
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of structural, electronic and magnetic properties of breathing metal–organic framework MIL-47(Mn): a first principles approach†
Mohammadreza Hosseini a,
Danny E. P. Vanpouckebc,
Paolo Giannozzide,
Masoud Berahmanf and
Nasser Hadipour*a
a,
Danny E. P. Vanpouckebc,
Paolo Giannozzide,
Masoud Berahmanf and
Nasser Hadipour*a
aDepartment of Physical Chemistry, Tarbiat Modares University, Tehran, Iran. E-mail: Hadipour@modares.ac.ir
bUHasselt, Institute for Materials Research (IMO-IMOMEC), Agoralaan, 3590 Diepenbeek, Belgium
cIMOMEC, IMEC vzw, 3590 Diepenbeek, Belgium
dDipartimento di Scienze Matematiche, Informatiche e Fisiche, Università degli Studi di Udine, Via delle Scienze 208, 33100 Udine, Italy
eCNR-IOM DEMOCRITOS, SISSA, Trieste, Italy
fDepartment of Electrical and Computer Engineering, Advanced Graduate University of Technology, Kerman, Iran
First published on 29th January 2020
Abstract
The structural, electronic and magnetic properties of the MIL-47(Mn) metal–organic framework are investigated using first principles calculations. We find that the large-pore structure is the ground state of this material. We show that upon transition from the large-pore to the narrow-pore structure, the magnetic ground-state configuration changes from antiferromagnetic to ferromagnetic, consistent with the computed values of the intra-chain coupling constant. Furthermore, the antiferromagnetic and ferromagnetic configuration phases have intrinsically different electronic behavior: the former is semiconducting, the latter is a metal or half-metal. The change of electronic properties during breathing posits MIL-47(Mn) as a good candidate for sensing and other applications. Our calculated electronic band structure for MIL-47(Mn) presents a combination of flat dispersionless and strongly dispersive regions in the valence and conduction bands, indicative of quasi-1D electronic behavior. The spin coupling constants are obtained by mapping the total energies onto a spin Hamiltonian. The inter-chain coupling is found to be at least one order of magnitude smaller than the intra-chain coupling for both large and narrow pores. Interestingly, the intra-chain coupling changes sign and becomes five times stronger going from the large pore to the narrow pore structure. As such MIL-47(Mn) could provide unique opportunities for tunable low-dimensional magnetism in transition metal oxide systems.
Introduction
Metal–Organic Frameworks (MOFs) are a class of novel crystalline materials composed of metal(-oxide) nodes and organic linker molecules1–3 This combination of nodes and linkers results in a crystal lattice with amazing properties such as high internal surface area, porosity and chemical4 and physical tunability.5 Due to these unique properties, MOFs are being developed in applications such as gas storage,6–8 water filtering,9,10 catalysis,11,12 photocatalysis,13 drug delivery,14,15 energy storage16 and also their luminescence properties have been attractive for some applications.17,18 MOFs are also exceedingly interesting for fundamental research focusing on exotic and low dimensional physics in materials science.19 Breathing MOFs are a subclass of MOFs having the ability to reversibly change their unit cell volume by as much as 50% under changes in temperature,20 guest molecules,21 or external pressure.22 Also, this volume change is reported for MOFs out of the breathing class.23 Several comprehensive reviews on their properties and applications are available in the literature,24,25 reporting the existence of multiple breathing MOFs: MIL-53(Al),26 COK-69(Ti),27 SHF61.28 Of these, several appear to have a MIL-47/MIL-53 type topology with different transition metal nodes. Much theoretical and experimental work is being performed to study their intrinsic properties and possible potential applications.29–31 In particular, the spin configuration was linked to the sample type: single crystal samples present mainly antiferromagnetic chains, while powder samples show mainly ferromagnetic configurations.32 The very weak inter-chain spin-coupling makes the MIL-47(V) also an interesting quasi-1D material.31 The small energy differences and barriers between different structures make their study very challenging, even for modern high-accuracy DFT calculations.33 Similar as the UiO-66(Zr),34 the electronic structure of the MIL-47(V) can be tuned by means of linker functionalization.4 In their theoretical study of a series of breathing MIL-53(X), with X = Fe, V, Sc, Cr, In, Ga, and Al, Ling and Slater observe a significant change of electronic properties, in particular of the band gap, as a result of the large-pore to narrow-pore transition.35 The electronic and magnetic properties of DUT-8 (Ni), another flexible MOF with MIL-47 topology, were investigated by Trepte et al.36 Their results show that the magnetic ordering of nodes can be tuned by altering the ligands which coordinate to the transition metals.Manganese is one of the elements with a very rich variety of electronic and magnetic properties in solid-state materials.37,38 Munn et al. investigated Mn-based MOFs with a MIL-47 topology, containing both 1,4-benzenedicarboxylate (BDC) and pyridine-N-oxide (PNO) linkers. Although the MOF is paramagnetic at room temperature, they observe anti-ferromagnetic behavior at low temperature.39 A high capacity electrode Mn-based material with high performance in energy storage field was introduced by Liu et al.40 and an ultra-layered Mn MOF was found to be a good candidate for lithium storage.41 The remarkable properties of breathing MOFs combined with the versatility of Mn, which is shown theoretically in previous literature,42 motivate us to study a breathing Mn-based MOF: MIL-47(Mn). Recently a new MOF, Mn(II) unsaturated metal nodes and BDC linker, has been synthesized and its structural change toward water adsorption was observed.43 It may be promising for future synthesis of MIL-47(Mn).
In this work, the structural, electronic and magnetic properties of the compound, MIL-47(Mn) are calculated for both of large (LP) and narrow pore (NP) morphology using periodic density functional theory (DFT). We compare the results for MIL-47(Mn) with MIL-47(V) examining in detail differences and similarities.
