DOI:
10.1039/C9RA09113K
(Paper)
RSC Adv., 2020,
10, 3872-3881
Synthesis of cucurbitacin IIa derivatives with apoptosis-inducing capabilities in human cancer cells†
Received
4th November 2019
, Accepted 10th January 2020
First published on 23rd January 2020
Abstract
Twenty-one cucurbitacin IIa derivatives were synthesized and screened for cytotoxic activity. Their structures were established using 1H NMR, 13C NMR, and LC-MS spectroscopic data. The absolute configuration of the derivatives was determined by single crystal diffraction. In sulforhodamine B (SRB) assays, nearly all compounds displayed low cytotoxicity toward normal human cells (HEK293). However, some derivatives displayed high cytotoxicity, in the low μM range, toward several human tumor cell lines (SKOV3, HT29, HEPG2, MCF-7, and LOVO). Low IC50 values were obtained, especially for acetyl-protected product 2, 2,4,6-trichlorophenylhydrazine derivative 4a, and 2-hydrazinopyridine derivative 4d. In particular, compounds 2 and 4d showed low IC50 values of 1.2 ± 0.01 and 2.2 ± 0.19 μM against SKOV3 cells. These compounds were submitted to extensive biological testing, which showed that compounds 2 and 4a did not inhibit tumor cells by influencing the cell cycle. Furthermore, compound 4a triggered the apoptotic pathway in cancer cells, showing high apoptosis ratios. This study mainly changed the structure of cucurbitacin tetracyclic triterpenoids and provided a novel tetracyclic skeleton derived from natural products that provided further references for the future modification of cucurbitacin tetracyclic triterpenoids.
Introduction
Cancer is among the most challenging diseases in modern medicine. Cancer is a presently incurable disease that affects hundreds of millions of families annually. Therefore, the discovery of effective anticancer drugs is an important medical research area. Chemotherapy is among the most common methods for treating cancer. With the extensive study of traditional Chinese medicine, compounds with obvious anticancer activities have been identified in many Chinese herbal medicines. Examples include camptothecins (lactone alkaloids from Camptotheca acuminata), taxanes (diterpenes first derived from the Pacific yew tree, Taxus brevifolia), and vinca alkaloids.1–3 Many natural products have provided skeletons and structural references for the invention of modern drugs.4
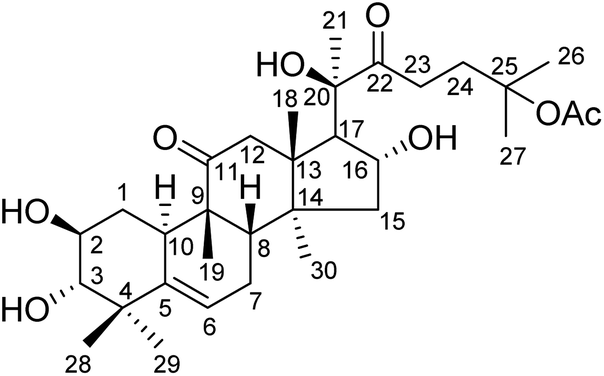
Hemsleya, known as ‘xue dan’ in China, belongs to the cucurbitaceous family. The roots of Hemsleya species are an especially rich source of triterpenoid saponins. Some triterpenoid saponins from Hemsleya have shown various biological activities, including anticarcinogenic, anti-inflammatory, cytotoxicity, and anti HBV activities. Cucurbitacin IIa (23,24-dihydroxycucurbitacin F-25-acetate), a natural pentacyclic triterpenoid, has five active sites, namely, C-2 hydroxyl, C-3 hydroxyl, C-16 hydroxyl, ring-B double bond, and C-22 aldehyde groups, which are amenable to a wide range of chemical transformations. The structure of cucurbitacin IIa is shown in Fig. 1. Cucurbitacin IIa mainly exists in the rhizomes of cucurbitaceous plant Hemsleya. This compound has bacteriostasis, antigastric ulcer, heat-clearing, detoxification, pain-relief and anti-HIV functions, is widely used in the treatment of hepatitis, coronary heart disease, and tracheitis,5–7 and has a prominent anticancer cell proliferation effect.8–11 The cytotoxicity and anti-hepatitis B virus properties of cucurbitacin IIa are of particular interest. In previous study, the C-2 hydroxyl, C-3 hydroxyl, and C-16 hydroxyl groups were used to prepare different types of ester. Most derivatives showed enhanced anti-HBV activities and exhibited significant inhibition of HBV DNA replication, comparable to that of positive control tenofovir. In this study we found that the IC50 values of cucurbitacin IIA toward HeLa and A549 cells were 0.389 and 0.108 μmol L−1, respectively.5 This strong evidence shows that cucurbitacin IIA and its derivatives have the potential to become new drugs.
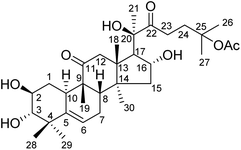 |
| | Fig. 1 Structure of cucurbitacin IIa. | |
Cucurbitacin IIa is found at high concentration in Hemsleya, making it easy to obtain.12 In this study, the C-2, C-3, and C-16 hydroxyl groups were protected with acetyl groups. Different types of oxime and hydrazone branches were designed by replacing the branched chain at C20–C22. Furthermore, we evaluated the inhibitory activities of these cucurbitacin IIa derivatives against five human cancer cell lines for the first time. The mechanism of cell apoptosis induced by these cucurbitacin IIa derivatives was also investigated.
Experimental
Materials and physical measurements
All commercial materials were used without further purification. Cucurbitacin IIa was extracted by our group8 and verified by single crystal diffraction. All solvents were of analytical grade. Proton and carbon nuclear magnetic resonance (1H and 13C NMR) spectra were recorded using a Bruker Avance DRX-400 instrument or a Bruker Avance DRX600 instrument with tetramethylsilane (TMS) as the internal standard. The chemical shifts (δ) are reported in parts per million (ppm) relative to the relevant solvent peak (CD3OD, CDCl3, or pyridine-d5). Coupling constants (J) are given in Hz. Mass spectrometry (MS) was performed using an API 4000 spectrometer. Thin-layer chromatography (TLC) was performed using silica gel GF254 plates (layer thickness, 0.2 mm). Column chromatography was performed using silica gel (200–300 mesh, Qingdao Marine Chemical Industry Factory, Qingdao, China).
