DOI:
10.1039/C9RA09245E
(Paper)
RSC Adv., 2020,
10, 4745-4754
Designing, docking and molecular dynamics simulation studies of novel cloperastine analogues as anti-allergic agents: homology modeling and active site prediction for the human histamine H1 receptor†
Received
7th November 2019
, Accepted 9th January 2020
First published on 29th January 2020
Abstract
The present study predicts a three-dimensional model for the histamine H1 receptor and the design of antihistamine inhibitors using cloperastine as the core molecule by docking studies. In this work, we predicted a three-dimensional structure of the histamine H1 receptor using the MODELLER9V7 software. The protein structure was developed based on the crystal structure of the histamine H1 receptor, the lysozyme chimera of Escherichia virus T4 (PDB ID: 3RZE_A) target collected from the PDB data bank. Using molecular dynamics simulation methods, the final predicted structure is obtained and further analyzed by VERIFY3D and PROCHECK programs, confirming that the final model is reliable. The drug derivatives of cloperastine were designed and docking was performed with the designed ligands along with the drug. The predicted model of the histamine H1 receptor structure is stable and confirms that it is a reliable structure for docking studies. The results indicate that MET 183, THR 184 and ILE 187 in the histamine H1 receptor are important determinant residues for binding as they have strong hydrogen bonding with cloperastine derivatives. The drug derivatives were docked to the histamine H1 receptor protein by hydrogen bonding interactions and these interactions played an important role in the binding studies. The molecule 1-{2-[(4-chlorophenyl) (phenyl) methoxy] ethyl}-4-methylenepiperidine showed the best docking results with the histamine H1 receptor. The docking results predicted the best compounds, which may act as better drugs than cloperastine and in the future, these may be developed for anti-allergy therapy.
1. Introduction
Histamine is a nitrogenous organic compound involved in immune responses such as allergy and it functions by combining with specific cellular histamine receptors such as H1, H2, H3 and H4, which are the members of the family of rhodopsin-like G protein-coupled receptors.1 The histamine H1 receptor (HRH1) is one of the four histamine receptors, and it plays an important role in different physiological functions such as inflammation, gastric acid secretion, mast cell-mediated chemotaxis and neurotransmitter release when bound to histamine.2,3 HRH1 is a member of class I G protein-coupled receptors (GPCRs) and it activates phospholipase C by interacting with the G proteins.4 GPCRs constitute the largest family of surface proteins with signal transduction functions.5,6 Among the available drugs blocking GPCRs, morphine (an opioid receptor antagonist), anti-allergic drugs inhibiting the histamine H1 and H2 receptors, antacids and antihypertensive beta-blockers are important.7 In various inflammatory effects such as negative inotropism, smooth muscle contraction, and depolarization, the HRH1 receptor is involved and is the important drug target for anti-allergic therapy, which is activated by endogenous histamine in allergic reactions. Antihistamine drugs block the functions of histamine by inhibiting it from binding to its receptors. The first histamine receptor antagonist was developed in 1937 and from then till now, more than 50 drugs have been introduced into the market.8 The first-generation drugs developed as antihistamines are clemastine, chlorpheniramine, diphenhydramine and triprolidine, and they cause marked sedation, central nervous system (CNS) dysfunction and anticholinergic adverse effects, which result in cognitive function impairment and therapy non-adherence.9 The second-generation antihistamine drugs such as astemizole, loratadine, terfenadine, cetirizine and fexofenadine are non-sedating antihistamines and were introduced into the market in the 1980s to reduce these side effects. Terfenadine and astemizole were removed from the market due to serious cardiovascular events related to torsade de pointes.10–12 Cloperastine is one of these drugs with different biological activities including GIRK channel inhibition,13–15 antihistamine activity and anticholinergic activity.16 Due to these properties, cloperastine shows some side effects such as sedation, somnolence and tussive effect.17 The precise mechanism of cloperastine is not fully known; in order to study the action of cloperastine on the histamine 1 receptor and to develop new anti-histamine drugs, we designed valid pharmacophore analogues of cloperastine for HRH1 inhibition agonists and used them in the screening for new lead analogues to find their interactions with the active residues of the HRH1 receptor by molecular docking. We modeled the HRH1 receptor via molecular dynamics-stabilized proteins and using a Ramachandran plot confirmed the reliability of the structure for docking. Later, active sites are predicted, indicating that MET 183, THR 184 and ILE 187 in the histamine H1 receptor are important amino acids involved in binding and the derivatives of cloperastine are docked into these active site residues to confirm the inhibitory action of the same. Here we studied the pharmacophore features that are essential for a histamine H1 inverse agonist and also designed new molecules with a greater affinity towards histamine H1 receptor.
2. Materials and methods
2.1. Domain identification and template search
All molecular simulations were performed on AMD 64 bits dual processing hi-end Linux desktop. Due to the lack of availability of the histamine H1 receptor 3D structure in the database, the amino acid sequence of the histamine H1 receptor from Homo sapiens (HRH1) (Uniprot_KB Accession Id: P35367) was obtained from the protein sequence databank. Histamine H1 receptor sequence was subjected to BLAST search against PDB to identify a suitable protein that shares similar structure of the query protein.18
The query sequence of Homo sapiens was submitted to the SBASE (http://pongor.itk.ppke.hu) server for domain selection. The predicted domains were searched by BLAST (http://blast.ncbi.nlm.nih.gov) against PDB to find out the related protein structure.19 The Swiss model search result, which showed the maximum sequence identity to the query protein HRH1, was selected as a template for further studies. The selected template protein was used as a reference structure for modeling the P35367 domain. Coordinates from the template protein to the structurally conserved regions, N-termini and C-termini and structurally variable regions (SVRs) were assigned to the template sequence based on satisfaction of spatial restraints. The sequence of the reference structures was extracted from the respective structure files and aligned with the target sequence using the default parameters in Clustal X2.
2.2. 3D model building of histamine H1 receptor
The crystal structure is not available for the histamine H1 receptor; thus, we have chosen the homology modeling methodology for three-dimensional structure building. The initial model of the histamine H1 receptor was built by using homology modeling methods and the MODELLER9V7 software,20 in which the program is based on comparative structure modeling. This software generated fifty models for the histamine H1 receptor and was based on the low objective function; the least energy model was selected for studies. Later, hydrogens were added to the three-dimensional structure stabilizing the protein by molecular dynamics simulation studies. The predicted model MD simulations were carried out using the NAMD 2.8 software using the CHARMM27 force field.21 The algorithm used is multiple-time-stepping with long-range electrostatics calculated every two steps and short-range forces calculated every step. The MD method used in this study is based on the Hamilton's equations of motion to get new velocities at the new positions. Finally, the model obtained is with new thermodynamic property information and stabilized in terms of RMSD.
2.3. Structure validation of histamine H1 receptor
The histamine H1 receptor structure with low Root Mean Square Deviation (RMSD) obtained from molecular dynamics studies is further analyzed by a Ramachandran plot to check the stereo chemical quality of the protein structures using the PROCHECK server and environment profile using the structure evaluation server ERRAT.22 Then, this protein can be used to predict active sites and for docking to the analogues.
2.4. Active site identification of histamine H1 receptor
After the final model was built, the possible binding sites of the histamine H1 receptor from Homo sapiens were searched based on the structural comparison of the template and the model build and also with the CASTp server (http://sts.bioe.uic.edu/castp). The final refined model of the histamine H1 receptor domain was developed using the SPDBV program based on the structure–structure comparison of the template.23
2.5. Docking studies with cloperastine derivatives
Docking studies were performed to gain insights into the binding conformation of pharmacophore analogues developed from the structural modifications of cloperastine using FRED (Open Eye Scientific Software, Santa Fe, NM). A set of 35 derivatives were designed for cloperastine and screened for minimal chemical criteria for docking studies using Molinspiration (http://www.molinspiration.com/). Free energy calculations were carried out using FRED 1.1 and the files generated were analyzed for their binding conformations.
2.6. Ligand molecules used for docking studies
Cloperastine was used as a parental compound for the construction of a library of lead molecules because cloperastine has several reactive centres in the molecule, which can be exploited in the synthesis of several derivatives and new ring systems for the purpose of evaluating their pharmaceutical potential by linking different amine groups; it was expected that the resulting derivatives may have interesting physiological properties. The docking results were validated by comparing the docking values with the experimental values of other drugs. The derivatives were designed in order to improve the molar refractivity and polarizability.