Computational details
DFT calculations
DFT calculations,44 in particular periodic DFT calculations with a plane wave basis set, are increasingly used to study MOFs.45,46 The advantage of a plane wave basis set is its ability to optimize the atomic positions as well as the shape and volume of the unit cell, while being unbiased and free from basis set superposition error.47 All calculations have been performed within DFT as implemented in the Quantum ESPRESSO (QE) package.48 We use ultrasoft pseudo-potentials, and the Perdew, Burke and Ernzerhof (PBE)49 exchange–correlation functional. Previous studies have shown that dispersive interactions (c.q., van der Waals forces) are crucial for the accurate optimization of the atomic structure and the relative stability of MOFs, especially in breathing ones.50 The DFT-D2 correction, formulated by Grimme et al.51 and implemented in the QE package, was applied to obtain more reliable structures. The kinetic cut-off energy for plane waves is set to 475 eV. The first Brillouin zone is sampled using a 2 × 2 × 6 and 2 × 6 × 6 Monkhorst–Pack k-point grid for LP and NP, respectively. The total and partial density of states are computed using a denser sampling grid of 3 × 3 × 9 and 3 × 9 × 9 k-points for LP and NP, respectively. Atomic charges are calculated from the electron density, using the Hirshfeld-I atoms-in-molecules partitioning scheme, which is know to provide consistent and high quality atomic charges in porous materials52–54 A spherical Lebedev grid of 1202 grid points is used in combination with a logarithmic radial grid.55,56 The threshold for convergence was set to 1.0 × 10−5 electron.Structural model of the MIL-47(Mn)
The initial atomic structures were constructed starting from the crystallography files for both the LP and NP MIL-47(V) CCDC 1419980 and 1419981,33 respectively. In these structures the V atoms were replaced by Mn. All schematic presentations of materials are done using the VESTA57 visualization software. In order to study the magnetic properties, spin-polarized calculations were performed. The Mn atoms were considered as the magnetic centers and different configurations were studied for both LP and NP. Fig. 1 shows typical ball-and-stick representation of our system. | ||
| Fig. 1 Structure of MIL-47(Mn): (a) large pore (b) narrow pore (c) metal oxide chain in which all of the inequivalent Mn–O bonds and octahedral related parameters are shown (d) pore angle. Atomic species: violet Mn, red O, brown C, white H atoms. | ||
Due to the relatively flat potential energy surface (PES) of breathing MOFs, accurate structure optimization can be a challenging task.33 We have therefore taken a two-step approach. In our first step, the geometry is optimized without taking the spin configuration into account, as it was shown, in case of the MIL-47(V), that the local spin configuration has only a minor influence on the geometry of the system.31 For the LP, first the atomic positions of MIL-47(Mn) were optimized at fixed volume. Then, a variable-cell relaxation was performed, in which both the atomic positions and the cell vectors were optimized. Also, the same procedure was performed for NP version. The resulting LP configuration had an orthorhombic lattice while the NP configuration had a monoclinic one.
Starting from these pre-converged geometries, spin-polarized structure optimizations were performed, as a second step, to obtain the geometries of the five possible inequivalent spin configurations. These spin configurations are schematically represented in Fig. 2: FMa and FMb (ferromagnetic chains), AFa and AFb (antiferromagnetic chains), and MIX (50% ferromagnetic and 50% antiferromagnetic chains). For each spin configuration, both fixed and variable cell optimizations are performed and compared. Due to nearly flat PES, the variable-cell relaxations are not sufficiently accurate for breathing MOFs, in agreement with earlier findings.33
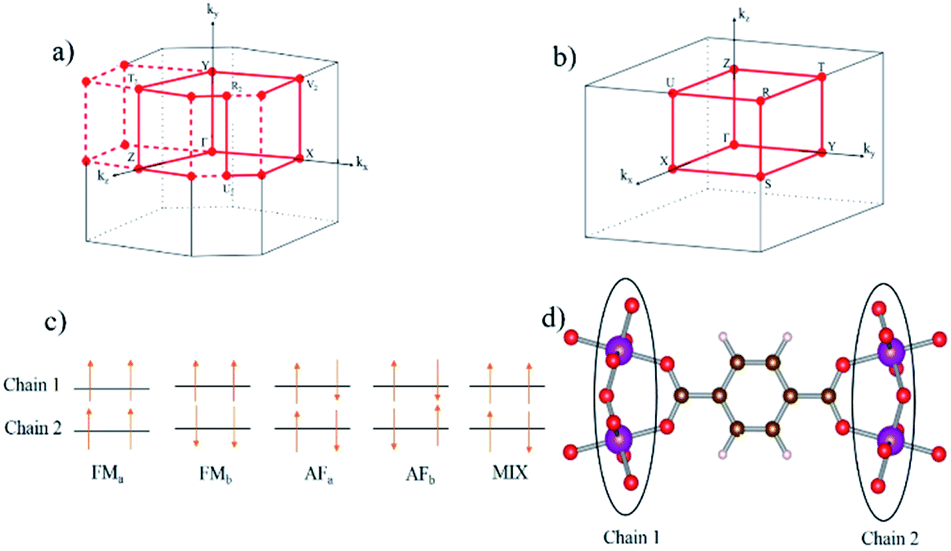 | ||
| Fig. 2 Reciprocal lattice and different high symmetry points of (a) monoclinic and (b) orthorhombic lattice is used to calculate the band structure for NP and LP geometry respectively. (c) Different magnetic configurations of our structure (d) magnetic centers in real space of two different Mn–O chains. | ||
The energetically most stable structure obtained in the fixed volume calculations, AFa, was selected and the optimal volume for the AFa spin configuration was obtained through a fitting to the Murnaghan equations of state (EOS).60 Then, starting from this structure, the atomic positions for the different spin configurations were obtained by optimizing the geometry under fixed volume constraints, after which the electronic and magnetic properties of the different spin configurations were computed. The difference between volumes obtained through EOS-fitting and variable-cell optimization is very small (∼0.05%). Therefore, we used the computationally less expensive variable-cell optimization for the NP.
Results and discussions
Structural properties
The cell parameters of the LP and NP structures are listed in Table 1. The computed volume is smaller than for MIL-47(V), which is used as initial geometry. This difference in volume originates mainly from the shorter c-axis. Table 1 indicates some structural features for MIL-47(Mn) like materials, MIL-53(Al)58 and (Ga)59 which have been reported experimentally. One can find MIL-47(Mn) has been optimized in smaller volume than them for both LP and NP crystal phase. As shown in Table 1, the breathing behavior results in a drastic change in volume. In addition, the symmetry changes from orthorhombic (LP) to monoclinic (NP). Such a volume change has been found to significantly alter physical and chemical properties such as the electronic structure.35 Let us therefore focus on the MnO6 complex and the metal-oxide chain part of the unit cell. Table 2 shows the energy and structure parameters of different magnetic configurations for both LP and NP phases. The global and local geometry of the MIL-47(Mn) MOF is shown in Fig. 1.| Structure | a | b | c | α | β | γ | Volume |
|---|---|---|---|---|---|---|---|
| LP-MIL-47(Mn) | 16.20 | 13.88 | 6.50 | 90 | 90 | 90 | 1460.70 |
| NP-MIL-47(Mn) | 20.98 | 6.51 | 6.44 | 90 | 113 | 90 | 806.76 |
| LP-MIL-47(V)33 | 16.23 | 13.97 | 6.85 | 90 | 90 | 90 | 1553.12 |
| NP-MIL-47(V)33 | 21.11 | 6.84 | 6.77 | 90 | 112.38 | 90 | 904.86 |
| Exp-LP-MIL-53(Al)58 | 16.91 | 12.67 | 6.62 | 90 | 90 | 90 | 1419.0 |
| Exp-NP-MIL-53(Al)58 | 20.82 | 6.87 | 6.61 | 90 | 119.95 | 90 | 863.9 |
| Exp-LP-MIL-53(Ga)59 | 16.68 | 13.21 | 6.72 | 90 | 90 | 90 | 1479.7 |
| Exp-NP-MIL-53(Ga)59 | 19.83 | 6.86 | 6.71 | 90 | 103.88 | 90 | 886.3 |
Comparing the relative energies for each of the magnetic configurations, we note that the spin ground state changes from an anti-ferromagnetic configuration to a ferromagnetic one upon collapse of the pores (LP to NP transition). The anti-ferromagnetic ground state for the LP phase makes this system similar to the MIL-47(V), while the FM ground state for the NP makes it different from the MIL-47(V). The relative energies, listed in Table S1,† indicate that the energy of transition between different magnetic configurations for LP is much smaller than for the phase transition between LP and NP. Here, we focus on the differences in geometry of distinct magnetic structures. We found that the magnetic configuration can affect some structural parameters, especially metal-oxide clusters ones.
