, 2005, 38, 6236–6239 CrossRef CAS.
-
(a) M. Wang, G. X. Zhang, D. Q. Zhang, D. B. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858–1867 RSC;
(b) Y. Dong, J. W. Y. Lam, A. Qin, J. Liu, Z. Li, B. Z. Tang, J. X. Sun and H. S. Kwok, Appl. Phys. Lett., 2007, 91, 011111 CrossRef;
(c) S. Kim, T. Y. Ohulchanskyy, H. E. Pudavar, R. K. Pandey and P. N. Prasad, J. Am. Chem. Soc., 2007, 129, 2669–2675 CrossRef CAS PubMed;
(d) Y. Q. Dong, J. W. Y. Lam and B. Z. Tang, J. Phys. Chem. Lett., 2015, 6, 3429–3436 CrossRef CAS PubMed;
(e) B. K. An, S. H. Gihm, J. W. Chung, C. R. Park, S. K. Kwon and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 3950–3957 CrossRef CAS PubMed;
(f) Z. P. Yu, Y. Y. Duan, L. H. Cheng, Z. L. Han, Z. Zheng, H. P. Zhou, J. Y. Wu and Y. P. Tian, J. Mater. Chem., 2012, 22, 16927–16932 RSC;
(g) M. D. Yang, D. L. Xu, W. G. Xi, L. K. Wang, J. Zheng, J. Huang, J. Y. Zhang, H. P. Zhou, J. Y. Wu and Y. P. Tian, J. Org. Chem., 2013, 78, 10344–10359 CrossRef CAS PubMed;
(h) Z. Zheng, Z. P. Yu, M. D. Yang, F. Jin, Q. Zhang, H. P. Zhou, J. Y. Wu and Y. P. Tian, J. Org. Chem., 2013, 78, 3222–3234 CrossRef CAS PubMed;
(i) L. K. Wang, Y. F. Shen, M. D. Yang, X. Z. Zhang, W. N. Xu, Q. J. Zhu, J. Y. Wu, Y. P. Tian and H. P. Zhou, Chem. Commun., 2014, 50, 8723–8726 RSC.
-
(a) S. Tao, Y. Zhou, C. S. Lee, S. T. Lee, D. Huang and X. Zhang, J. Phys. Chem. C, 2008, 112, 14603–14606 CrossRef CAS;
(b) X. Li, L. Yang, L. Zhao, X. L. Wang, K. Z. Shao and Z. M. Su, Cryst. Growth Des., 2016, 16, 4374–4382 CrossRef CAS.
- S. Kim, Q. Zheng, G. S. He, D. J. Bharali, H. E. Pudavar, A. Baev and P. N. Prasad, Adv. Funct. Mater., 2006, 16, 2317–2323 CrossRef CAS.
-
(a) Y. J. Dong, B. Xu, J. B. Zhang, X. Tan, L. J. Wang, J. L. Chen, H. G. Lv, S. P. Wen, B. Li, L. Ye, B. Zou and W. J. Tian, Angew. Chem., Int. Ed., 2012, 124, 10940–10943 CrossRef;
(b) S. Kim, H. E. Pudavar, A. Bonoiu and P. N. Prasad, Adv. Mater., 2007, 19, 3791–3795 CrossRef CAS.
- M. D. Yang, Y. Zhang, W. J. Zhu, H. Z. Wang, J. Huang, L. H. Cheng, H. P. Zhou, J. Y. Wu and Y. P. Tian, J. Mater. Chem. C, 2015, 3, 1994–2002 RSC.
- B. Xu, J. He, Y. Dong, F. Chen, W. Yu and W. J. Tian, Chem. Commun., 2011, 47, 6602–6604 RSC.
- G. Heinrich, S. Schoof and H. Gusten, J. Photochem., 1974, 3, 315–320 CrossRef CAS.
-
(a) A. Kivrak, Ö. F. Er, H. Kivrak, Y. Topal, M. Kus and Y. Çamlısory, Opt. Mater., 2017, 73, 206–212 CrossRef CAS;
(b) J. H. Ho, Y. H. Chen, L. T. Chou, P. W. Lai and P. S. Chen, Tetrahedron Lett., 2014, 55, 5727–5731 CrossRef CAS.
- X. Zhang, Z. Chi, X. Zhou, S. Liu, Y. Zhang and J. Xu, J. Phys. Chem. C, 2012, 116, 23629–23638 CrossRef CAS.
-
(a) Z. L. Wang, L. L. Yan, L. Zhang, Y. J. Chen, H. Li, J. B. Zhang, Y. Zhang, X. Li, B. Xu, X. Q. Fu, Z. C. Sun and W. J. Tian, Polym. Chem., 2014, 5, 7013–7020 RSC;
(b) H. Li, Z. Chi, X. Zhang, B. J. Xu, S. Liu, Y. Zhang and J. Xu, Chem. Commun., 2011, 47, 11273–11275 RSC;
(c) B. J. Xu, Z. G. Chi, X. F. Li, H. Y. Li, W. Zhou, X. Q. Zhang, C. C. Wang, Y. Zhang, S. W. Liu and J. R. Xu, J. Fluoresc., 2011, 21, 433–441 CrossRef CAS PubMed.
- H. Y. Li, Z. G. Chi, B. J. Xu, X. Q. Zhang, X. F. Li, S. W. Liu, Y. Zhang and J. R. Xu, J. Mater. Chem., 2011, 21, 3760–3770 RSC.
- H. C. Weiss, D. Blaser, R. Boese, B. M. Doughan and M. M. Haley, Chem. Commun., 1997, 1703–1704 RSC.
- W. J. Feast, P. W. Lövenich, H. Puschmann and C. Taliani, Chem. Commun., 2001, 5, 505–506 RSC.
- J. T. He, B. Xu, F. P. Chen, H. J. Xia, K. P. Li, L. Ye and W. J. Tian, J. Phys. Chem. C, 2009, 113, 9892–9899 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 29, 4332–4353 RSC.
- T. Chen, Z. Q. Chen, W. L. Gong, C. Li and M. Q. Zhu, Mater. Chem. Front., 2017, 1, 1841–1846 RSC.
- R. J. Wang, L. Diao, Q. Ren, G. Liu and S. Z. Pu, ACS Omega, 2019, 4, 309–3198 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1874463, 1874464, 1874465 and 1887433. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra09290k |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Yunfei Liang‡,
Gang Liu
,
Yunfei Liang‡,
Gang Liu