Syntheses and characterization
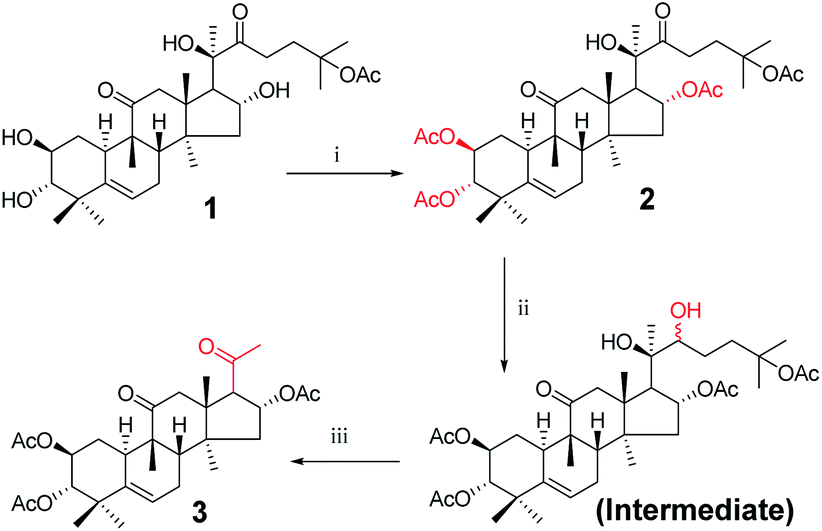
Synthesis of compound 3. Acetic anhydride (2 mL) and DMAP (22 mg, 0.18 mmol) were added to a solution of cucurbitacin IIa (500 mg, 8.9 × 10−4 mol) in pyridine (15 mL) at 25 °C. The resulting solution was stirred for 10 min. The mixture was concentrated under reduced pressure, diluted with EtOAc, filtered, and concentrated under reduced pressure to obtain 2. Sodium borohydride (112 mg, 3.48 × 10−3 mol) was then added to a solution of 2 in methanol (5 mL). After stirring at 0 °C for 6 h, the reaction was quenched with water (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phase was concentrated under reduced pressure. The resultant residue was dissolved in water and tetrahydrofuran (50 mL, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v)), and reacted overnight with sodium periodate (355 mg, 1.65 mmol) at room temperature. The reaction was quenched with water (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phase was concentrated under reduced pressure and the resultant residue was subjected to purification by silica gel column chromatography (eluent: CH2Cl2/CH3OH = 200:1, v/v) to afford 3 (392 mg, 83%) as a white solid.13 [α]25D = +102.0 (c = 0.1, MeOH). 1H NMR (400 MHz, pyridine-d5, ppm) δ 6.02 (t, J = 7.7 Hz, 1H, C-16-H), 5.72 (d, J = 5.6 Hz, 1H, C-6-H), 5.51–5.42 (m, 1H, C-2-H), 5.07 (d, 1H, C-3-H), 3.51 (d, J = 6.7 Hz, 1H, C-17-H), 3.18 (d, J = 14.6 Hz, 1H, C-12a-H), 2.77 (d, J = 13.2 Hz, 1H, C-8-H), 2.51 (d, J = 14.5 Hz, 1H, C-12b-H), 2.35–2.26 (m, 2H, C-7a-H and C-1a-H), 2.16 (s, 3H, CH3), 2.15 (s, 3H, CH3), 2.10 (s, 3H, CH3), 2.05 (m, 1H, C-15a-H), 2.01 (s, 3H, CH3), 1.86 (m, 2H, C-10-H and C-7b-H), 1.59 (m, 1H, C-15b-H), 1.43 (m, 1H, C-1b-H), 1.24 (s, 3H, CH3), 1.24 (s, 3H, CH3), 1.19 (s, 3H, CH3), 1.14 (s, 3H, CH3), 0.73 (s, 3H, CH3). 13C NMR (100 MHz, pyridine-d5, ppm) δ 213.08, 142.35, 118.64, 81.51, 81.33, 80.09, 70.92, 70.30, 58.90, 51.00, 49.18, 48.75, 48.64, 46.33, 43.03, 42.74, 35.31, 34.59, 34.34, 32.10, 25.97, 25.88, 25.43, 25.40, 24.08, 22.37, 22.13, 20.34, 20.30, 19.09. HRMS (ESI) m/z calculated for C30H42NaO8 (M + H)+: 553.2777, found: 553.2704.
:1 (v/v)), and reacted overnight with sodium periodate (355 mg, 1.65 mmol) at room temperature. The reaction was quenched with water (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phase was concentrated under reduced pressure and the resultant residue was subjected to purification by silica gel column chromatography (eluent: CH2Cl2/CH3OH = 200:1, v/v) to afford 3 (392 mg, 83%) as a white solid.13 [α]25D = +102.0 (c = 0.1, MeOH). 1H NMR (400 MHz, pyridine-d5, ppm) δ 6.02 (t, J = 7.7 Hz, 1H, C-16-H), 5.72 (d, J = 5.6 Hz, 1H, C-6-H), 5.51–5.42 (m, 1H, C-2-H), 5.07 (d, 1H, C-3-H), 3.51 (d, J = 6.7 Hz, 1H, C-17-H), 3.18 (d, J = 14.6 Hz, 1H, C-12a-H), 2.77 (d, J = 13.2 Hz, 1H, C-8-H), 2.51 (d, J = 14.5 Hz, 1H, C-12b-H), 2.35–2.26 (m, 2H, C-7a-H and C-1a-H), 2.16 (s, 3H, CH3), 2.15 (s, 3H, CH3), 2.10 (s, 3H, CH3), 2.05 (m, 1H, C-15a-H), 2.01 (s, 3H, CH3), 1.86 (m, 2H, C-10-H and C-7b-H), 1.59 (m, 1H, C-15b-H), 1.43 (m, 1H, C-1b-H), 1.24 (s, 3H, CH3), 1.24 (s, 3H, CH3), 1.19 (s, 3H, CH3), 1.14 (s, 3H, CH3), 0.73 (s, 3H, CH3). 13C NMR (100 MHz, pyridine-d5, ppm) δ 213.08, 142.35, 118.64, 81.51, 81.33, 80.09, 70.92, 70.30, 58.90, 51.00, 49.18, 48.75, 48.64, 46.33, 43.03, 42.74, 35.31, 34.59, 34.34, 32.10, 25.97, 25.88, 25.43, 25.40, 24.08, 22.37, 22.13, 20.34, 20.30, 19.09. HRMS (ESI) m/z calculated for C30H42NaO8 (M + H)+: 553.2777, found: 553.2704.
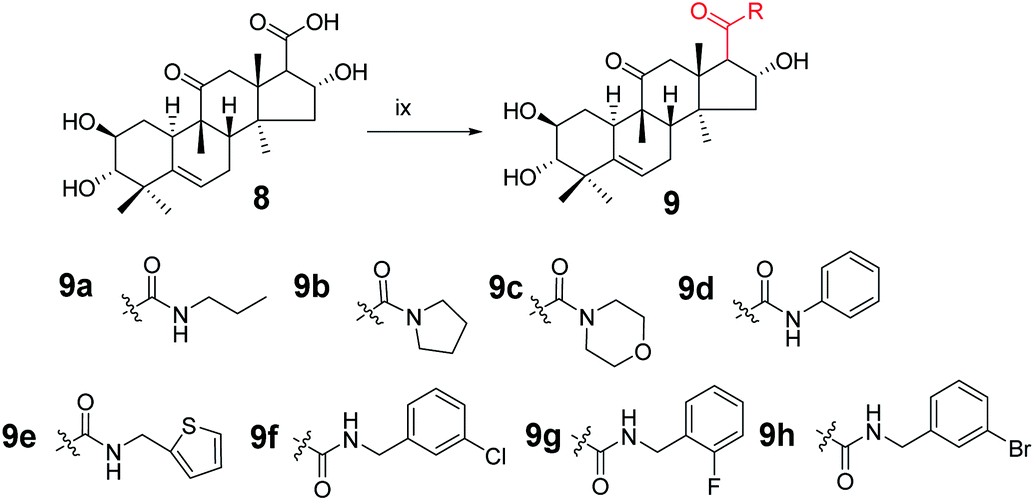
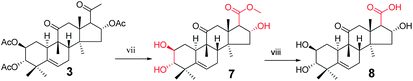
Synthesis of compound 8. To a solution of 3 (940 mg, 1.5 × 10−3 mol) in methanol (10 mL) was added sodium hypochlorite (6 mL) dropwise at 25 °C. The resulting solution was stirred for 30 min. Complete consumption of the starting material was detected by TLC. The reaction liquid was concentrated under reduced pressure, the resultant residue was quenched with water (50 mL) and extracted with EtOAc (3 × 30 mL), and the combined organic phase was concentrated under reduced pressure. The crude product and sodium hydroxide (320 mg, 8 × 10−3 mol) were added to methanol (30 mL). The resulting solution was stirred at 80 °C for 5 h.14,15 Complete consumption of the starting material was detected by TLC. After cooling to ambient temperature, the reaction was neutralized with 1 M HCl and extracted with EtOAc (3 × 20 mL). The combined organic phase was concentrated under reduced pressure and the resultant residue was subjected to purification by silica gel column chromatography (eluent: CH2Cl2/CH3OH = 10:1, v/v) to afford 8 (475 mg, 78%) as a white solid.