3. Results and discussion
3.1. Homology modeling of histamine H1 receptor domain
The most reliable and acceptable technique for predicting protein structures is homology modeling. It provides the geometry of one or more template proteins when sufficient sequence identities are given. If the sequence identity between the template and target protein is high enough, the resulting model may even be sufficiently accurate to perform structure-based drug design.24 The histamine H1 receptor contains two discontinuous rhodopsin-like GPCR superfamily-like domains from 45 to 162 amino acids in the sequence and 178 to 461 amino acids (ESI Fig. 1†). The BLAST search against PDB indicated that only 3RZE had a high level of sequence identity with the histamine H1 receptor domain. Structurally conserved regions for the predicted structure and template were determined by the multiple sequence alignment of the two sequences and superimposition of the two structures.
3.2. Identification of template
A high level of sequence identity should guarantee a more accurate alignment between the target sequence and template structure. The template 3RZE_A is a histamine receptor membrane protein, lysozyme chimera of Escherichia virus T4, and the source of this protein is Homo sapiens and bacteriophage T4.25 The results of BLAST analysis showed that only one reference protein 3RZE A (chain A, histamine H1 receptor, lysozyme chimera of Escherichia virus T4) showed maximum identity with the histamine H1 receptor domain. Here, 3RZE was selected as a template and used for further studies. The structure of the template 3RZE was obtained from PDB (ESI Fig. 2†). The sequence identity between the target sequence (HRH1) and template structure (3RZE_A) was found to be 67%. The characterization of the 3RZE A protein was described by predicting the secondary structure of the template sequence, indicating that the protein 3RZE A is a histamine H1 receptor, lysozyme chimera of Escherichia virus T4. It is a polypeptide chain consisting of 493 amino acids and is classified as a membrane protein and the source of this protein is Homo sapiens and bacteriophage T4. The secondary structure showed that it consists of 68% helical (25 helices; 308 residues) and 3% beta sheets (4 strands; 15 residues). Similarly the best template was obtained through the PSI-BLAST search of the PDB database for the selection of a template to model the histamine H1 receptor.
3.3. Sequence alignment
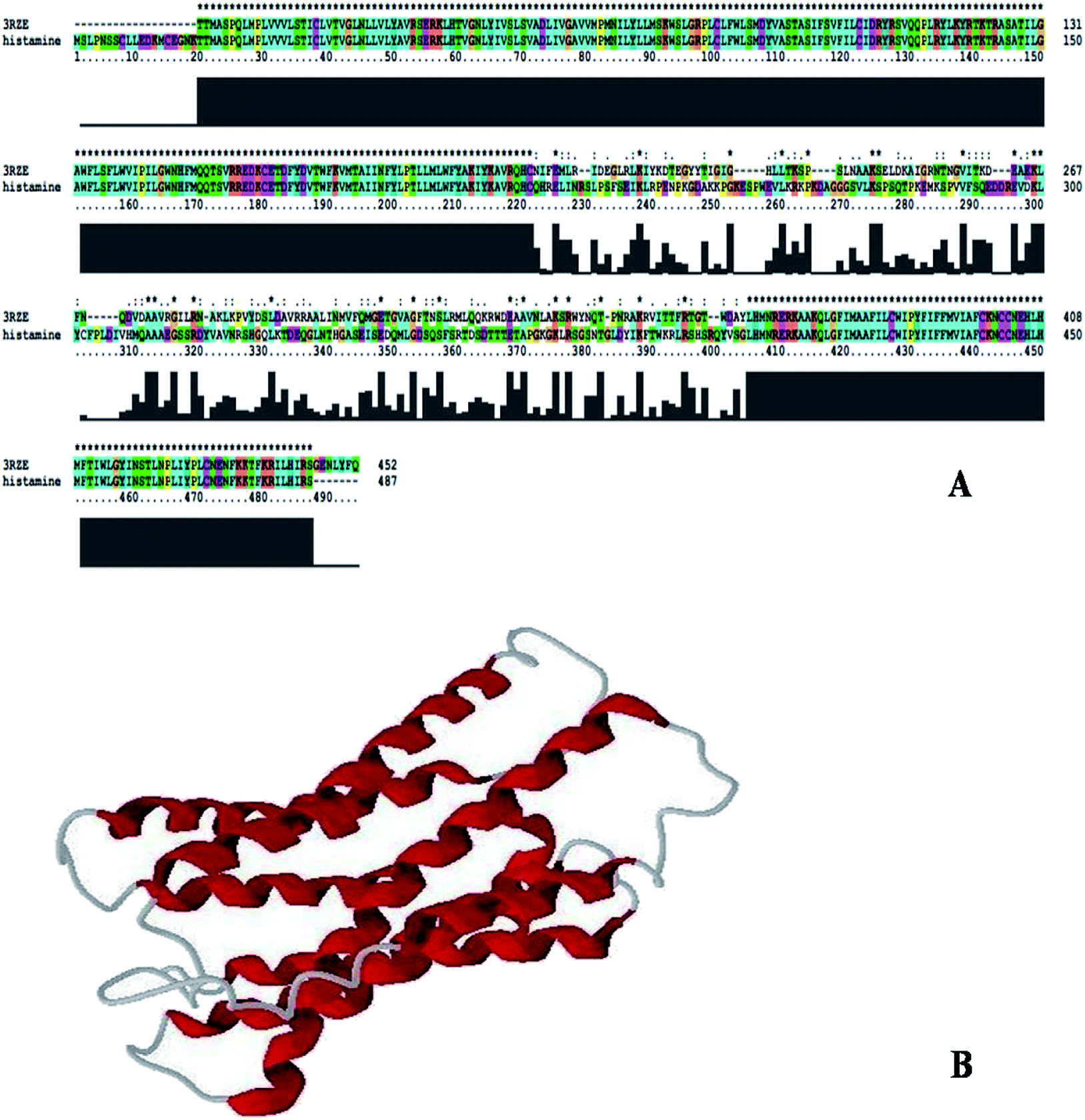
Sequence identity is very important in comparative protein modeling for assuring a more accurate alignment between the target protein and the template structure.26 First, a template search is conducted to develop a structure and for homology modeling, a minimum of 30% identity is needed between the sequence and the template.27 The results of the amino acid sequence alignment of the template protein and the target protein are shown in Fig. 1A and the alignment developed between the histamine H1 receptor and the template protein indicates that both the proteins are closely related in origin. Likewise, the sequence alignment of the histamine H1 receptor and 3RZE shares 67% sequence identity.
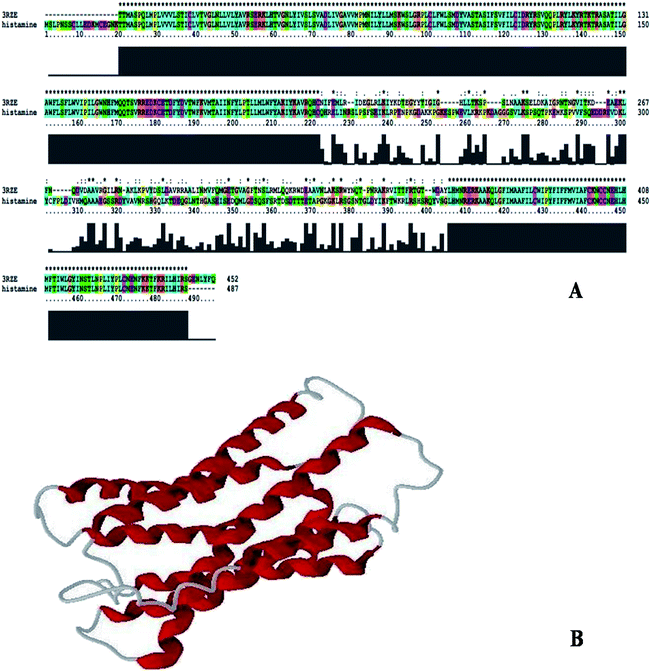 |
| | Fig. 1 (A) Alignment of histamine H1 receptor with template 3RZE. (B) 3D structure of histamine H1 receptor generated by Modeller9v7. | |
3.4. Homology modeling
In order to develop new inhibitors, a three-dimensional model of the histamine H1 receptor domain using MODELLER9V7 and the final refined model was obtained after energy minimization.28 With all the available data, the structure of the histamine H1 receptor protein was generated with MODELLER9V7 by using the 3RZE_A structure as the template (Fig. 1B). The 3D homology model of the target protein sequence was predicted using the crystal structural coordinates of the templates on the basis of the sequence alignment, whereas homology modeling and refinement were carried out through MODELLER 9V7 using base line commands specified by the software supplier. The secondary structural analysis of the developed histamine H1 receptor from Homo sapiens revealed that there was an expected similarity in the relative abundances of α-helix, β-sheet and other secondary structural conformations between the predicted model of the histamine H1 receptor and the template 3RZE was subjected to SBASE for domain identification and analysis.