Generally, all clusters show deviations from a perfectly octahedral complex, due to the presence of different ligands around the Mn atom (O2− in axial direction and BDC− in plane of complex). Mn–O bonds can be divided into two categories: the bonds d2 between Mn and the oxygen atoms of the BDC organic linkers, forming the plane of the octahedral complex, and the bonds d1 between Mn and oxygen atoms of metal oxide chains (cf. Fig. 1). Interestingly, in contrast to MIL-47(V)—where two different bond lengths along c-axis had been reported,31 we observe only one bond length along the MnO chain. As a result, we also find a volume difference between MIL-47(Mn) and MIL-47(V).
The +4 oxidation state of the metal in the MIL-47 framework leaves the V and Mn with one and three unpaired electrons in t2g orbitals, respectively. So, the presence of a Jahn–Teller effect is expected for the V version of MIL-47 in addition to a larger volume compared to MIL-47(Mn). The obtained length for d1 is in good agreement with previous works, presenting a length typically observed in bulk manganese oxide phases.61 On the other hand, d2 length is sufficiently close to the obtained value for the Mn–O bond length in organometallic environments.62
As shown in Table S1,† there is a difference of almost 0.2 angstrom between them. This difference in bond length results from the different strength of two ligands O2− and BDC− which induce different fields of strength. To characterize the MnO chains further, we study two relevant angles of the MnO6 complexes (cf. Fig. 1). First, the “super-exchange” angle σ, is defined as the Mn–O–Mn angle in the metal-oxide chain; the second, the O–Mn–O angle, φ, is part of octahedral complex. The super-exchange angle obtained for NP is smaller than for LP. Also, in contrast to MIL-47(V) the ferromagnetic configuration results in smaller σ in both crystal structures which can be originated from difference in number of unpaired electrons between Mn and V. This phenomena could be investigated in terms of the Goodenough–Kanamori rules.63 In this context, super-exchange angles σ = 90 and 180 degrees are indicative of ferromagnetic and antiferromagnetic coupling, respectively.
Accordingly, in this research antiferromagnetic coupling was observed for the larger angles, while the smaller ones show the ferromagnetic behavior. The angle φ of the octahedral complex is nearly identical for both LP and NP, and present a small deviation from the perfect octahedral configuration in which this angle is 90°.
As mentioned before, breathing occurs through the change of cell shape and volume. The opening angle, ω, is defined as the angle made by three Mn atoms and follows the pore opening (cf. Fig. 1d) angle can be considered as a quantitative parameter for breathing MOFs and has been computed as 81.2° and 37.2° for LP and NP, respectively which is within 1° of the opening angles for LP and NP configurations of the MIL-47(V).33 It should be noted that although DFT+U could be considered as a suitable functional correction for describing the electronic structure of transition metal oxides, the U-dependence of other materials properties, including the lattice parameters, makes it less suited for the investigation of the atomic structure. With regard to the system at hand, previous reports on manganese-oxide compounds have shown that PBE+U tends to overestimate some structural features.64