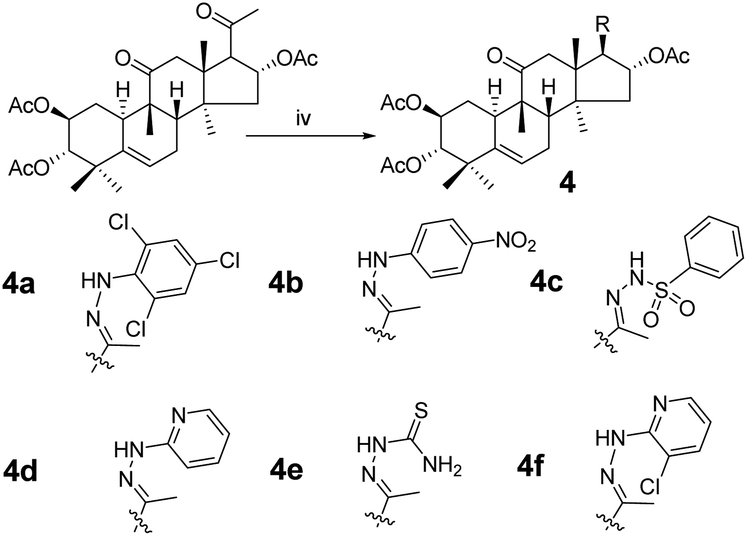
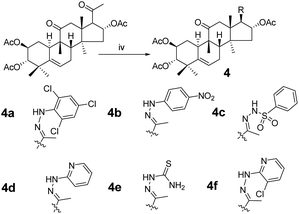
Synthesis of compound 4a. Under a nitrogen atmosphere a mixture of 3 (40 mg, 7.5 × 10−5 mol) and 2,4,6-trichlorophenylhydrazine (32 mg, 1.5 × 10−4 mol) in acetic acid (4.0 mL) was stirred at ambient temperature for 24 h. The reaction was then quenched with water (15 mL) and extracted with EtOAc (3 × 15 mL). The combined organic phase was concentrated under reduced pressure and the resultant residue was subjected to purification by silica gel column chromatography (eluent: CH2Cl2/CH3OH = 80:1, v/v) to afford 4a as a white solid (43 mg, 80%). [α]25D = +31.5 (c = 0.1, MeOH). 1H NMR (400 MHz, chloroform-d, ppm) δ 7.28 (s, 2H, C-3′-H and C-5′-H), 7.02 (s, 1H, NH), 5.84–5.74 (m, 2H, C-16-H and C-6-H), 5.07–4.97 (m, 1H, C-2-H), 4.69 (d, J = 10.0 Hz, 1H, C-3-H), 3.24–3.14 (m, 2H, C-17-H and C-12a-H), 2.51–2.37 (m, 2H, C-8-H and C-7a-H), 2.34 (d, J = 14.3 Hz, 1H, C-12b-H), 2.06 (s, 3H, CH3), 2.05–2.00 (m, 2H, C-1a-H and C-15a-H), 1.99 (s, 6H, 2 × CH3), 1.94 (s, 3H, CH3), 1.92–1.82 (m, 2H, C-10-H and C-7b-H), 1.46 (d, J = 14.2 Hz, 1H, C-15b-H), 1.30 (m, 1H, C-1b-H), 1.26 (s, 3H, CH3), 1.09 (s, 3H, CH3), 1.06 (s, 3H, CH3), 1.06 (s, 3H, CH3), 0.67 (s, 3H, CH3). 13C NMR (100 MHz, chloroform-d, ppm) δ 211.43, 170.54, 170.39, 170.74, 147.27, 139.16, 138.51, 128.82, 128.82, 127.48, 126.37, 120.30, 78.01, 77.16, 74.45, 71.04, 58.77, 49.80, 48.90, 47.60, 47.20, 43.55, 43.37, 41.92, 33.47, 30.71, 24.50, 23.91, 22.68, 21.28, 21.13, 20.98, 20.12, 19.95, 18.94, 16.61. HRMS (ESI) m/z calculated for C36H46Cl3N2O7 (M + H)+: 723.2370, found: 723.2359.Compounds 4b–4f were synthesized from 3 using the same method as above with different hydrazines.
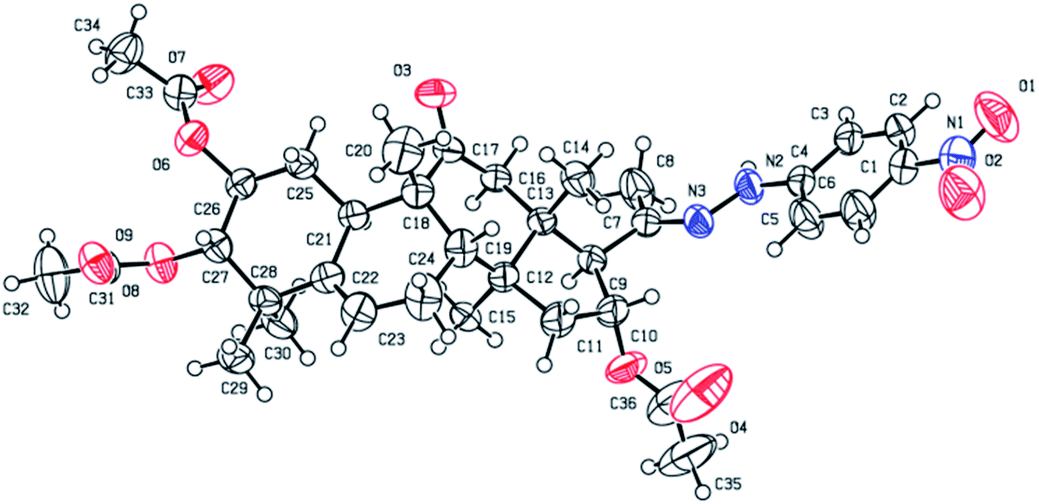
Compound 4b. Yellow solid, 83% yield, [α]25D = +20.6 (c = 0.1, MeOH). 1H NMR (400 MHz, pyridine-d5, ppm) δ 10.50 (s, 1H, NH), 8.21 (d, J = 8.8 Hz, 2H, C-3′-H and C-5′-H), 7.37 (d, J = 8.8 Hz, 2H, C-2′-H and C-6′-H), 6.51 (t, J = 8.1 Hz, 1H, C-16-H), 5.77–5.68 (m, 1H, C-6-H), 5.45 (td, J = 11.0, 4.3 Hz, 1H, C-2-H), 5.06 (d, J = 10.0 Hz, 1H, C-3-H), 3.45 (d, J = 7.2 Hz, 1H, C-17-H), 3.22 (d, J = 14.5 Hz, 1H, C-12a-H), 2.79 (d, J = 12.9 Hz, 1H, C-8-H), 2.36–2.19 (m, 4H, C-12b-H, C-7a-H, C-1a-H, and C-15a-H), 2.16 (s, 3H, CH3), 2.14 (s, 3H, CH3), 1.98 (s, 6H, 2 × CH3), 1.95–1.83 (m, 2H, C-10-H and C-7b-H), 1.63 (d, J = 13.9 Hz, 1H, C-15b-H), 1.42 (d, J = 12.4 Hz, 1H, C-1b-H), 1.32 (s, 3H, CH3), 1.23 (s, 3H, CH3), 1.18 (s, 3H, CH3), 1.14 (s, 3H, CH3), 0.79 (s, 3H, CH3). 13C NMR (100 MHz, pyridine-d5, ppm) δ 211.38, 170.73, 170.41, 170.28, 152.50, 147.58, 139.56, 139.41, 126.35, 126.35, 120.47, 112.06, 112.06, 78.37, 74.40, 71.64, 59.36, 50.05, 48.79, 47.97, 47.43, 43.59, 43.30, 42.14, 33.45, 31.18, 24.61, 23.94, 22.56, 21.15, 20.84, 20.73, 20.19, 19.88, 18.82, 17.92. HRMS (ESI) m/z calculated for C36H47N3NaO9 (M + Na)+: 688.3210, found: 688.3190 (Fig. 2).