3.5. Structural validation
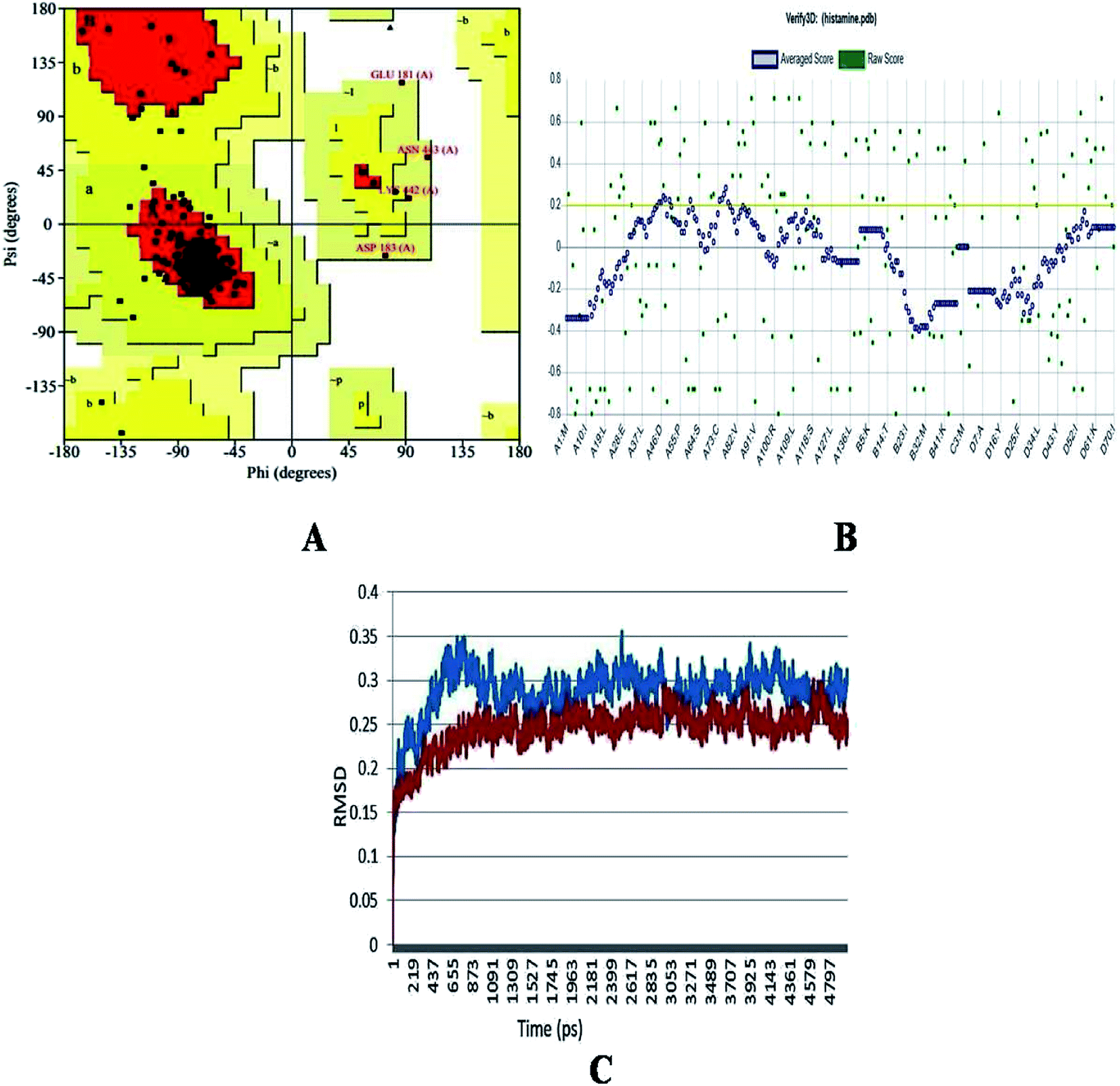
The generated structure of the histamine H1 receptor from Homo sapiens was studied using different parameters for validation purpose. The φ (psi) and ψ (pi) distributions of each residue of the histamine H1 receptor obtained from the Ramachandran plot showed that 91.1% residues were in the preferred region and 8.9% residues were in the additionally allowed region (Table 1 and Fig. 2A). The reliability confirmation of the generated structure was analyzed using PROCHECK (Fig. 2B). Expect glycine, there were no residues in the disallowed regions. The amino acids in the favored region indicate that the predicted model of the histamine H1 receptor is well built and more reliable. The RMSD analysis of the predicted structure was calculated by means of deviation with its template structure using molecular dynamics. The Cα RMSD and backbone RMS deviations for the structure and template were 0.25 Å and 0.3 Å, respectively (Fig. 2C). The final predicted structure was checked by the PROCHECK and Verify 3D programs and based on the results, the model reliability was confirmed. This difference in RMS deviations between the previous structure and after MD simulations confirmed that our predicted structure can be securely used in the consequent structural actions.29
Table 1 Ramachandran plot analysis of residues in favoured and allowed regions
| S. No |
Residues in plot |
Percentage |
| 1 |
Number of residues in favoured region |
216 (91.1%) |
| 2 |
Number of residues in allowed region |
17 (7.2%) |
| 3 |
Number of residues in generously allowed region |
4 (1.7%) |
| 4 |
Number of residues in outlier region |
0 (0.0%) |
| 5 |
% of non-glycine and non-proline residues |
237 (100.0%) |
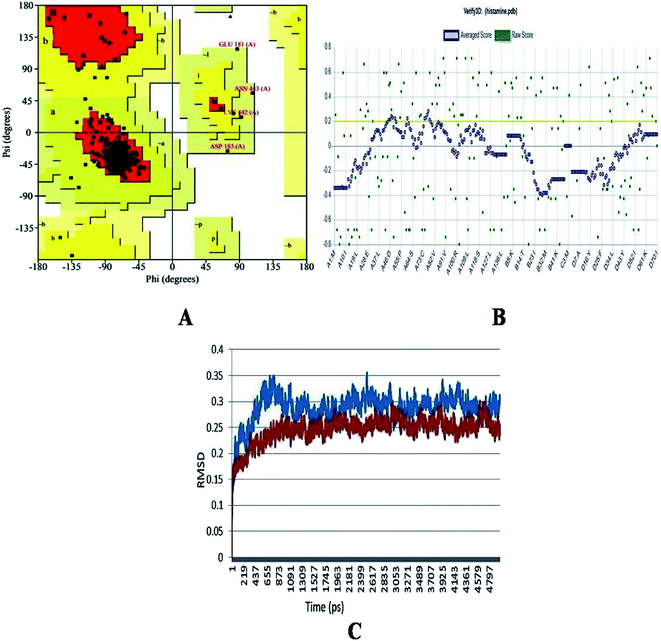 |
| | Fig. 2 (A) Ramachandran plot, (B) Verify_3D analysis and (C) RMSD studies of the histamine H1 receptor before molecular dynamics (blue) and after studies (red) for predicted structure of histamine H1 receptor. | |
3.6. Superimposition
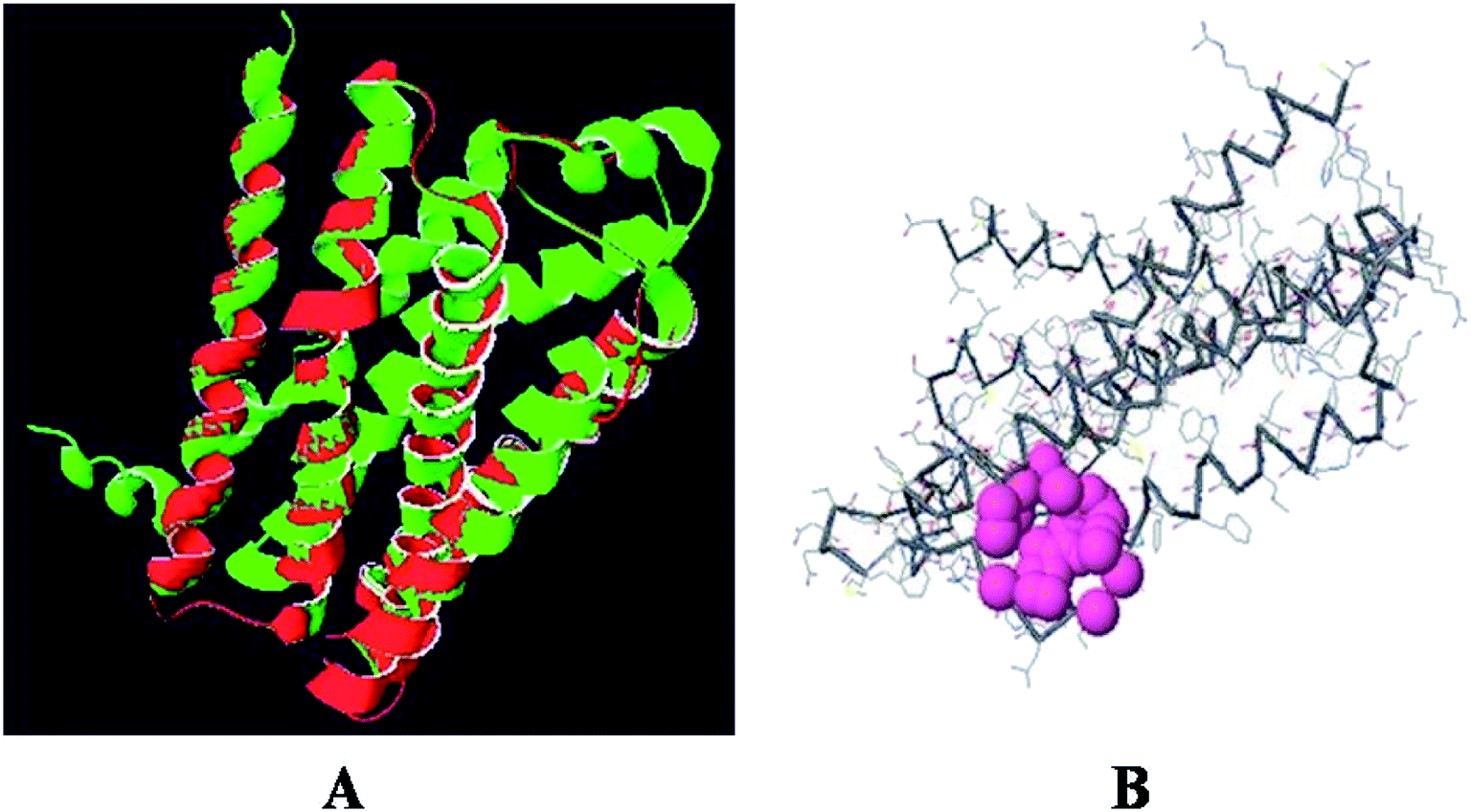
The superimposition of the histamine H1 receptor and the template 3RZE is shown in Fig. 3A. The calculated root mean square deviation of the trace between these structures is 0.85 Å. This predicted structure of the histamine H1 receptor was further used for the identification of active sites and for docking studies.
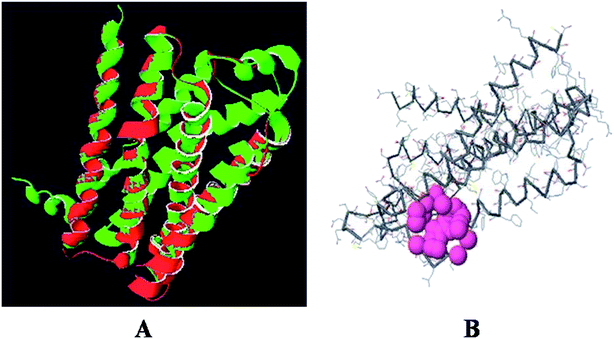 |
| | Fig. 3 (A) Superposition of the histamine H1 receptor (red) and template 3RZE (green). (B) Active site of histamine H1 receptor. | |
3.7. Active site identification
The predicted model binding sites need to be identified for docking studies. Due to the unavailability of crystal structure in the database, these active site residues need to be predicted and based on the highest pocket size, the binding region is selected to dock derivatives.30 Here, the active site of the histamine H1 receptor is predicted and docked with 36 cloperastine derivatives for the histamine H1 receptor antagonist activity. Since, the histamine H1 receptor and 3RZE are well conserved in both sequence and structure, their biological functions will be identical. The abundance of amino acid residues in the active sites of the histamine H1 receptor shows that the secondary structures are highly conserved and the residues are TRP 148, ILE 150, ILE 152, PHE 180, MET 183, THR 184, and ILE 187 (Fig. 3B).
3.8. Pharmacophore designing of cloperastine derivatives