Electronic properties
The electronic band structures for all magnetic configurations are calculated along the high-symmetry lines of the first Brillouin zone of LP and NP structures, shown in Fig. 2. Densities of states (DOS) and DOS projected on atomic orbitals (PDOS) are reported for both of large and narrow pore which crystallize into orthorhombic and monoclinic lattice systems, respectively. With our unit cell containing four Mn centers, five inequivalent magnetic configurations are possible (cf. Fig. 2). Although the ground state is antiferromagnetic AFa, investigation of other spin configurations may be worthwhile to gain a deeper understanding of the electronic behavior. In the case of MIL-47 topology, the coordination environment requires a +4 oxidation state of the transition metal. The electronic structure of Mn4+ contains three unpaired d electrons, giving rise to a 3/2 spin per Mn center. The PDOS of 4s and 3d with different ml numbers for Mn of LPAFa (presented in S5†) confirm the Mn4+ configuration. Fig. 3 presents the electronic band structure of the zero total-magnetization configurations, FMb and AFa systems, in the LP geometry (AFb is similar to AFa so this is presented in ESI†). One immediately notices an important difference between these two systems. The AFa configuration presents a small bandgap, while in the FMb system several bands cross the Fermi level, making it a metallic system. A similar behavior is observed for AFb, shown in Fig. S1,† and FMa, presented in Fig. 5. For the ferromagnetic configurations, the valence bands cross the Fermi energy along Γ–Z, Z–Y, Y–T and U–X, and S–R lines which all are oriented along the c-axis of the system (Mn–O chains). Along the orthogonal directions the bands near the Fermi level are almost perfectly flat, indicative of quasi-1D behavior, similar to what was observed for the MIL-47(V) system.31 There are minor differences between FMa and FMb spin up configurations (but also between AFa and AFb), showing that some coupling between the chains is present but it is small. Both antiferromagnetic systems have the same band gap, about 0.5 eV, at the same position in the first Brillouin zone. Closer comparison with the electronic band structure of MIL-47(V) shows, in case of the antiferromagnetic configurations, the presence of two almost parallel bands just below the Fermi-level. These bands are associated with the additional unpaired d electrons of Mn4+ (when compared to V4+).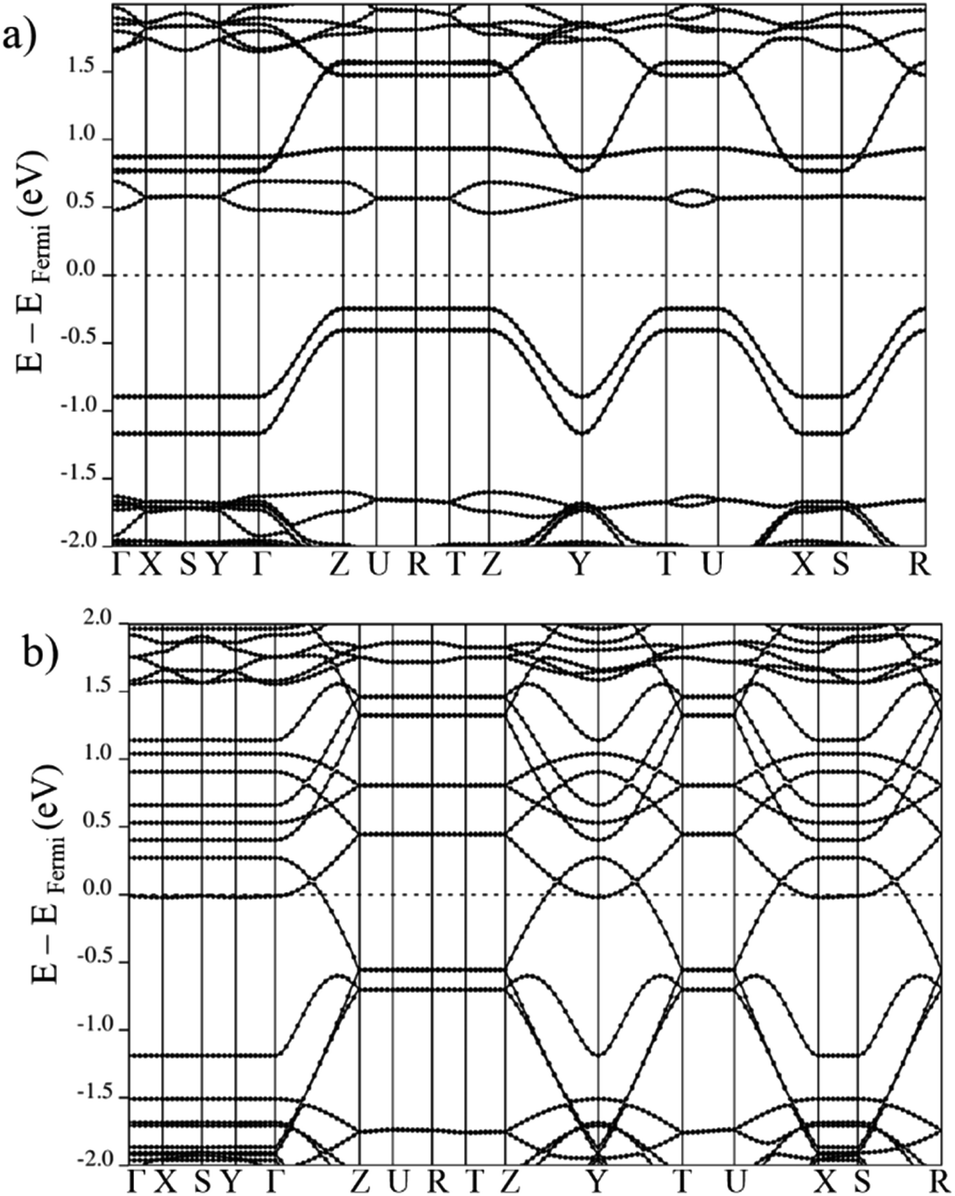 | ||
| Fig. 3 Electronic band structure of large pore geometry in (a) AFa (b) FMb magnetic configuration around Fermi level. | ||
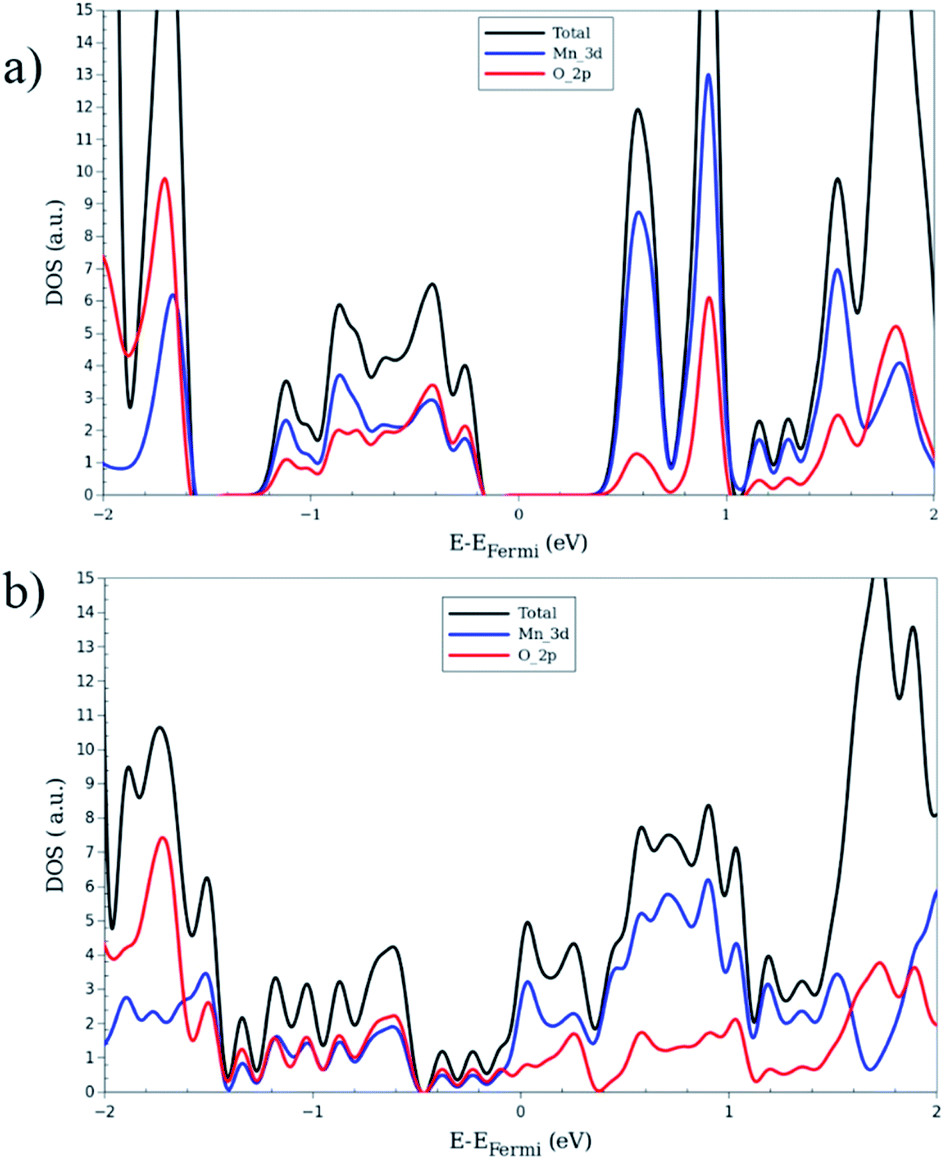 | ||
| Fig. 4 Total and projected density of states of large-pore geometry in (a) AFa (b) FMb magnetic configuration. The black, blue and red lines represent total, Mn 3d and O 2p contributions respectively. | ||
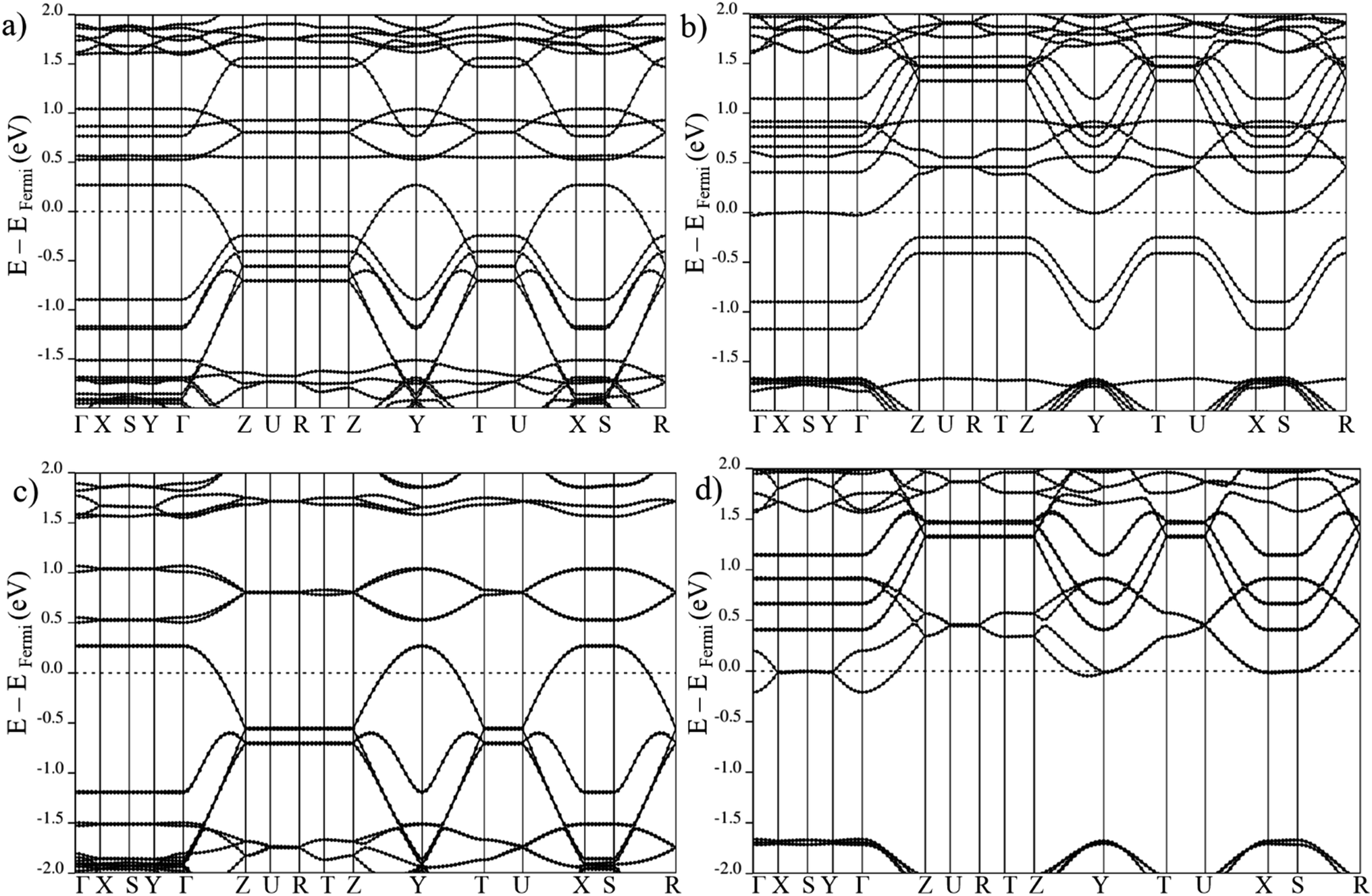 | ||
| Fig. 5 Electronic band structure of large pore geometry in (a) MIX spin up, (b) MIX spin down, (c) FMa spin up and (d) FMa spin down magnetic configuration around Fermi level. | ||