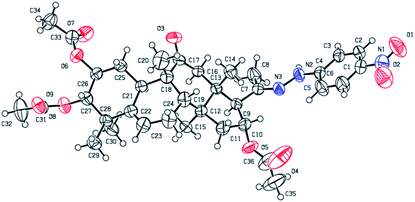 |
| | Fig. 2 Molecular structure of derivative 4b. | |
Compound 4c. White solid, 74% yield, [α]25D = +53.5 (c = 0.1, MeOH). 1H NMR (400 MHz, pyridine-d5, ppm) δ 11.55 (s, 1H, NH), 8.27–8.21 (m, 2H, C-2′-H and C-6′-H), 7.51 (m, 3H, C-3′-H, C-4′-H and C-5′-H), 6.20 (t, J = 8.0 Hz, 1H, C-16-H), 5.71 (m, 1H, C-6-H), 5.42 (m, 1H, C-2-H), 5.02 (m, 1H, C-3-H), 3.32 (dd, J = 7.2, 2.1 Hz, 1H, C-17-H), 3.09 (d, J = 14.6 Hz, 1H, C-12a-H), 2.73 (d, J = 13.0 Hz, 1H, C-8-H), 2.32–2.22 (m, 3H, C-12b-H, C-7a-H and C-1a-H), 2.12 (s, 3H, CH3), 2.07 (s, 3H, CH3), 2.03 (m, 1H, C-15a-H), 1.97 (s, 3H, CH3), 1.93 (s, 3H, CH3), 1.83–1.77 (m, 2H, C-10-H and C-7b-H), 1.55 (d, J = 13.9 Hz, 1H, C-15b-H), 1.37 (m, 1H, C-1b-H), 1.22 (s, 3H, CH3), 1.19 (s, 3H, CH3), 1.16 (s, 3H, CH3), 1.09 (s, 3H, CH3), 0.56 (s, 3H, CH3). 13C NMR (100 MHz, pyridine-d5) δ 211.22, 170.52, 170.41, 170.28, 154.90, 140.17, 139.38, 132.91, 129.12, 129.12, 128.53, 128.53, 120.41, 78.35, 74.55, 71.61, 58.74, 49.79, 48.69, 47.86, 47.32, 43.60, 43.12, 42.11, 33.42, 31.14, 24.60, 23.86, 22.57, 21.14, 20.84, 20.73, 20.12, 19.46, 18.75, 18.42. HRMS (ESI): m/z calculated: C36H49N2O9S (M + H)+: 685.3159, found: 685.3135.
Compound 4d. White solid, 84% yield, [α]25D = +36.0 (c = 0.1, MeOH). 1H NMR (400 MHz, pyridine-d5, ppm) δ 9.76 (s, 1H, NH), 8.28 (d, J = 4.8 Hz, 1H, C-6′-H), 7.62–7.51 (m, 2H, C-4′-H and C-5′-H), 6.71 (t, J = 6.0 Hz, 1H, C-3′-H), 6.49 (t, J = 8.0 Hz, 1H, C-16-H), 5.73 (d, J = 5.5 Hz, 1H, C-6-H), 5.46 (m, 1H, C-2-H), 5.04 (d, J = 10.0 Hz, 1H, C-3-H), 3.41 (d, J = 7.1 Hz, 1H, C-17-H), 3.19 (d, J = 14.5 Hz, 1H, C-12a-H), 2.79 (d, J = 13.0 Hz, 1H, C-8-H), 2.37 (d, J = 14.7 Hz, 1H, C-12b-H), 2.33–2.15 (m, 3H, C-7a-H, C-1a-H and C-15a-H), 2.13 (s, 3H, CH3), 2.12 (s, 3H, CH3), 2.02 (s, 3H, CH3), 1.98 (s, 3H, CH3), 1.91 (d, J = 7.9 Hz, 1H, C-10-H), 1.86 (dd, J = 19.4, 5.9 Hz, 1H, C-7b-H), 1.61 (d, J = 13.9 Hz, 1H, C-15b-H), 1.41 (m, 1H, C-1b-H), 1.31 (s, 3H, CH3), 1.23 (s, 3H, CH3), 1.17 (s, 3H, CH3), 1.12 (s, 3H, CH3), 0.76 (s, 3H, CH3). 13C NMR (100 MHz, pyridine-d5, ppm) δ 211.44, 170.73, 170.42, 170.29, 158.94, 148.23, 144.29, 139.43, 138.05, 120.46, 115.38, 107.65, 78.38, 74.41, 71.65, 59.13, 49.97, 48.79, 47.90, 47.42, 43.66, 43.33, 42.14, 33.45, 31.19, 24.62, 23.95, 22.59, 21.15, 20.86, 20.74, 20.18, 19.81, 18.82, 17.40. HRMS (ESI): m/z calculated: C35H48N3O7 (M + H)+: 622.3492, found: 622.3763.
Compound 4e. White solid, 90% yield, [α]25D = +27.0 (c = 0.1, MeOH). 1H NMR (400 MHz, pyridine-d5, ppm) δ 10.73 (s, 1H, NH), 9.69 (s, 1H, NH2), 8.47 (s, 1H, NH2), 6.49 (t, J = 8.1 Hz, 1H, C-16-H), 5.71 (s, 1H, C-6-H), 5.43 (td, J = 11.0, 4.4 Hz, 1H, C-2-H), 5.05 (s, 1H, C-3-H), 5.01 (s, 1H), 3.33 (d, J = 7.1 Hz, 1H, C-17-H), 3.14 (d, J = 14.5 Hz, 1H, C-12a-H), 2.74 (d, J = 12.9 Hz, 1H, C-8-H), 2.36–2.22 (m, 4H, C-12b-H, C-7a-H, C-1a-H and C-15a-H), 2.13 (s, 3H, CH3), 2.11 (s, 3H, CH3), 2.02 (s, 3H, CH3), 1.98 (s, 3H, CH3), 1.82 (m, 2H, C-10-H and C-7b-H), 1.52 (d, J = 13.8 Hz, 1H, C-15b-H), 1.39 (d, J = 12.5 Hz, 1H, C-1b-H), 1.26 (s, 3H, CH3), 1.20 (s, 3H, CH3), 1.15 (s, 3H, CH3), 1.09 (s, 3H, CH3), 0.70 (s, 3H, CH3). 13C NMR (100 MHz, pyridine-d5, ppm) δ 211.17, 181.22, 170.86, 170.43, 170.33, 149.29, 139.41, 120.40, 78.33, 73.51, 71.64, 59.69, 50.14, 48.75, 47.97, 47.29, 43.15, 43.10, 42.13, 33.42, 31.14, 24.60, 23.93, 22.59, 21.21, 20.87, 20.76, 20.13, 19.69, 18.75, 17.99. HRMS (ESI): m/z calculated: C31H46N3O7S (M + H)+: 604.3056, found: 604.3035.
Compound 4f. White solid, 77% yield, [α]25D = +22.8 (c = 0.1, MeOH). 1H NMR (400 MHz, chloroform-d, ppm) δ 8.24 (dd, J = 4.9, 1.5 Hz, 1H, C-6′-H), 7.56 (dd, J = 7.7, 1.5 Hz, 1H, C-4′-H), 6.74–6.69 (m, 1H, C-5′-H), 5.90 (t, J = 7.9 Hz, 1H, C-16-H), 5.77 (d, J = 5.6 Hz, 1H, C-6-H), 5.02 (m, 1H, C-2-H), 4.69 (d, J = 10.0 Hz, 1H, C-3-H), 3.37 (d, J = 7.0 Hz, 1H, C-17-H), 3.18 (d, J = 14.6 Hz, 1H, C-12a-H), 2.45 (m, 1H, C-8-H), 2.30 (d, J = 14.4 Hz, 1H, C-12b-H), 2.17–2.10 (m, 3H, C-7a-H, C-1a-H, C-15a-H), 2.07 (s, 3H, CH3), 2.00 (s, 6H, CH3, CH3), 1.98 (s, 4H, CH3, C-10-H), 1.87 (m, 1H, C-7b-H), 1.57 (d, J = 14.1 Hz, 1H, C-15b-H), 1.28 (s, 3H, CH3), 1.25 (m, 1H, C-1b-H), 1.09 (s, 3H, CH3), 1.06 (m, 6H, 2 × CH3), 0.74 (s, 3H, CH3). 13C NMR (100 MHz, chloroform-d, ppm) δ 211.62, 170.78, 170.53, 170.27, 150.43, 148.38, 146.37, 139.14, 137.35, 120.12, 115.73, 115.07, 74.90, 70.98, 59.01, 50.68, 48.84, 47.42, 46.60, 43.42, 43.32, 41.81, 33.39, 30.62, 24.41, 23.94, 22.52, 21.24, 21.02, 20.89, 20.85, 20.11, 20.04, 18.84, 16.21. HRMS (ESI): m/z calculated: C35H47ClN3O7 (M + H)+: 656.3103, found: 656.3087.