Cloperastine and its derivatives were drawn based on the selected substituted scaffolds of the drug using the Chemsketch software. The QSAR structures were constructed for 35 compounds from the predicted descriptors (molar refractivity, polarizability, index of refraction, density, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (o/w), polar surface area, van der Waals volume, and molecular weight). The pharmacophore properties of these derivatives are given in Table 2. From the results, it is suggested that with small structural changes in cloperastine, new potentially active derivatives against the histamine H1 receptor can be developed. As the molar refractivity and index of refraction are associated with molecular volume and molecular weight, higher values increase the activity. Molecular polarizability is the capability of the molecular electronic system to be distorted by an external field, and this plays a major role in designing derivatives and their properties in biological activities.31 The aqueous solubility of the compound depends on its polarizability. The high polarizability of the molecule is expected to result in strong bondings with other molecules, which are measured by van der Waals interactions. Molar refractivity is related to molecular polarizability and inverse of molar volume.32 Molar refractivity (MR) is used to study the steric factor and is designated as the measure of the volume occupied by an individual atom or a group of atoms. Polarizability and molar refractivity increased with the size and the molecular weight of the studied cloperastine derivatives (Table 2). This is in agreement with the Lorentz-Lorenz formula, which states the relationship between the molar refractivity, polarizability and volume.33 Volume depends on density indirectly and low-density molecules are preferred for transport properties such as the blood–brain barrier penetration of the compounds. Bulkiness is related to the molecular weight of the derivatives. The absorption, diffusion and transportation of the derivatives are good for low-molecular-weight compounds when compared to those for high-molecular-weight compounds. The Lipinski's rule of five states that the permeation of a molecule is high when the molar refractivity is below 130, the molecular weight is under 500 g mol−1, logP is lower than 5, and the total number of atoms are between 20 and 70 with at least 5 H-donor and 10 H-acceptor atoms.34 The designed derivatives showed acceptable values of molar refraction, polarizability, density and index of refraction. Then, the designed derivatives were checked for molecular properties and bioavailability using the Molinspiration server (ESI Tables 1 and 2†). The MlogP calculation shows the lipophilic nature of the derivatives and is important in absorption. Transport properties were studied by using the topological polar surface area (TPSA) score by considering all polar atoms such as hydrogen, nitrogen, and oxygen. Good binding affinity and flexibility depend on the rotatable and non-rotatable bonds of the derivatives and here, all the derivatives have good score. Biological activity is measured by the bioactivity score predicting the physiological effect of the derivatives, i.e., whether they are biologically active.35 From these results, the derivatives with high binding activity and fewer side effects are predicted and can be used to develop new functional drugs.