On the other hand, in case of the ferromagnetic spin configuration, with non-zero total magnetization (MIX and FMa), a remarkable difference between majority and minority spin is observed. In the case of MIX (cf. Fig. 5), the valence band in the spin up crosses the Fermi level but for spin down the conduction band with the Fermi level. Same behavior is seen for the FMa configuration with the down spin conduction band coinciding with the Fermi level while the spin up valence band crosses it. In the FMa case, the degeneracy of the coinciding conduction band is even lifted by weak in-plane coupling of the Mn centers of neighboring chains.
To investigate the electronic structure from a different perspective, the DOS and PDOS are calculated for all configurations. Fig. 4 shows them for FMb and AFa as representative systems of zero total magnetization, Fig. S3† exhibits the plots for MIX and FMa of non-zero magnetization case. For the systems with zero global magnetization: FMb, AFa and AFb, the spin-up and spin-down DOS's are identical so only the up component is shown. Comparison of Fig. 4 and S2† shows that AFa and AFb present a nearly identical DOS, in stark contrast with the FMa and FMb systems, which results from the difference in the global magnetization of the latter. A near identical DOS can be an additional indication for weak inter-chain interaction. In order to obtain a more detailed insight in the electronic properties of the LP structures, we consider the projection of the DOS onto atomic orbitals. The main contribution around the Fermi level comes from the metal-oxide complex. For this reason, the PDOS analysis is only performed for Mn and O atoms. In Fig. 4, it can be seen that the valence band contains nearly the same contribution for both Mnd and Op electrons, showing a strong hybridization between them and formation of chemical bonding. In contrast, the conduction band presents mainly a Mnd character. Looking at the band diagram for AFa and MIX into detail, some nearly flat bands are observed along the whole Brillouin zone, in the energy range from 0.5 to 1.0 eV above the Fermi level. These give rise to the sharp Mnd peak in the corresponding DOS plots.
So far, the electronic properties of the LP in different magnetic configurations were described. Let us now focus on the electronic features of the NP structure. Ling and Slater35 noted that the band gap of a NP is usually reduced with respect to the LP, in case of MIL-53. This reduction is attributed to a smaller volume and a larger interaction between organic linkers. In this context, we investigate the AFb and FMa magnetic configurations for the NP as two case of studies. Fig. 6 depicts the band structure, DOS and the projected (PDOS) for AFb magnetic configuration of NP crystal. In contrast to the LP, the NP DOS also presents a significant Cp character in addition to the Op and Mnd contributions near the Fermi level especially in valence band region. As a result of the crystal volume reduction and of the increase of interactions between the organic linkers, the contribution of carbon atoms around Fermi level is enhanced compared to the LP.