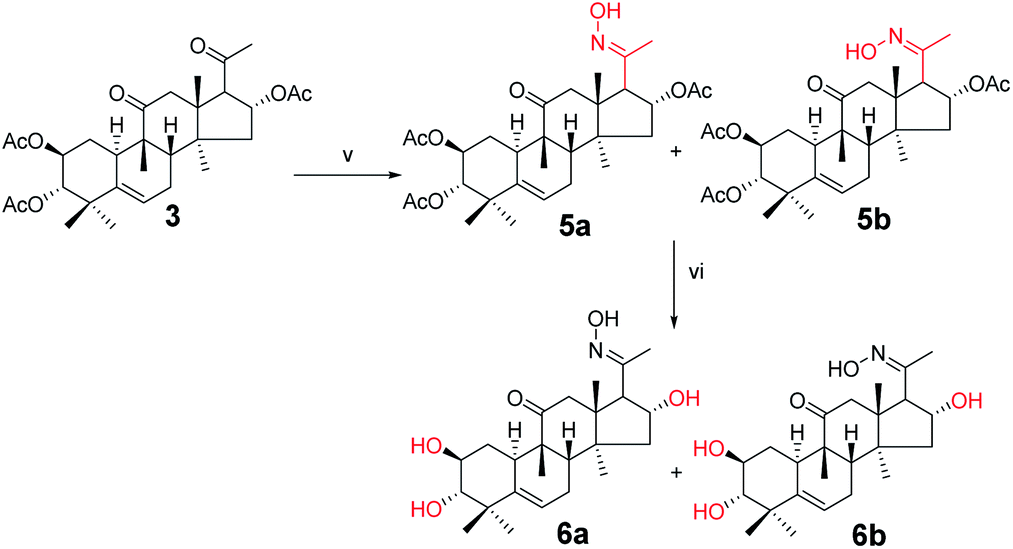
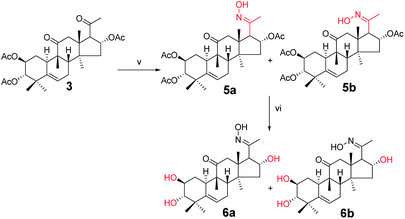
Synthesis of compounds 5a and 5b. Under a nitrogen atmosphere, a mixture of 3 (100 mg, 1.9 × 10−4 mol) and hydroxylamine hydrochloride (26.2 mg, 3.8 × 10−4 mol) in pyridine (8.0 mL) was stirred at 50 °C for 10 h. After cooling to ambient temperature, the reaction was quenched with water (30 mL) and extracted with EtOAc (3 × 30 mL). The combined organic phase was concentrated under reduced pressure and the resultant residue was subjected to purification by silica gel column chromatography (eluent: CH2Cl2/CH3OH = 80:1, v/v) to afford 5a (57 mg, 55%) and 5b (43 mg, 42%) as white solids.
Compound 5a. [α]25D = +51.5 (c = 0.1, MeOH). 1H NMR (400 MHz, pyridine-d5, ppm) δ 12.88 (s, 1H, N–OH), 6.33 (t, J = 8.1 Hz, 1H, C-16-H), 5.71 (d, J = 5.6 Hz, 1H, C-6-H), 5.46 (m, 1H, C-2-H), 5.04 (d, J = 10.1 Hz, 1H, C-3-H), 3.36 (d, J = 7.3 Hz, 1H, C-17-H), 3.10 (d, J = 14.6 Hz, 1H, C-12a-H), 2.76 (d, J = 13.1 Hz, 1H, C-8-H), 2.39 (d, J = 14.6 Hz, 1H, C-12b-H), 2.35–2.27 (m, 2H, C-1a-H and C-7a-H), 2.13 (s, 3H, CH3), 2.10 (s, 1H, C-15a-H), 2.07 (s, 3H, CH3), 2.05 (s, 3H, CH3), 1.98 (s, 3H, CH3), 1.86–1.79 (m, 2H, C-10-H and C-7b-H), 1.57 (d, J = 13.9 Hz, 1H, C-15b-H), 1.41 (d, J = 12.4 Hz, 1H, C-1b-H), 1.26 (s, 3H, CH3), 1.23 (s, 3H, CH3), 1.16 (s, 3H, CH3), 1.11 (s, 3H, CH3), 0.79 (s, 3H, CH3). 13C NMR (100 MHz, pyridine-d5, ppm) δ 211.42, 170.39, 170.25, 152.78, 139.41, 120.43, 78.38, 74.23, 71.62, 57.03, 49.75, 48.74, 47.63, 47.33, 43.70, 43.28, 42.12, 33.45, 31.17, 29.95, 24.60, 23.92, 22.56, 21.08, 20.83, 20.72, 20.15, 19.83, 18.77, 15.55. HRMS (ESI): m/z calculated: C30H44NO8 (M + H)+: 546.3067, found: 546.3052.
Compound 5b. [α]25D = +38.1 (c = 0.1, MeOH). 1H NMR (400 MHz, pyridine-d5, ppm) δ 12.70 (s, 1H, OH), 5.87 (t, J = 8.1 Hz, 1H, C-16-H), 5.72 (d, J = 5.6 Hz, 1H, C-6-H), 5.48 (td, J = 10.8, 4.4 Hz, 1H, C-2-H), 5.02 (m, 1H, C-3-H), 4.61 (d, J = 7.4 Hz, 1H, C-17-H), 3.60 (d, J = 15.4 Hz, 1H, C-12a-H), 2.83 (d, J = 13.0 Hz, 1H, C-8-H), 2.70 (d, J = 15.4 Hz, 1H, C-12b-H), 2.39–2.18 (m, 3H, C-1a-H, C-7a-H and C-15a-H), 2.13 (s, 3H, CH3), 2.12 (s, 3H, CH3), 2.06 (s, 3H, CH3), 1.97 (s, 3H, CH3), 1.91 (d, J = 8.5 Hz, 1H, C-10-H), 1.84 (m, 1H, C-7b-H), 1.65 (d, J = 14.0 Hz, 1H, C-15b-H), 1.38 (s, 3H, CH3), 1.25 (m, 1H, C-1b-H), 1.19 (s, 3H, CH3), 1.16 (s, 6H, 2 × CH3), 0.87 (s, 3H, CH3). 13C NMR (100 MHz, pyridine-d5, ppm) δ 212.35, 170.58, 170.44, 170.20, 154.33, 139.46, 120.39, 78.58, 75.57, 71.63, 52.21, 49.41, 48.75, 48.46, 47.69, 44.00, 43.28, 42.11, 33.51, 31.30, 24.64, 24.09, 22.44, 21.22, 21.00, 20.85, 20.74, 20.58, 20.36, 19.08. HRMS (ESI): m/z calculated: C30H44NO8 (M + H)+: 546.3067, found: 546.3047.
Synthesis of compound 6a. Under a nitrogen atmosphere, a mixture of 5a (50 mg, 0.09 mmol) and sodium hydroxide (18.3 mg, 0.46 mmol) in methanol (4.0 mL) was stirred at ambient temperature for 6 h. The reaction was then quenched with water (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phase was concentrated under reduced pressure and the resultant residue was subjected to purification by silica gel column chromatography (eluent: CH2Cl2/CH3OH = 25:1, v/v) to afford 6a as a white solid (34.3 mg, 91%). [α]25D = +105.8 (c = 0.1, MeOH). 1H NMR (400 MHz, pyridine-d5, ppm) δ 5.73 (d, J = 5.6 Hz, 1H, C-6-H), 5.52 (t, J = 7.8 Hz, 1H, C-2-H), 4.10 (m, 1H, C-16-H), 3.39 (dd, J = 17.6, 7.9 Hz, 2H, C-12a-H and C-17-H), 3.29 (d, J = 14.3 Hz, 1H, C-3-H), 2.77 (d, J = 13.0 Hz, 1H, C-8-H), 2.48 (d, J = 14.3 Hz, 1H, C-12b-H), 2.45–2.29 (m, 2H, C-7a-H and C-1a-H), 2.07 (s, 3H, CH3), 2.04–1.84 (m, 3H, C-15a-H, C-10-H, and C-7b-H), 1.59 (s, 3H, CH3), 1.53 (d, J = 12.0 Hz, 1H, C-15b-H), 1.49 (s, 3H, CH3), 1.30 (s, 4H, C-1b-H and CH3), 1.23 (s, 3H, CH3), 0.89 (s, 3H, CH3). 13C NMR (100 MHz, pyridine-d5, ppm) δ 212.35, 154.44, 142.39, 118.54, 81.28, 70.88, 70.78, 61.23, 50.72, 49.26, 48.05, 47.57, 45.98, 43.91, 42.72, 34.57, 34.27, 25.37, 24.19, 22.27, 20.28, 20.08, 19.18, 15.92. HRMS (ESI) m/z calculated for C24H38NO5 (M + H)+: 420.2750, found: 420.2740.