P (o/w), polar surface area, van der Waals volume, and molecular weight). The pharmacophore properties of these derivatives are given in Table 2. From the results, it is suggested that with small structural changes in cloperastine, new potentially active derivatives against the histamine H1 receptor can be developed. As the molar refractivity and index of refraction are associated with molecular volume and molecular weight, higher values increase the activity. Molecular polarizability is the capability of the molecular electronic system to be distorted by an external field, and this plays a major role in designing derivatives and their properties in biological activities.31 The aqueous solubility of the compound depends on its polarizability. The high polarizability of the molecule is expected to result in strong bondings with other molecules, which are measured by van der Waals interactions. Molar refractivity is related to molecular polarizability and inverse of molar volume.32 Molar refractivity (MR) is used to study the steric factor and is designated as the measure of the volume occupied by an individual atom or a group of atoms. Polarizability and molar refractivity increased with the size and the molecular weight of the studied cloperastine derivatives (Table 2). This is in agreement with the Lorentz-Lorenz formula, which states the relationship between the molar refractivity, polarizability and volume.33 Volume depends on density indirectly and low-density molecules are preferred for transport properties such as the blood–brain barrier penetration of the compounds. Bulkiness is related to the molecular weight of the derivatives. The absorption, diffusion and transportation of the derivatives are good for low-molecular-weight compounds when compared to those for high-molecular-weight compounds. The Lipinski's rule of five states that the permeation of a molecule is high when the molar refractivity is below 130, the molecular weight is under 500 g mol−1, logP is lower than 5, and the total number of atoms are between 20 and 70 with at least 5 H-donor and 10 H-acceptor atoms.34 The designed derivatives showed acceptable values of molar refraction, polarizability, density and index of refraction. Then, the designed derivatives were checked for molecular properties and bioavailability using the Molinspiration server (ESI Tables 1 and 2†). The MlogP calculation shows the lipophilic nature of the derivatives and is important in absorption. Transport properties were studied by using the topological polar surface area (TPSA) score by considering all polar atoms such as hydrogen, nitrogen, and oxygen. Good binding affinity and flexibility depend on the rotatable and non-rotatable bonds of the derivatives and here, all the derivatives have good score. Biological activity is measured by the bioactivity score predicting the physiological effect of the derivatives, i.e., whether they are biologically active.35 From these results, the derivatives with high binding activity and fewer side effects are predicted and can be used to develop new functional drugs.
Table 2 Chemical properties of the cloperastine derivatives
| S. no |
Compounds (molecular formula) |
Formula weight |
Molar refractivity cm3 |
Index of refraction |
Density g cm−3 |
Polarisability cm3 |
| 1 |
Cloperastine (C20H24ClNO) |
329.86 |
96.11 ± 0.3 |
1.566 ± 0.02 |
1.120 ± 0.06 |
38.10 ± 0.510−24 |
| 2 |
Derivative 1 (C19H22ClNO2) |
331.83 |
93.10 ± 0.3 |
1.565 ± 0.02 |
1.162 ± 0.06 |
36.91 ± 0.510−24 |
| 3 |
Derivative 2 (C37H35Cl2NO2) |
596.58 |
173.74 ± 0.3 |
1.615 ± 0.02 |
1.198 ± 0.06 |
68.87 ± 0.510−24 |
| 4 |
Derivative 3 (C40H41Cl2NO4) |
670.66 |
191.73 ± 0.3 |
1.597 ± 0.02 |
1.192 ± 0.06 |
76.00 ± 0.510−24 |
| 5 |
Derivative 4 (C21H20ClNO) |
337.84 |
100.65 ± 0.3 |
1.624 ± 0.02 |
1.186 ± 0.06 |
39.90 ± 0.510−24 |
| 6 |
Derivative 5 (C22H22ClNO) |
351.86 |
105.48 ± 0.3 |
1.618 ± 0.02 |
1.168 ± 0.06 |
41.81 ± 0.510−24 |
| 7 |
Derivative 6 (C23H30N2O2) |
366.49 |
110.04 ± 0.3 |
1.575 ± 0.02 |
1.101 ± 0.06 |
43.62 ± 0.510−24 |
| 8 |
Derivative 7 (C21H26ClNO) |
343.89 |
100.41 ± 0.3 |
1.553 ± 0.02 |
1.097 ± 0.06 |
39.80 ± 0.510−24 |
| 9 |
Derivative 8 (C21H25ClN2O3) |
388.88 |
105.94 ± 0.3 |
1.589 ± 0.02 |
1.237 ± 0.06 |
41.99 ± 0.510−24 |
| 10 |
Derivative 9 (C21H28ClNO) |
345.90 |
102.13 ± 0.4 |
1.572 ± 0.02 |
1.11 ± 0.1 |
40.49 ± 0.510−24 |
| 11 |
Derivative 10 (C16H20N2) |
240.34 |
75.91 ± 0.3 |
1.556 ± 0.02 |
1.018 ± 0.06 |
30.09 ± 0.510−24 |
| 12 |
Derivative 11 (C17H21NO) |
255.35 |
79.56 ± 0.3 |
1.551 ± 0.02 |
1.024 ± 0.06 |
31.54 ± 0.510−24 |
| 13 |
Derivative 12 (C32H39NO2) |
469.65 |
144.65 ± 0.4 |
1.590 ± 0.03 |
1.09 ± 0.1 |
57.34 ± 0.510−24 |
| 14 |
Derivative 13 (C18H22BrNO) |
348.27 |
91.46 ± 0.3 |
1.561 ± 0.02 |
1.234 ± 0.06 |
36.25 ± 0.510−24 |
| 15 |
Derivative 14 (C32H39NO4) |
501.65 |
145.86 ± 0.3 |
1.596 ± 0.02 |
1.171 ± 0.06 |
57.82 ± 0.510−24 |
| 16 |
Derivative 15 (C16H19ClN2) |
274.78 |
80.80 ± 0.3 |
1.565 ± 0.02 |
1.107 ± 0.06 |
32.03 ± 0.510−24 |
| 17 |
Derivative 16 (C17H22N2O) |
270.36 |
81.86 ± 0.3 |
1.554 ± 0.02 |
1.043 ± 0.06 |
32.45 ± 0.510−24 |
| 18 |
Derivative 17 (C22H29N3O4S) |
431.54 |
117.69 ± 0.3 |
1.598 ± 0.02 |
1.252 ± 0.06 |
46.65 ± 0.510−24 |
| 19 |
Derivative 18 (C25H27ClN2) |
390.94 |
118.03 ± 0.3 |
1.617 ± 0.02 |
1.159 ± 0.06 |
46.79 ± 0.510−24 |
| 20 |
Derivative 19 (C13H22N4O3S) |
314.40 |
85.64 ± 0.3 |
1.558 ± 0.02 |
1.184 ± 0.06 |
33.95 ± 0.510−24 |
| 21 |
Derivative 20 (C19H28N2O4) |
348.43 |
95.66 ± 0.3 |
1.532 ± 0.02 |
1.130 ± 0.06 |
37.92 ± 0.510−24 |
| 22 |
Derivative 21 (C15H24N4S) |
292.44 |
84.56 ± 0.4 |
1.614 ± 0.03 |
1.20 ± 0.1 |
33.52 ± 0.510−24 |
| 23 |
Derivative 22 (C13H15ClN4O) |
278.73 |
75.19 ± 0.3 |
1.662 ± 0.02 |
1.371 ± 0.06 |
29.80 ± 0.510-24 |
| 24 |
Derivative 23 (C21H23NO3) |
337.41 |
99.62 ± 0.3 |
1.640 ± 0.02 |
1.221 ± 0.06 |
39.49 ± 0.510−24 |
| 25 |
Derivative 24C21H25N3O2S |
383.50 |
110.15 ± 0.5 |
1.652 ± 0.05 |
1.27 ± 0.1 |
43.66 ± 0.510−24 |
| 26 |
Derivative 25 (C22H23ClN2O2) |
382.88 |
105.85 ± 0.3 |
1.614 ± 0.02 |
1.261 ± 0.06 |
41.96 ± 0.510−24 |
| 27 |
Derivative 26 (C19H19ClN2) |
310.82 |
90.05 ± 0.3 |
1.625 ± 0.02 |
1.221 ± 0.06 |
35.69 ± 0.510−24 |
| 28 |
Derivative 27 (C20H24N2) |
292.41 |
92.23 ± 0.3 |
1.587 ± 0.02 |
1.065 ± 0.06 |
36.56 ± 0.510−24 |
| 29 |
Derivative 28 (C26H34N2O3) |
422.55 |
123.24 ± 0.3 |
1.555 ± 0.02 |
1.101 ± 0.06 |
48.85 ± 0.510−24 |
| 30 |
Derivative 29 (C22H22N2O) |
330.42 |
100.29 ± 0.4 |
1.641 ± 0.03 |
1.18 ± 0.1 |
39.75 ± 0.510−24 |
| 31 |
Derivative 30 (C9H16N6S) |
240.32 |
66.09 ± 0.5 |
1.645 ± 0.05 |
1.31 ± 0.1 |
26.20 ± 0.510−24 |
| 32 |
Derivative 31 (C16H18N2O2) |
270.32 |
76.43 ± 0.3 |
1.613 ± 0.02 |
1.231 ± 0.06 |
30.30 ± 0.510−24 |
| 33 |
Derivative 32 (C14H17ClN4S) |
308.82 |
85.45 ± 0.5 |
1.645 ± 0.05 |
1.31 ± 0.1 |
33.87 ± 0.510−24 |
| 34 |
Derivative 33 (C8H15N7O2S3) |
337.44 |
79.06 ± 0.5 |
1.808 ± 0.05 |
1.83 ± 0.1 |
31.34 ± 0.510−24 |
| 35 |
Derivative 34 (C14H16ClN3O) |
277.74 |
77.10 ± 0.3 |
1.655 ± 0.05 |
1.332 ± 0.06 |
30.56 ± 0.510−24 |
| 36 |
Derivative 35 (C17H20N2S) |
284.41 |
87.81 ± 0.3 |
1.615 ± 0.02 |
1.131 ± 0.06 |
34.81 ± 0.510−24 |
3.9. Docking studies of cloperastine and its derivatives with histamine H1 receptor
Previous studies show that docking is the best method to conduct protein-ligand binding, protein–protein binding and protein-compound binding studies.36–41 From the docking results, it was concluded that the conserved amino acid residues of the histamine H1 receptor played an important role in conserving a functional conformation and were involved in donor-substrate binding. The docking studies between the histamine H1 receptor and cloperastine derivatives are suitable for understanding the possible mechanisms of domain and inhibitor binding. The results of docking analysis are based on the free energy of binding and calculated RMSD values (Table 3). Among the designed cloperastine derivatives, all the 35 analogues satisfied the Lipinski's rule of five with zero violations for docking with the histamine H1 receptor structure.