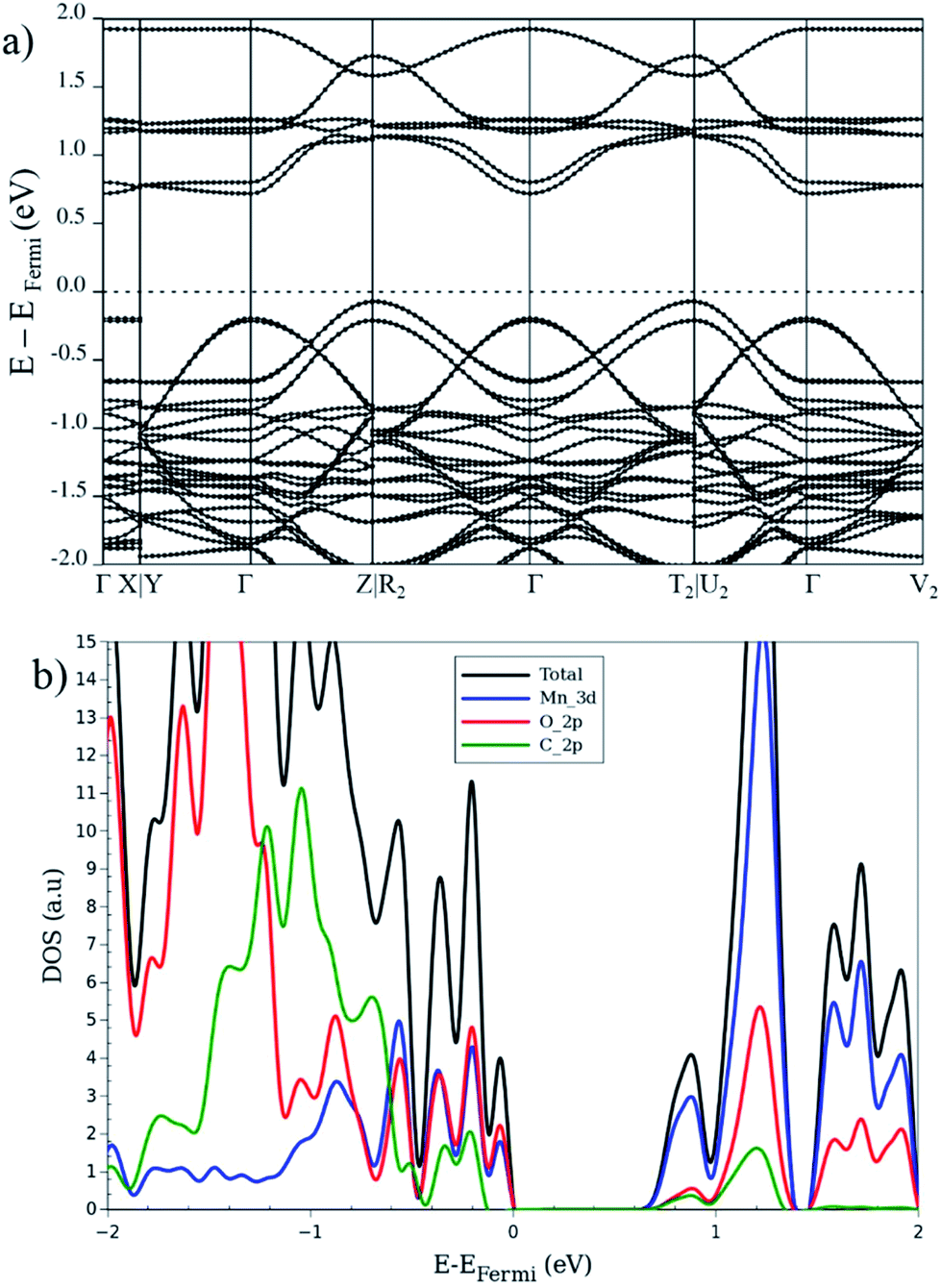 | ||
| Fig. 6 (a) Band structure, (b) total and projected DOS, for the narrow-pore geometry in the AFb magnetic configuration. | ||
From band structure analysis, in contrast to the same configuration in LP, no flat bands in valence region can be seen and electronic dispersion is present throughout the first Brillouin zone. On the other hand, for the FMa configuration, like in the same configuration for LP, the band diagram for the spin-up states crosses the Fermi level and a band gap, about 1.2 eV at the Γ point appears for the spin-down states. In the latter structure, the carbon contribution is not observable below the Fermi level and only appears above it. In contrast, the AFa and AFb configurations present a larger contribution of carbon atoms both below and above the Fermi level. In case of the FMa and MIX configurations with a global non-zero magnetization, the up and down spins show remarkable differences. The electronic band structure of AFb presents a strong dispersion of the valence electrons along the high-symmetry lines of the first Brillouin zone for the NP (cf. Fig. 6). In contrast, for FMa, as shown in Fig S4,† a flat band in the XY plane is seen while the dispersion along the paths parallel to c-axis remain. Furthermore, the conduction band is a combination of several (almost entirely) flat bands originated from Mnd, Op and Cp electrons. This dependency of electronic properties on the magnetic configuration makes the investigation of the spin–orbit coupling within this MOF of interest for future study. Recently, spin spirals have been observed in manganese oxide chains experimentally65 and confirmed by theoretical methods. This behavior can also occur for the MIL-47(Mn) system due to presence of 1-D Mn–O chain. However, this is beyond the scope of the current study and will be considered for future work.
Charge density analysis
Hirshfeld-I charges were calculated for the different spin configurations of the LP and NP geometry of the MIL-47(Mn).52,53 The different inequivalent sites are indicated in Fig. 7, while average values are presented in Table S2.† The standard deviation on these values is, with the exception of the H atoms in the NP geometry, below 0.01e, and in most cases even below 0.001e. As can be seen in Table S2,† the average charges show no significant variation between the various spin configurations, indicating the different spin configurations do not give rise to a different local chemistry. Even more, also the transition of the NP to the LP has only minor impact on the atomic charges. The largest relative variation is seen in the atomic charge of the Cc atoms (about 7%), which could be related to the bending of the terephthalate linker.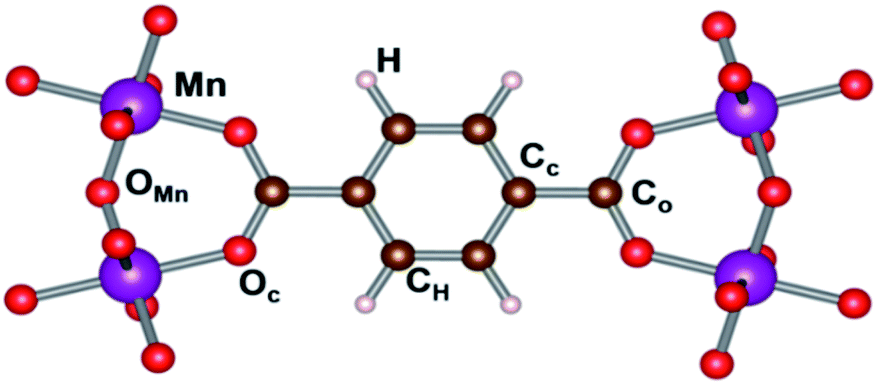 | ||
| Fig. 7 Inequivalent atomic positions in MIL-47(Mn). | ||
Interestingly, if we compare the charge of the Mn to that of V in a MIL-47(X) topology, we see that both elements have nearly the same charge, with the Mn one, slightly smaller than the 2.44e observed for V, supporting the assumption Mn to be in a +4 oxidation state.31 In contrast, the O ions of the metal-oxide chain have a significantly larger negative charge of −1.16e of OMn instead of −1.01 for OV in MIL-47(V), while the O atoms in the plane of the MnO6 octahedra have the same charge as in the VO6 case. This shows the Oc and OMn atoms present a different bonding with the Mn atom. Furthermore, it shows the transition-metal oxide chains to have a stronger polarization in case of Mn, in line with the electronegativity of the elements. In contrast, the polarization between the linker and the transition metal oxide chain is significantly smaller in than for the MIL-47(V), with the linkers having a charge of about −1.25e, for MIL-47(Mn), compared to −1.41e, for MIL47(V).33 This shows us that the polarization within the framework can be tuned by selecting different metals. Furthermore, as charges in these MOFs are rather local, polarization gradients could be designed in mixed metal compounds.