Synthesis of compound 6b. Under a nitrogen atmosphere, a mixture of 5b (50 mg, 0.09 mmol) and sodium hydroxide (18.3 mg, 0.46 mmol) in methanol (4.0 mL) was stirred at ambient temperature, the reaction was quenched with water (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phase was concentrated under reduced pressure and the resultant residue was subjected to purification by silica gel column chromatography (eluent: CH2Cl2/CH3OH = 20:1, v/v) to afford 6b as a white solid (35.1 mg, 93%). [α]25D = +39.10 (c = 0.1, MeOH). 1H NMR (400 MHz, chloroform-d, ppm) δ 5.75 (d, J = 5.4 Hz, 1H, C-6-H), 3.95 (d, J = 7.3 Hz, 1H, C-2-H), 3.55 (m, 1H, C-16-H), 3.40–3.33 (m, 2H, C-12a-H and C-17-H), 3.26 (d, J = 15.3 Hz, 1H, C-3-H), 2.89 (d, J = 9.3 Hz, 1H, C-8-H), 2.48–2.37 (m, 2H, C-12b-H and C-7a-H), 2.26 (m, 1H, C-1a-H), 2.03–1.96 (m, 4H, C-15a-H and CH3), 1.91 (t, J = 6.0 Hz, 1H, C-10-H), 1.84–1.78 (m, 1H, C-7b-H), 1.59 (d, J = 4.6 Hz, 1H, C-15b-H), 1.37 (d, J = 4.4 Hz, 3H, CH3), 1.28 (d, J = 4.0 Hz, 1H, C-1b-H), 1.20 (d, J = 4.3 Hz, 3H, CH3), 1.09 (d, J = 4.7 Hz, 3H, CH3), 0.97 (d, J = 4.3 Hz, 3H, CH3), 0.75 (d, J = 4.3 Hz, 3H, CH3). 13C NMR (100 MHz, chloroform-d, ppm) δ 213.72, 157.94, 141.80, 119.42, 81.26, 72.62, 71.06, 52.89, 52.46, 49.75, 49.00, 48.32, 46.20, 44.18, 42.74, 34.45, 33.89, 25.02, 24.48, 22.00, 21.33, 20.37, 20.27, 19.89. HRMS (ESI) m/z calculated for C24H38NO5 (M + H)+: 420.2750, found: 420.2736.
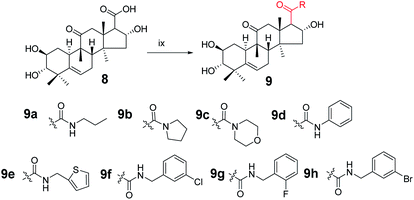
Synthesis of compound 9a. Under a nitrogen atmosphere, a mixture of 8 (50 mg, 0.123 mmol), N-propylamine (11.1 μL, 0.135 mmol), 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride (DMTMM; 37.4 mg, 0.135 mmol), and N-methylmorpholine (30 μL) in methanol (5.0 mL) was stirred at ambient temperature for 10 h. The reaction was then quenched with water (20 mL) and extracted with EtOAc (3 × 10 mL).17,18 The combined organic phase was concentrated under reduced pressure and the resultant residue was subjected to purification by silica gel column chromatography (eluent: CH2Cl2/CH3OH = 20:1, v/v) to afford 9a as a white solid (51.1 mg, 93%). [α]25D = +111.4 (c = 0.1, MeOH). 1H NMR (600 MHz, chloroform-d, ppm) δ 6.59 (s, 1H, NH), 5.72 (d, J = 4.1 Hz, 1H, C-6-H), 4.82 (m, 1H, C-2-H), 3.58–3.49 (m, 1H, C-16-H), 3.25–3.05 (m, 3H, C-1′-H and C-12a-H), 2.91 (dd, J = 9.3, 3.1 Hz, 1H, C-17-H), 2.83 (dd, J = 6.8, 1.9 Hz, 1H, C-3-H), 2.44–2.37 (m, 1H, C-8-H), 2.35–2.31 (m, 1H, C-12b-H), 2.28 (dd, J = 14.5, 3.6 Hz, 1H, C-7a-H), 1.99–1.89 (m, 2H, C-1a-H and C-15a-H), 1.84–1.77 (m, 1H, C-10-H), 1.57–1.45 (m, 3H, C-7b-H and C-2′-H), 1.27–1.21 (m, 4H, C-15b-H and CH3), 1.18 (s, 3H, CH3), 1.07 (d, J = 2.8 Hz, 3H, CH3), 1.06–1.00 (m, 1H, C-1b-H), 0.93 (m, 6H, 2 × CH3), 0.72 (m, 3H, CH3). 13C NMR (150 MHz, chloroform-d, ppm) δ 213.14, 172.18, 140.87, 118.86, 80.50, 72.24, 70.55, 59.61, 49.81, 49.02, 48.12, 46.43, 45.13, 43.27, 42.01, 41.49, 33.81, 33.20, 24.65, 23.89, 22.87, 21.59, 20.09, 19.66, 18.99, 11.50. HRMS (ESI) m/z calculated for C26H42NO5 (M + H)+: 448.3063, found: 448.3015.Compounds 9b–9h were synthesized from 8 using the same method as above with different amines.
Compound 9b. White solid, 78% yield, [α]25D = +91.4 (c = 0.1, MeOH). 1H NMR (600 MHz, pyridine-d5, ppm) δ 6.92 (d, J = 3.7 Hz, 1H, OH), 6.48 (s, 1H, OH), 6.25 (s, 1H, OH), 5.75 (d, J = 5.6 Hz, 1H, C-6-H), 5.63 (t, J = 7.8 Hz, 1H, C-2-H), 4.13 (m, 1H, C-16-H), 3.52 (d, J = 13.9 Hz, 1H, C-17-H), 3.48–3.42 (m, 2H, C-3-H and C-2′-H), 3.42–3.37 (m, 1H, C-5′-H), 3.28 (dd, J = 9.8, 6.9 Hz, 1H, C-2′-H), 3.21–3.16 (m, 1H, C-5′-H), 2.89 (d, J = 13.0 Hz, 1H, C-8-H), 2.48–2.42 (m, 1H, C-7a-H), 2.35 (m, 1H, C-1a-H), 2.32 (d, J = 14.0 Hz, 1H, C-12b-H), 2.11 (dd, J = 12.9, 9.2 Hz, 1H, C-15a-H), 1.97 (m, 2H, C-7b-H and C-10-H), 1.88 (d, J = 12.9 Hz, 1H, C-15b-H), 1.68 (s, 3H), 1.61–1.55 (m, 1H, C-1b-H), 1.54–1.36 (m, 7H, CH3, C-3′-H and C-4′-H), 1.33 (s, 3H), 1.25 (s, 3H), 1.07 (s, 3H). 13C NMR (150 MHz, pyridine-d5, ppm) δ 212.09, 170.71, 142.46, 118.61, 81.42, 73.71, 70.88, 58.97, 51.40, 49.42, 49.28, 47.70, 46.87, 46.23, 46.17, 43.83, 42.87, 34.72, 34.29, 26.23, 25.47, 24.35, 24.19, 22.35, 21.12, 20.30, 19.35. HRMS (ESI) m/z calculated for C27H42NO5 (M + H)+: 460.3063, found: 460.3060.
Compound 9c. White solid, 75% yield, [α]25D = +95.0 (c = 0.1, MeOH). 1H NMR (600 MHz, chloroform-d, ppm) δ 5.73 (d, J = 5.5 Hz, 1H, C-6-H), 5.01–4.96 (m, 1H, C-2-H), 3.74–3.24 (m, 10H, morpholine, C-16-H and C-12a-H), 3.12 (d, J = 13.7 Hz, 1H, C-17-H), 2.91 (d, J = 9.2 Hz, 1H, C-3-H), 2.45–2.37 (m, 1H, C-7a-H), 2.33 (d, J = 12.2 Hz, 1H, C-8-H), 2.12 (d, J = 13.7 Hz, 1H, C-12b-H), 2.01–1.94 (m, 2H, C-7b-H and C-15a-H), 1.92 (d, J = 7.9 Hz, 1H, C-10-H), 1.82–1.76 (m, 1H, C-1a-H), 1.53 (d, J = 1.7 Hz, 1H, C-15b-H), 1.31 (s, 3H, CH3), 1.19 (s, 3H, CH3), 1.08 (s, 3H, CH3), 1.04 (m, 1H, C-1b-H), 0.94 (s, 3H, CH3), 0.71 (s, 3H, CH3). 13C NMR (150 MHz, chloroform-d, ppm) δ 212.17, 171.03, 140.79, 118.90, 77.16, 73.39, 70.46, 66.94, 66.83, 55.20, 50.80, 49.09, 48.76, 46.81, 46.60, 44.94, 43.39, 42.78, 41.99, 33.79, 33.01, 24.59, 23.82, 21.54, 20.25, 20.00, 19.07. HRMS (ESI): m/z calculated: C27H42NO6 (M + H)+: 476.3012, found: 476.3005.