Table 3 Docking scores of cloperastine derivatives with histamine H1 receptor
| Compounds |
No. of hydrogen bonds |
Chem score |
Chem guass |
PLP |
Screen score |
Total (kcal mol−1) |
| Cloperastine |
1 |
−17.38 |
−51.37 |
−44.18 |
−87.11 |
−200.04 |
| Derivative 1 |
1 |
−14.00 |
−50.15 |
−40.71 |
−91.25 |
−196.11 |
| Derivative 2 |
— |
— |
— |
— |
— |
— |
| Derivative 3 |
— |
— |
— |
— |
— |
— |
| Derivative 4 |
2 |
−20.72 |
−52.89 |
−52.83 |
−130.45 |
−256.89 |
| Derivative 5 |
1 |
−18.99 |
−51.76 |
−46.18 |
−95.91 |
−212.84 |
| Derivative 6 |
1 |
−7.71 |
−53.08 |
−30.97 |
−64.69 |
−156.45 |
| Derivative 7 |
1 |
−16.29 |
−48.23 |
−39.81 |
−77.39 |
−181.72 |
| Derivative 8 |
1 |
−4.78 |
−51.15 |
−37.59 |
−86.63 |
−180.15 |
| Derivative 9 |
1 |
−12.88 |
−42.02 |
−34.32 |
−71.14 |
−160.36 |
| Derivative 10 |
1 |
−14.35 |
−39.92 |
−38.16 |
−78.47 |
−170.90 |
| Derivative 11 |
1 |
−15.38 |
−41.73 |
−42.20 |
−82.47 |
−181.72 |
| Derivative 12 |
— |
— |
— |
— |
— |
— |
| Derivative 13 |
1 |
−12.25 |
−46.07 |
−30.87 |
−72.19 |
−161.38 |
| Derivative 14 |
— |
— |
— |
— |
— |
— |
| Derivative 15 |
1 |
−13.22 |
−40.47 |
−36.42 |
−72.83 |
−162.94 |
| Derivative 16 |
1 |
−12.11 |
−42.41 |
−38.91 |
−80.45 |
−173.88 |
| Derivative 17 |
1 |
+8.67 |
−45.41 |
+2.38 |
−13.81 |
−52.93 |
| Derivative 18 |
1 |
−19.04 |
−58.85 |
−46.87 |
−102.77 |
−227.53 |
| Derivative 19 |
1 |
−5.14 |
−46.05 |
−41.05 |
−88.16 |
−180.4 |
| Derivative 20 |
1 |
−9.14 |
−59.14 |
−43.15 |
−81.32 |
−192.77 |
| Derivative 21 |
— |
— |
— |
— |
— |
— |
| Derivative22 |
— |
— |
— |
— |
— |
— |
| Derivative 23 |
1 |
−11.31 |
−50.83 |
−30.32 |
−76.17 |
−168.63 |
| Derivative 24 |
1 |
−7.73 |
−41.89 |
−32.37 |
−72.89 |
−154.88 |
| Derivative 25 |
1 |
−9.17 |
−54.89 |
−29.85 |
−69.27 |
−163.18 |
| Derivative 26 |
1 |
−15.30 |
−48.15 |
−36.67 |
−90.39 |
−190.51 |
| Derivative 27 |
1 |
−9.16 |
−48.52 |
−30.68 |
−63.50 |
−151.86 |
| Derivative 28 |
1 |
−7.55 |
−48.92 |
−29.89 |
−71.02 |
−157.38 |
| Derivative 29 |
1 |
−14.19 |
−42.14 |
−42.97 |
−79.34 |
−178.64 |
| Derivative 30 |
— |
— |
— |
— |
— |
— |
| Derivative 31 |
— |
— |
— |
— |
— |
— |
| Derivative 32 |
— |
— |
— |
— |
— |
— |
| Derivative 33 |
1 |
+1.18 |
−39.33 |
−30.92 |
86.43 |
−155.5 |
| Derivative 34 |
— |
— |
— |
— |
— |
— |
| Derivative 35 |
1 |
−15.91 |
−43.38 |
−37.71 |
−84.83 |
−181.83 |
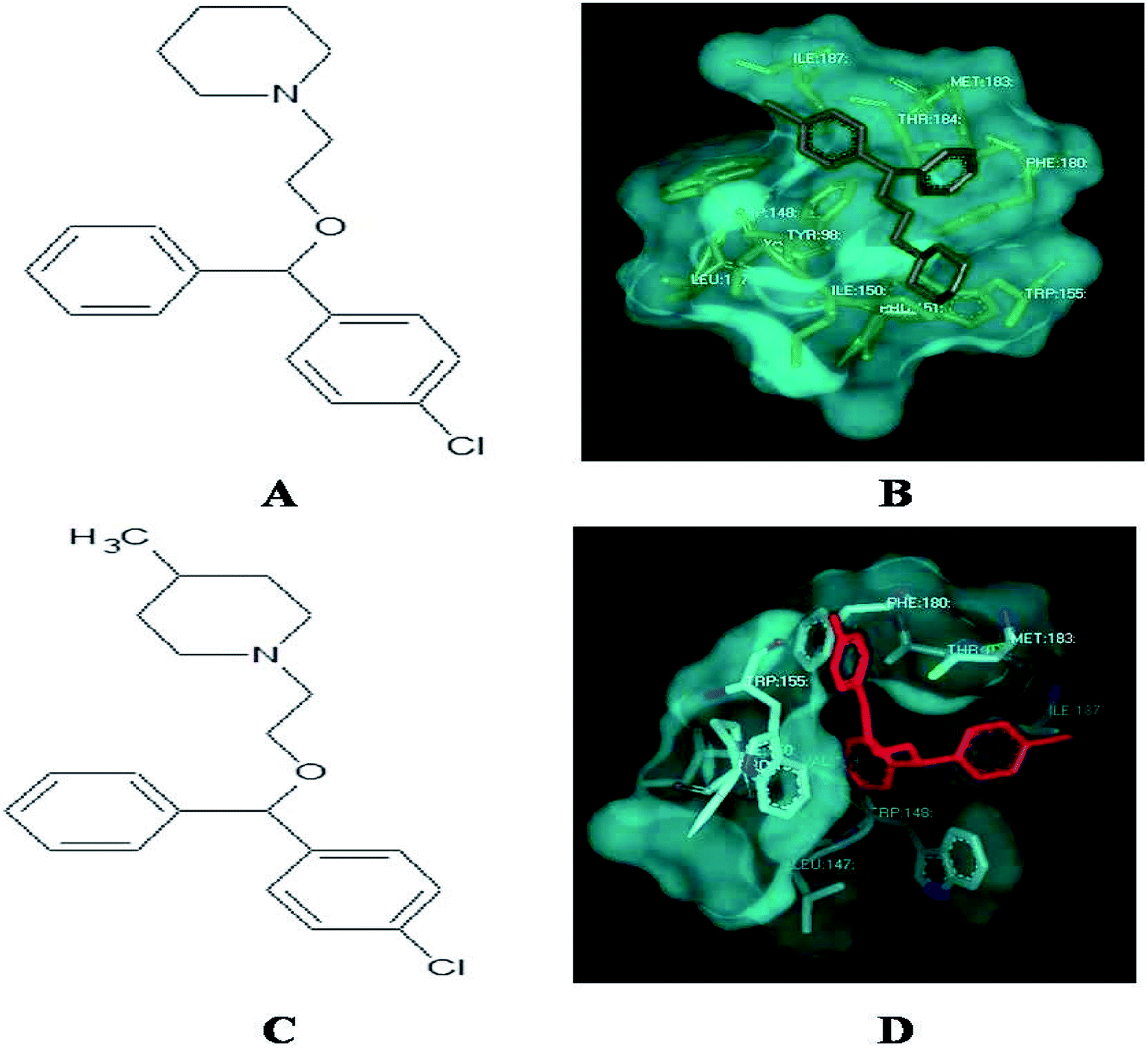
Among the derivatives docked, 2, 3, 12, 14, 21, 22, 30, 31, 32 and 33 showed no bonding with the histamine H1 receptor, whereas 4, 5 and 18 showed better docking energy values than cloperastine (Table 3). The docking confirmations of cloperastine (Fig. 4A and B) and all 35 molecules confirmed the inhibitory activity on the histamine H1 receptor (ESI Fig. 3a–3z†). Cloperastine formed one hydrogen bond with the histamine H1 receptor in docking, and N12 formed a hydrogen bond with the 3rd hydrogen of THR184 of the histamine H1 receptor, whereas the derivative 4 showed the best docking by forming two hydrogen bonds: one hydrogen bond was formed between N12 of the derivative 4 and the 6th hydrogen atom of ILE187, and the second hydrogen bond was formed between the 2nd hydrogen atom of the substituted methyl group and N28 of ILE187 in the histamine H1 receptor. Among all docking conformations, the derivative 4 (1-{2-[(4-chlorophenyl) (phenyl)methoxy]ethyl}-4-methylenepiperidine) showed better predicted binding free energy of −256.89 kcal mol−1 to the histamine H1 receptor than cloperastine (Fig. 4C and D).
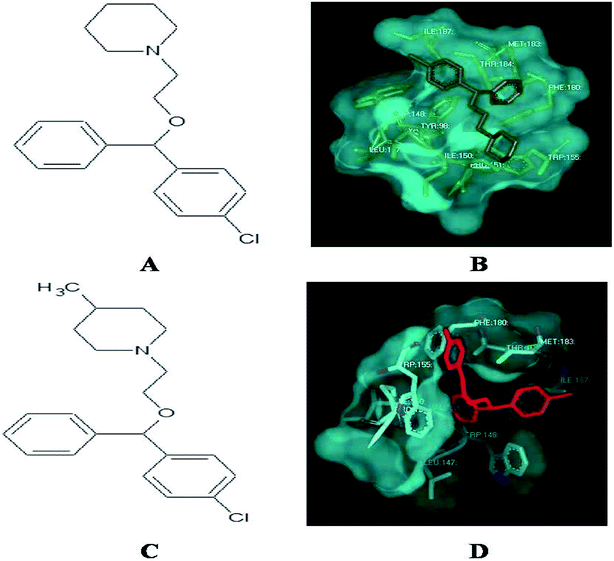 |