Magnetic properties
The magnetic properties of transition metals, owing to their unpaired electrons, are of great interest to chemistry and physics.66,67 In this context, MOFs can be viewed as a large lattice of organic linkers and widely separated transition metals centers. Such a configuration is very well suited for the observation of low-dimensional behavior. As the spin ground state is different for the LP and NP structures, this could also give rise interesting magnetic transitions. The computed local charges on the Mn centers of the different magnetic configurations (both LP and NP) indicate the same oxidation state is present for all configurations: in this case +4 which is typical one for MIL-47.In our system, we define two coupling constants: the intra-chain coupling Jc and inter-chain coupling Ji. We calculated the coupling constants using an Ising type model, eqn (1). Such a model was used successfully before with the MIL-47(V).31
 | (1) |
The resulting coupling constants are listed in Table 2, for both LP and NP geometry.
| Structure | Jc (meV) | Ji (meV) |
|---|---|---|
| LP | −0.5808 | 0.0432 |
| NP | 2.8533 | 0.0749 |
There is a significant difference between the two crystalline structures from the point of view of magnetism. In comparison to MIL-47(V), we notice a significantly weaker interaction between magnetic centers.31 This indicates that MIL-47(Mn) can switch much easier than MIL-47(V) version between different magnetic configurations. The negative value of Jc of the LP structure is indicative of an antiferromagnetic ground state, corroborating our earlier observations. On the other hand, the NP shows a positive Jc value, indicating a ferromagnetic ground state. Quantitively, the computed intra-chain coupling value for the NP is about 6 times the one for LP, showing the intrachain spin coupling to strongly depend on the pore size despite the fact that the pore geometry has only a minor influence on the MnO-chain geometry. The inter-chain coupling is comparable for the NP and LP case, and at least one order of magnitude smaller than the intra-chain coupling, in agreement with quasi-1D magnetic behavior.
Conclusion
In this work, structural, electronic and magnetic properties of MIL-47(Mn) as a breathing metal–organic framework was studied using periodic DFT calculations and the results were compared to its analogues, MIL-47(V). From the structural point of view, the obtained volumes for both large and narrow pore geometries were smaller than those reported for MIL-47(V), which can originate from the absence of Jahn–Teller effect in Mn. Also, MnO6 complex analysis shows that a deviation from octahedral symmetry, due to the presence of ligands with different field strength around the metal, can be found. Total energy data reveal an antiferromagnetic ground state for the large-pore geometry, but the system tends to move toward ferromagnetic state after breathing: the narrow-pore structure was found to have a ferromagnetic configuration. Electronic band structures show a strong dependency of electronic properties on spin configuration. In all cases, for both large- and narrow-pore crystals, the ferromagnetic configurations results in metallic behavior while the antiferromagnetic ones show semiconducting one. Also, a distinct behavior for bands parallel and orthogonal to the MnO chain is obtained. In the large-pore case, both valence and conduction bands show dispersion along the MnO chain and flat shape orthogonal to this. On the other hand, in narrow-pore geometry a dispersion can be detected even orthogonal to the MnO chain in the antiferromagnetic configuration. Calculated magnetic coupling constants confirm the stability of large- and narrow-pore structures. According to computed coupling constants, inter-chain interaction increases after volume reduction, but similarly to what reported in MIL-47(V)31 the inter-chain coupling is much smaller than intra-chain one, a reliable evidence for 1-D behavior in this material.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. G. acknowledges support from the European Union through the MaX Centre of Excellence (Grant no. 824143). D. E. P. V. gratefully acknowledges the computational resources provided by the VSC (Flemish Supercomputer Center), funded by the Research Foundation – Flanders (FWO) and the Flemish Government – department EWI, for the calculation of atomic charges.References
- A. Schoedel, M. Li, D. Li, M. O'Keeffe and O. M. Yaghi, Chem. Rev., 2016, 116, 12466–12535 CrossRef CAS PubMed.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Introduction to Metal–Organic Frameworks, ACS Publications, 2012 Search PubMed.
- Y. Noori and K. Akhbari, RSC Adv., 2017, 7, 1782–1808 RSC.
- D. E. P. Vanpoucke, J. Phys. Chem. C, 2017, 121, 8014–8022 CrossRef.
- Y. Tian, A. Stroppa, Y. Chai, L. Yan, S. Wang, P. Barone, S. Picozzi and Y. Sun, Sci. Rep., 2014, 4, 6062 CrossRef CAS PubMed.
- J. J. Gutiérrez-Sevillano, A. Martín-Calvo, D. Dubbeldam, S. Calero and S. Hamad, RSC Adv., 2013, 3, 14737–14749 RSC.
- D. Alezi, Y. Belmabkhout, M. Suyetin, P. M. Bhatt, Ł. J. Weseliński, V. Solovyeva, K. Adil, I. Spanopoulos, P. N. Trikalitis and A.-H. Emwas, J. Am. Chem. Soc., 2015, 137, 13308–13318 CrossRef CAS PubMed.
- S. Ma and H.-C. Zhou, Chem. Commun., 2010, 46, 44–53 RSC.
- N. Goel and N. Kumar, RSC Adv., 2018, 8, 10746–10755 RSC.
- S. Ramanayaka, M. Vithanage, A. Sarmah, T. An, K.-H. Kim and Y. S. Ok, RSC Adv., 2019, 9, 34359–34376 RSC.
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- Y.-B. Huang, J. Liang, X.-S. Wang and R. Cao, Chem. Soc. Rev., 2017, 46, 126–157 RSC.
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC.
- M. X. Wu and Y. W. Yang, Adv. Mater., 2017, 29, 1606134 CrossRef PubMed.
- C. Orellana-Tavra, E. F. Baxter, T. Tian, T. D. Bennett, N. K. Slater, A. K. Cheetham and D. Fairen-Jimenez, Chem. Commun., 2015, 51, 13878–13881 RSC.
- L. Wang, Y. Han, X. Feng, J. Zhou, P. Qi and B. Wang, Coord. Chem. Rev., 2016, 307, 361–381 CrossRef CAS.
- Y. Y. Wang, Q. Jin, S. X. Liu, C. Guo, Y. Y. Liu, B. Ding, X. X. Wu, Y. Li and Z. Z. Zhu, RSC Adv., 2015, 5, 35238–35251 RSC.
- J.-Z. Gu, Y.-H. Cui, J. Wu and A. M. Kirillov, RSC Adv., 2015, 5, 78889–78901 RSC.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- V. Finsy, C. E. Kirschhock, G. Vedts, M. Maes, L. Alaerts, D. E. De Vos, G. V. Baron and J. F. Denayer, Chem.–Eur. J., 2009, 15, 7724–7731 CrossRef CAS PubMed.
- A. Boutin, M. A. Springuel-Huet, A. Nossov, A. Gedeon, T. Loiseau, C. Volkringer, G. Ferey, F. X. Coudert and A. H. Fuchs, Angew. Chem., Int. Ed. Engl., 2009, 48, 8314–8317 CrossRef CAS PubMed.
- P. G. Yot, Q. Ma, J. Haines, Q. Yang, A. Ghoufi, T. Devic, C. Serre, V. Dmitriev, G. Férey and C. Zhong, Chem. Sci., 2012, 3, 1100–1104 RSC.
- J. Joo, H. Kim and S. S. Han, Phys. Chem. Chem. Phys., 2013, 15, 18822–18826 RSC.
- M. Alhamami, H. Doan and C.-H. Cheng, Materials, 2014, 7, 3198–3250 CrossRef PubMed.
- L. R. Parent, C. H. Pham, J. P. Patterson, M. S. Denny Jr, S. M. Cohen, N. C. Gianneschi and F. Paesani, J. Am. Chem. Soc., 2017, 139, 13973–13976 CrossRef CAS PubMed.