Compound 9d. White solid, 90% yield, [α]25D = +131.4 (c = 0.1, MeOH). 1H NMR (400 MHz, acetone-d6, ppm) δ 8.92 (s, 1H, NH), 7.69–7.64 (m, 2H, C-2′-H and C-6′-H), 7.29 (t, J = 7.8 Hz, 2H, C-3′-H and C-5′-H), 7.05 (t, J = 7.4 Hz, 1H, C-4′-H), 5.75 (d, J = 6.2 Hz, 1H, C-6-H), 4.93 (d, J = 8.6 Hz, 1H, C-2-H), 4.35 (d, J = 4.0 Hz, 1H, OH), 3.80 (s, 1H, OH), 3.62 (m, 1H, OH), 3.51 (t, J = 5.5 Hz, 1H, C-16-H), 3.32 (d, J = 14.4 Hz, 1H, C-12a-H), 3.15 (m, 1H, C-17-H), 2.85 (m, 2H, C-8-H and C-3-H), 2.50–2.40 (m, 2H, C-7a-H and C-1a-H), 2.26 (d, J = 14.4 Hz, 1H, C-12b-H), 2.05–2.00 (m, 2H, C-15a-H and C-10-H), 1.78–1.71 (m, 1H, C-7b-H), 1.57–1.51 (m, 1H, C-15b-H), 1.36 (s, 3H, CH3), 1.19 (s, 3H, CH3), 1.05 (s, 3H, CH3), 1.01–0.94 (m, 4H, C-1b-H and CH3), 0.79 (s, 3H). 13C NMR (100 MHz, acetone-d6, ppm) δ 211.83, 171.15, 142.75, 140.41, 129.62, 129.62, 124.18, 120.20, 120.20, 119.35, 81.66, 73.16, 71.22, 61.68, 51.15, 49.71, 49.33, 47.28, 46.50, 44.46, 42.83, 34.77, 34.46, 25.40, 24.78, 22.18, 20.59, 20.44, 19.65. HRMS (ESI): m/z calculated: C29H40NO5 (M + H)+: 482.2906, found: 488.2888.
Compound 9e. White solid, 84% yield, [α]25D = +81.4 (c = 0.1, MeOH). 1H NMR (600 MHz, chloroform-d, ppm) δ 7.22–7.20 (m, 1H, C-5′-H), 6.96 (d, J = 3.3 Hz, 1H, C-6′-H), 6.94–6.91 (m, 1H, C-4′-H), 5.71 (dd, J = 5.9, 2.6 Hz, 1H, C-6-H), 4.87–4.82 (m, 1H, C-2-H), 4.66 (dd, J = 15.3, 3.7 Hz, 1H, C-1′-H), 4.49 (dd, J = 15.3, 4.4 Hz, 1H, C-1′-H), 3.53–3.48 (m, 1H, C-16-H), 3.06 (d, J = 14.6 Hz, 1H, C-12a-H), 2.88 (m, 2H, C-17-H and C-3-H), 2.39 (m, 1H, C-7a-H), 2.30 (m, 2H, C-12b-H and C-8-H), 1.98–1.88 (m, 3H, C-7b-H, C-15a-H and C-10-H), 1.79 (m, 1H, C-1a-H), 1.48 (d, J = 13.4 Hz, 1H, C-15b-H), 1.26 (m, 1H, C-1b-H), 1.22 (s, 3H, CH3), 1.17 (s, 3H, CH3), 1.06 (s, 3H, CH3), 1.03 (d, J = 12.3 Hz, 1H), 0.91 (s, 3H, CH3), 0.72 (s, 3H, CH3). 13C NMR (150 MHz, chloroform-d, ppm) δ 213.31, 172.06, 141.16, 140.80, 126.72, 125.73, 124.99, 118.74, 80.44, 72.11, 70.43, 59.27, 49.95, 48.90, 48.09, 46.23, 45.04, 43.17, 41.94, 38.14, 33.67, 33.05, 24.54, 23.80, 21.44, 19.99, 19.63, 18.82. HRMS (ESI): m/z calculated: C28H40NO5S (M + H)+: 502.2627, found: 502.2619.
Compound 9f. White solid, 84% yield, [α]25D = +102.0 (c = 0.1, MeOH). 1H NMR (600 MHz, chloroform-d, ppm) δ 7.32–7.15 (m, 5H, benzene ring and NH), 5.71 (m, 1H, C-6-H), 4.85–4.79 (m, 1H, C-16-H), 4.42–4.32 (m, 2H, C-7-H), 3.50 (m, 2H, C-2-H), 3.07 (d, J = 14.5 Hz, 1H, C-12a-H), 2.89 (d, J = 8.6 Hz, 2H, C-3-H and C-17-H), 2.42–2.24 (m, 3H, C-8-H, C-12b-H and C-7a-H), 1.97–1.86 (m, 3H, C-1a-H, C-15a-H and C-10-H), 1.78 (dt, J = 12.4, 4.1 Hz, 1H, C-7b-H), 1.48 (d, J = 13.4 Hz, 1H, C-15b-H), 1.22 (s, 3H, CH3), 1.17 (s, 3H, CH3), 1.06 (d, J = 3.8 Hz, 3H, CH3), 1.01 (m, 1H, C-1b-H), 0.91 (s, 3H, CH3), 0.72 (s, 3H, CH3). 13C NMR (150 MHz, chloroform-d, ppm) δ 213.11, 172.34, 140.81, 140.45, 134.29, 129.94, 129.91, 127.67, 127.44, 127.38, 127.26, 125.73, 125.72, 125.39, 118.76, 80.46, 72.30, 70.49, 59.32, 49.84, 48.93, 48.07, 46.36, 45.11, 43.18, 42.90, 41.94, 33.71, 33.11, 24.57, 23.81, 21.50, 20.02, 19.60, 18.90. HRMS (ESI): m/z calculated: C30H41ClNO5 (M + H)+: 530.2673, found: 530.2662.
Compound 9g. White solid, 78% yield, [α]25D = +101.9 (c = 0.1, MeOH). 1H NMR (400 MHz, chloroform-d, ppm) δ 7.36–7.01 (m, 4H, benzene ring), 5.70 (d, J = 5.6 Hz, 1H, C-6-H), 4.83 (t, J = 7.9 Hz, 1H, C-2-H), 4.53–4.39 (m, 2H, C-7′-H), 3.54–3.42 (m, 1H, C-16-H), 3.06 (d, J = 14.5 Hz, 1H, C-3-H), 2.88 (m, 2H, C-17-H and C-12a-H), 2.45–2.23 (m, 3H, C-7a-H, C-1a-H and C-8-H), 1.98–1.87 (m, 3H, C-12b-H, C-15a-H and C-10-H), 1.79 (dd, J = 12.5, 3.9 Hz, 1H, C-7b-H), 1.47 (d, J = 13.4 Hz, 1H, C-15b-H), 1.22 (s, 3H, CH3), 1.17 (s, 3H, CH3), 1.08–0.98 (m, 4H, C-1b-H and CH3), 0.91 (s, 3H, CH3), 0.68 (s, 3H, CH3). 13C NMR (100 MHz, chloroform-d, ppm) δ 213.04, 172.22, 160.95, 140.85, 129.61, 125.17, 124.18, 118.72, 115.39, 115.18, 80.48, 72.21, 70.48, 59.34, 49.83, 48.90, 48.05, 46.24, 45.07, 43.20, 41.91, 37.49, 33.71, 33.06, 24.53, 23.79, 21.42, 19.98, 19.50, 18.86. HRMS (ESI): m/z calculated: C30H41FNO5 (M + H)+: 514.2969, found: 514.2956.