| | Fig. 4 (A) Structure of cloperastine. (B) Docking studies of histamine H1 receptor with cloperastine. (C) Structure of cloperastine derivative 4. (D) Docking studies of histamine H1 receptor with cloperastine derivative 4. | |
4. Conclusion
The histamine H1 receptor is one of the G protein-coupled receptors and plays an important role in allergy. It is involved in a variety of physiological actions such as inflammation, gastric acid secretion, neurotransmitter release and mast cell-mediated chemotaxis upon binding to histamine. In conclusion, here, a new model was generated to study the drug inhibition by using the homology modeling method and validation studies confirmed that the predicted structure was reliable. Till now, no model is available in the database for the selected histamine H1 receptor. Multiple sequence alignment has been done to study the secondary conserved regions of the sequences using Clustalx and the identity is 67%. As is well-known, hydrogen bonds play important roles for the structure and functions of biological molecules. In this study, it was found that TRP 148, ILE 150, ILE 152, PHE 180, MET 183, THR 184, and ILE 187 are important residues involved in docking studies with derivatives through strong hydrogen bonding. Also, new analogues of cloperastine are designed and checked for inhibitory activity. Among the designed derivatives, the derivative 4 shows good binding energy calculations; it has a methyl group at C4 of the cyclohexane ring of clopestarine and can be used as a drug in the histamine H1 receptor-involved diseases in the future. The molecule 1-{2-[(4-chlorophenyl) (phenyl)methoxy]ethyl}-4-methylenepiperidine showed the best docking results with the histamine H1 receptor.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are thankful to Akshaya Biologicals, Hyderabad for the help and support in running software used in this work.
References
- G. W. Canonica and M. Blaiss, Antihistaminic, anti-inflammatory, and antiallergic properties of the nonsedating second-generation antihistamine desloratadine: a review of the evidence, World Allergy Organ. J., 2011, 4, 47–53 CrossRef CAS PubMed.
- H. H. Andersen, J. Elberling and L. Arendt-Nielsen, Human surrogate models of histaminergic and non-histaminergic itch, Acta Derm.-Venereol., 2015, 95, 771–777 CrossRef CAS PubMed.
- M. M. Wouters, M. Vicario and J. Santos, The role of mast cells in functional GI disorders, Gut, 2015, 65, 155–168 CrossRef PubMed.
- A. Krishnan, M. S. Almén, R. Fredriksson and H. B. Schiöth, The origin of GPCRs: identification of mammalian like Rhodopsin, Adhesion, Glutamate and Frizzled GPCRs in fungi, PLoS One, 2012, 7, e29817 CrossRef CAS PubMed.
- A. R. Thomsen, B. Plouffe, T. J. Cahill, A. K. Shukla, J. T. Tarrasch, A. M. Dosey, A. W. Kahsai, R. T. Strachan, B. Pani, J. P. Mahoney, L. Huang, B. Breton, F. M. Heydenreich, R. K. Sunahara, G. Skiniotis, M. Bouvier and R. J. Lefkowitz, GPCR-G Protein-β-Arrestin Super-Complex Mediates Sustained G Protein Signaling, Cell, 2016, 166, 907–919 CrossRef CAS PubMed.
- B. Trzaskowski, D. Latek, S. Yuan, U. Ghoshdastider, A. Debinski and S. Filipek, Action ofmolecular switches in GPCRs—theoretical and experimental studies, Curr. Med. Chem., 2012, 19, 1090–1109 CrossRef CAS.
- A. S. Hauser, S. Chavali, I. Masuho, L. J. Jahn, K. A. Martemyanov, D. E. Gloriam and M. M. Babu, Cell, 2018, 172, 41–54.e19 CrossRef CAS PubMed.
- J. A. Allen and B. L. Roth, Strategies to discover unexpected targets for drugs active at Gprotein–coupled receptors, Annu. Rev. Pharmacol. Toxicol., 2011, 51, 117–144 CrossRef CAS PubMed.