- X. Qian, B. Yadian, R. Wu, Y. Long, K. Zhou, B. Zhu and Y. Huang, Int. J. Hydrogen Energy, 2013, 38, 16710–16715 CrossRef CAS.
- B. Bueken, F. Vermoortele, D. E. P. Vanpoucke, H. Reinsch, C. C. Tsou, P. Valvekens, T. De Baerdemaeker, R. Ameloot, C. E. Kirschhock and V. Van Speybroeck, Angew. Chem., Int. Ed., 2015, 54, 13912–13917 CrossRef CAS PubMed.
- E. J. Carrington, C. A. McAnally, A. J. Fletcher, S. P. Thompson, M. Warren and L. Brammer, Nat. Chem., 2017, 9, 882 CrossRef CAS PubMed.
- F. Salles, A. Ghoufi, G. Maurin, R. G. Bell, C. Mellot-Draznieks and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 8487–8491 CrossRef CAS PubMed.
- G. Xu, X. Zhang, P. Guo, C. Pan, H. Zhang and C. Wang, J. Am. Chem. Soc., 2010, 132, 3656–3657 CrossRef CAS PubMed.
- D. E. P. Vanpoucke, J. W. Jaeken, S. De Baerdemacker, K. Lejaeghere and V. Van Speybroeck, Beilstein J. Nanotechnol., 2014, 5, 1738–1748 CrossRef PubMed.
- T. Bogaerts, L. Vanduyfhuys, D. E. P. Vanpoucke, J. Wieme, M. Waroquier, P. Van Der Voort and V. Van Speybroeck, CrystEngComm, 2015, 17, 8612–8622 RSC.
- D. E. P. Vanpoucke, K. Lejaeghere, V. Van Speybroeck, M. Waroquier and A. Ghysels, J. Phys. Chem. C, 2015, 119, 23752–23766 CrossRef CAS.
- K. Hendrickx, D. E. P. Vanpoucke, K. Leus, K. Lejaeghere, A. Van Yperen-De Deyne, V. Van Speybroeck, P. Van Der Voort and K. Hemelsoet, Inorg. Chem., 2015, 54, 10701–10710 CrossRef CAS PubMed.
- S. Ling and B. Slater, J. Phys. Chem. C, 2015, 119, 16667–16677 CrossRef CAS.
- K. Trepte, S. Schwalbe and G. Seifert, Phys. Chem. Chem. Phys., 2015, 17, 17122–17129 RSC.
- S. S. Essaoud, Z. Charifi, H. Baaziz, G. Uğur and Ş. Uğur, J. Magn. Magn. Mater., 2019, 469, 329–341 CrossRef CAS.
- Y. Moritomo, Y. Tomioka, A. Asamitsu, Y. Tokura and Y. Matsui, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 3297 CrossRef CAS PubMed.
- A. S. Munn, G. J. Clarkson, F. Millange, Y. Dumont and R. I. Walton, CrystEngComm, 2013, 15, 9679 RSC.
- X. Wang, X. Liu, H. Rong, Y. Song, H. Wen and Q. Liu, RSC Adv., 2017, 7, 29611–29617 RSC.
- C. Li, X. Hu, W. Tong, W. Yan, X. Lou, M. Shen and B. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 29829–29838 CrossRef CAS PubMed.
- X. Wang, G. Gou, D. Wang, H. Xiao, Y. Liu, M. Zhang, B. Dkhil, X. Ren and X. Lou, RSC Adv., 2016, 6, 48779–48787 RSC.
- K. Adpakpang, W. Pratanpornlerd, P. Ponchai, W. Tranganphaibul, S. Thongratkaew, K. Faungnawakij, S. Horike, T. Siritanon, A. Rujiwatra, M. Ogawa and S. Bureekaew, Inorg. Chem., 2018, 57, 13075–13078 CrossRef CAS PubMed.
- P. J. Hasnip, K. Refson, M. I. Probert, J. R. Yates, S. J. Clark and C. J. Pickard, Philos. Trans. R. Soc., A, 2014, 372, 20130270 CrossRef PubMed.
- R. Mercado, B. Vlaisavljevich, L.-C. Lin, K. Lee, Y. Lee, J. A. Mason, D. J. Xiao, M. I. Gonzalez, M. T. Kapelewski and J. B. Neaton, J. Phys. Chem. C, 2016, 120, 12590–12604 CrossRef CAS.
- T. Watanabe and D. S. Sholl, J. Chem. Phys., 2010, 133, 094509 CrossRef PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- R. Demuynck, S. M. Rogge, L. Vanduyfhuys, J. Wieme, M. Waroquier and V. Van Speybroeck, J. Chem. Theory Comput., 2017, 13, 5861–5873 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- D. E. P. Vanpoucke, P. Bultinck and I. Van Driessche, J. Comput. Chem., 2013, 34, 405–417 CrossRef CAS PubMed.
- D. E. P. Vanpoucke, I. Van Driessche and P. Bultinck, J. Comput. Chem., 2013, 34, 422–427 CrossRef CAS PubMed.
- J. J. Wolffis, D. E. P. Vanpoucke, A. Sharma, K. V. Lawler and P. M. Forster, Microporous Mesoporous Mater., 2019, 277, 184–196 CrossRef CAS.
- V. I. Lebedev and D. Laikov, A quadrature formula for the sphere of the 131st algebraic order of accuracy, 1999 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1988, 88, 2547–2553 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- Y. Liu, J.-H. Her, A. Dailly, A. J. Ramirez-Cuesta, D. A. Neumann and C. M. Brown, J. Am. Chem. Soc., 2008, 130, 11813–11818 CrossRef CAS PubMed.
- C. Volkringer, T. Loiseau, N. Guillou, G. Férey, E. Elkaïm and A. Vimont, Dalton Trans., 2009, 2241–2249 RSC.
- F. Murnaghan, Proc. Natl. Acad. Sci. U. S. A., 1944, 30, 244–247 CrossRef CAS PubMed.
- M. J. Han, T. Ozaki and J. Yu, J. Chem. Phys., 2005, 123, 34306 CrossRef PubMed.
- R. Gupta, T. Taguchi, B. Lassalle-Kaiser, E. L. Bominaar, J. Yano, M. P. Hendrich and A. S. Borovik, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5319–5324 CrossRef CAS PubMed.
- H. Weihe and H. U. Güdel, Inorg. Chem., 1997, 36, 3632–3639 CrossRef CAS PubMed.
- C. Franchini, R. Podloucky, J. Paier, M. Marsman and G. Kresse, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 195128 CrossRef.
- M. Schmitt, P. Moras, G. Bihlmayer, R. Cotsakis, M. Vogt, J. Kemmer, A. Belabbes, P. M. Sheverdyaeva, A. K. Kundu and C. Carbone, Nat. Commun., 2019, 10, 2610 CrossRef PubMed.
- H. Nakotte, M. Shrestha, S. Adak, M. Boergert, V. S. Zapf, N. Harrison, G. King and L. L. Daemen, Journal of Science: Advanced Materials and Devices, 2016, 1, 113–120 Search PubMed.
- A. Ondo and T. Ishida, AIP Conf. Proc., 2017, 1807, 020023 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09196c |
| This journal is © The Royal Society of Chemistry 2020 |