- R. J. Hoagland, E. N. Deitz, P. W. Myers and H. C. Cosand, Antihistaminic drugs for colds; evaluation based on a controlled study, J. Am. Med. Assoc., 1950, 143, 157–160 CrossRef CAS PubMed.
- I. C. Camelo-Nunes, New antihistamines: a critical view, Jornal de Pediatria, 2006, 82, S173–S180 CrossRef.
- R. Leurs, M. K. Church and M. Taglialatela, H1-antihistamines: inverse agonism, anti-inflammatory actions and cardiac effects, Clin. Exp. Allergy, 2002, 32, 489–498 CrossRef CAS.
- F. Soeda, Y. Fujieda, M. Kinoshita, T. Shirasaki and K. Takahama, Centrally acting nonnarcotic antitussives prevent hyperactivity in mice: Involvement of GIRKchannels, Pharmacol., Biochem. Behav., 2016, 144, 26–32 CrossRef CAS PubMed.
- G. Yamamoto, F. Soeda, T. Shirasaki and K. Takahama, Is the GIRK Channel a PossibleTarget in the Development of a Novel Therapeutic Drug of Urinary Disturbance?, Yakugaku Zasshi, 2011, 131, 523–532 CrossRef CAS PubMed.
- K. Kawaura, S. Honda, F. Soeda, T. Shirasaki and K. Takahama, A Novel AntidepressantlikeAction of Drugs Possessing GIRK Channel Blocking Action in Rats, Yakugaku Zasshi, 2010, 130, 699–705 CrossRef CAS.
- E. Gregori-Puigjane, V. Setola, J. Hert, B. A. Crews, J. J. Irwin, E. Lounkine, L. Marnett, B. L. Roth and B. K Shoichet, Identifying mechanism-of-action targets for drugs and probes, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11178–11183 CrossRef CAS PubMed.
- I. Svízenská, P. Dubový and A. Sulcová, Cannabinoid Receptors 1 and 2 (CB1 and CB2),Their Distribution, Ligands and Functional Involvement in Nervous System Structures —A Short Review, Pharmacol., Biochem. Behav., 2008, 90, 501–511 CrossRef PubMed.
- S. Cuzzocrea and M. Catania, Pharmacological and clinical overview of cloperastine intreatment of cough, Ther. Clin. Risk Manage., 2011, 7, 83 CrossRef PubMed.
- R. Leinonen, F. G. Diez, D. Binns, W. Fleischmann, R. Lopez and R. Apweiler, UniProt Archive, Bioinformatics, 2004, 20, 3236–3237 CrossRef CAS PubMed.
- G. M. Boratyn, A. A. Schäffer, R. Agarwala, S. F. Altschul, D. J. Lipman and T. L. Madden, Domain enhanced lookup time accelerated BLAST, Biol. Direct, 2012, 7, 12 CrossRef CAS PubMed.
- J. Ko, H. Park, L. Heo and C. Seok, GalaxyWEB server for protein structure prediction andrefinement, Nucleic Acids Res., 2012, 40, W294–W297 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu and S. Zhong, CHARMM generalforce field: a force field for drug-like molecules compatible with the CHARMM all-atomadditive biological force fields, J. Comput. Chem., 2010, 31, 671–690 CAS.
- J. R. Daddam, M. R. Dowlathabad, S. Panthangi and P. Jasti, Molecular docking and Pglycoproteininhibitory activity of flavonoids, Interdiscip. Sci.: Comput. Life Sci., 2014, 6, 167–175 CrossRef CAS PubMed.
- M. Kurjogi, P. Satapute, S. Jogaiah, M. Abdelrahman, J. R. Daddam, V. Ramu and L.-S. P. Tran, Computational Modeling of the Staphylococcal Enterotoxins and Their Interactionwith Natural Antitoxin Compounds, Int. J. Mol. Sci., 2018, 19, 133 CrossRef PubMed.
- P. Narendra Kumar, T. H. Swapna, M. Y. Khan, J. R. Daddam and H. Bee, Moleculardynamics and protein interaction studies of lipopeptide (Iturin A) on α- amylase ofSpodoptera litura, J. Theor. Biol., 2017, 415, 41–47 CrossRef PubMed.
- J. Zhang, C. H. Luan and G. V. W. Johnson, Identification of the N-terminal functionaldomains of Cdk5 by molecular truncation and computer modeling, Proteins: Struct., Funct., Genet., 2002, 48, 447–453 CrossRef CAS PubMed.
- G. Raghava, S. M. Searle, P. C. Audley, J. D. Barber and G. J. Barton, OXBench: A benchmarkfor evaluation of protein multiple sequence alignment accuracy, BMC Bioinf., 2003, 4, 47 CrossRef CAS PubMed.
- W. Willy and S. Klaus, Investigating a back door mechanism of actin phosphate release bysteeredmolecular dynamics, Proteins: Struct., Funct., Bioinf., 1999, 35, 262–273 CrossRef.
- T. Shimamura, M. Shiroishi, S. Weyand, H. Tsujimoto, G. Winter, V. Katritch, R. Abagyan, V. Cherezov, W. Liu, G. W. Han, T. Kobayashi, R. C. Stevens and S. Iwata, Structure of the human histamine H1 receptor complex with doxepin, Nature, 2011, 475, 65–70 CrossRef CAS PubMed.
- N. K. Singh, B. C. Pakkkianathan, M. Kumar, J. R. Daddam, S. Jayavel, M. Kannan, G. C. Pillai and M. Krishnan, Computational studies on molecular interactions of 6- thioguanosineanalogs with anthrax toxin receptor 1, Interdiscip. Sci.: Comput. Life Sci., 2012, 4, 183–189 CrossRef CAS PubMed.
- D. J. Rayalu, C. Selvaraj, S. K. Singh, R. Ganeshan, N. U. Kumar and P. Seshapani, Homologymodeling, active site prediction, and targeting the anti – hypertension activity throughmolecular docking on endothelin – B receptor domain, Bioinformation, 2012, 8, 81–86 CrossRef PubMed.
- P. Pacák, Molar refractivity and interactions in solutions I. Molar refractivity of some monovalent ions in aqueous and dimethyl sulfoxide solutions, Chem. Pap., 1989, 43, 289–500 Search PubMed.
- P. Ertl, B. Rohde and P. Selzer, Fast calculation of molecular polar surface area as a sum of fragment based contributions and its application to the prediction of drug transport properties, J. Med. Chem., 2000, 43, 3714–3717 CrossRef CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23, 4–25 CrossRef.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- J. Tolls, M. Muller and A. Willing, A new concept for the environmental risk assessment of poorly water soluble compounds and its application to consumer products, Integr. Environ. Assess. Manage., 2009, 5, 374–378 CrossRef CAS PubMed.
- J. F. Wang, D. Q. Wei, L. Li and S. Y. Zheng, 3D structure modeling of cytochrome P4502C19 and its implication for personalized drug design, Biochem. Biophys. Res. Commun., 2007, 355, 513–519 CrossRef CAS.
- B. Webb and A. Sali, Protein structure modeling with MODELLER, Methods Mol. Biol., 2014, 1137, 1–15 CrossRef CAS PubMed.
- J. F. Wang, Insights into the Mutation-Induced HHH Syndrome from Modeling HumanMitochondrial Ornithine Transporter-1, PLoS One, 2012, 7, e3104 Search PubMed.
- A. Sircar, K. A. Sanni, J. Shi and J. J. Gray, Analysis and Modeling of the Variable Region ofCamelid Single Domain Antibodies, J. Immunol., 2011, 186, 6357–6367 CrossRef CAS PubMed.
- B. D. Weitzner, J. R. Jeliazkov, S. Lyskov, N. Marze, D. Kuroda, R. Frick, J. Adolf-Bryfogle, N. Biswas, R. L. Dunbrack and J. J. Gray, Modeling and docking of antibodystructures with Rosetta, Nat. Protoc., 2017, 12, 401–416 CrossRef CAS PubMed.
- P. Kotha, J. Rayalu Daddam, S. V. R. Sai Gopal Divi, S. R. Dakinedi and M. Dowlathabad, Modelling simulation phylogenetics of leukemia FMS tyrosine kinase 3 (FLT3), Onl. J. Vet. Res, 2015, 16, 8–17 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09245e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *d
*d