Compound 9h. White solid, 85% yield, [α]25D = +102.5 (c = 0.1, MeOH). 1H NMR (400 MHz, chloroform-d, ppm) δ 7.43 (s, 1H, C-2′-H), 7.41–7.36 (m, 1H, C-4′-H), 7.23–7.19 (m, 2H, C-5′-H and C-6′-H), 5.71 (d, J = 5.6 Hz, 1H, C-6-H), 4.82 (t, J = 7.9 Hz, 1H, C-2-H), 4.37 (q, J = 15.0 Hz, 2H, C-7′-H), 3.51 (m, 1H, C-16-H), 3.07 (d, J = 14.5 Hz, 1H, C-8-H), 2.89 (m, 2H, C-17-H and C-3-H), 2.45–2.36 (m, 1H, C-1a-H), 2.30 (t, J = 12.1 Hz, 2H, C-7a-H and C-12a-H), 2.00–1.89 (m, 3H, C-15a-H, C-12b-H and C-10-H), 1.79 (dt, J = 12.4, 4.2 Hz, 1H, C-7b-H), 1.48 (d, J = 13.3 Hz, 1H, C-15b-H), 1.23 (s, 3H, CH3), 1.17 (s, 3H, CH3), 1.07 (s, 4H, C-1b-H and CH3), 0.91 (s, 3H, CH3), 0.73 (s, 3H, CH3). 13C NMR (100 MHz, chloroform-d, ppm) δ 213.11, 172.38, 140.90, 140.79, 130.64, 130.41, 130.23, 126.23, 122.56, 118.80, 80.55, 72.34, 70.53, 59.37, 49.88, 48.97, 48.10, 46.40, 45.17, 43.27, 42.89, 41.97, 33.79, 33.12, 24.60, 23.86, 21.49, 20.06, 19.61, 18.93. HRMS (ESI): m/z calculated: C30H41BrNO5 (M + H)+: 574.2168, found: 574.2165.
Bioassay
Cell culture. LOVO, SKOV3, HT-29, MCF-7, and HepG2 (purchased from the Cell Bank of the Chinese Academy of Sciences, Shanghai, China) were cultured in minimum essential medium (modified) with 1.5 mM L-glutamine adjusted to contain 2.2 g L−1 sodium bicarbonate (90%) and fetal bovine serum (10%). All cells were cultured in a humidified atmosphere containing 5% CO2 at 37 °C.
Antiproliferative activity assay on tumor cell lines
The antiproliferative activities of all synthesized cucurbitacin IIa derivatives against SKOV3 (ovarian carcinoma), LOVO (colon carcinoma), HT-29 (colon carcinoma), MCF-7 (breast carcinoma), and HepG2 (hepatic carcinoma) cells were measured using the sulforhodamine B (SRB) assay with cisplatin as reference. Cells were seeded in 96-well plates and then treated with different drug concentrations. After incubation for 72 h, cells were fixed with 10% trichloroacetic acid for 1 h at 4 °C, washed five times with tap water, and air-dried. Cells that survived were stained with 0.4% (w/v) sulforhodamine B (SRB) for 20 min at room temperature and washed five times with 1% acetic acid. Bound SRB was solubilized with 10 mM Tris and absorbance was measured at 540 nm.19
Cell cycle
SKOV3 cells at the density of 1 × 106 cells per well were seeded in six-well plates and treated with 4a (1, 2, 4, and 8 μM) and control for 48 h at 37 °C, the supernatant was removed and the cells were washed with cold PBS three times. The cells were then harvested and fixed with 70% precooled ethanol and cultured overnight at −20 °C. SKOV3 cells were then treated with RNase A and stained with propidium iodide (PI). Finally, the suspended cell was analyzed using a flow cytometer (FACS Calibur, BD Biosciences).
Cell apoptosis
SKOV3 cells at a density of 1 × 106 cells per well were seeded into each well of a six-well plate and allowed to grow for 48 h. The cells were then treated with 4a (1, 4, 6, 10, and 20 μM) for 48 h, while cells without treatment were used as a control group. The treated cells were trypsinized and washed with cold PBS three times, and centrifuged at 1200 rpm for 5 min. The supernatants were discarded, and the cells were stained using an Annexin-V-fluorescein isothiocyanate (FITC) kit with binding buffer for 15 min. Subsequently, the cells were labeled with PI and the apoptotic cells were measured using a flow cytometer (FACSCalibur BD, USA). Data were analyzed with ModFit Lt Mac V3.0.
Kinase inhibitory activities
Kinases CDK1/cyclinB, AMPKα2, EGFR, GSK3α, JAK2, and PKBα were diluted with a buffer comprising 20 mM MOPS, 1 mM EDTA, 0.01% Brij-35, 5% glycerol, 0.1% β-mercaptoethanol, and 1 mg per mL BSA before adding to the reaction mixture. Kinase mTOR was diluted with a buffer comprising 500 mM HEPES, 10 mM EGTA, and 0.1% Tween20 before adding to the reaction mixture. Kinase MAPK1 was diluted with a buffer comprising 50 mM TRIS, 0.1 mM EGTA, 0.1 mM Na3VO4, 0.1% β-mercaptoethanol, and 1 mg per mL BSA before adding to the reaction mixture. CDK1/cyclinB(h) was incubated with 8 mM MOPS (pH 7.0), 0.2 mM EDTA, 0.1 mg per mL histone H1, 10 mM magnesium acetate, and [γ-33P]-ATP (specific activity and concentration as required). The reaction was initiated by adding the Mg/ATP mix. After incubating for 40 min at room temperature, the reaction was stopped by adding phosphoric acid to a concentration of 0.5%. A 10 μL sample of the reaction was then spotted onto a P30 filtermat and washed with 0.425% phosphoric acid four times for 4 min, and once in methanol, prior to drying and scintillation counting.
Results and discussion
Twenty-one cucurbitacin IIa derivatives were synthesized through a series of reactions, as outlined in Schemes 1–5. All compounds were obtained in different yields. Cucurbitacin IIa (1) was extracted from Hemsleya by our research group8 and verified by single-crystal X-ray diffraction. For compound 1, the signals for H-2, H-3, and H-6 were observed at δ 4.10 (1H, ddd, J = 11.4, 9.1, 4.1 Hz), 3.43 (1H, d, J = 9.1 Hz), and 5.74 (1H, dt, J = 6.2, 2.1 Hz), respectively. The related carbon signals were also evident in the 13C NMR spectra, with C-2, C-3, and C-6 observed at δ 80.0, 81.4, and 118.7, respectively. Compound 3 was synthesized according to the synthetic route outlined in Scheme 1. Cucurbitacin IIa (1) was reacted with 0.2 equiv. of DMAP and 6 equiv. of acetic anhydride in the presence of pyridine at 25 °C to afford compound 2. Next, the intermediate was prepared by reacting 2 with sodium borohydride in methanol at 0 °C, followed by reacting with 2 equiv. of sodium periodate in tetrahydrofuran and water as mixed solvent to obtain compound 3 in 83% yield.12 In the 1H NMR spectrum for compound 3, the H-2 proton signal was observed at δ 5.47 instead of δ 4.10.
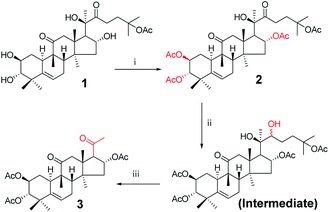 |
| | Scheme 1 Synthetic route to target compounds: (i) acetic anhydride, DMAP, pyridine, rt, 10 min; (ii) methanol, sodium borohydride (NaBH4), rt, 5 h; (iii) sodium periodate, tetrahydrofuran (THF), rt, 16 h. | |
 |
| | Scheme 2 Synthetic route to target compounds: (iv) hydrazine, acetic acid (AcOH), rt, 18 h. | |
 |
| | Scheme 3 Synthetic route to target compounds: (v) hydroxylamine hydrochloride, pyridine, 50 °C, 10 h; (vi) sodium hydroxide (NaOH), methanol, 80 °C, 6 h. | |
 |
| | Scheme 4 Synthetic route to target compounds: (vii) sodium hypochlorite (NaClO), methanol, rt, 1 h; (viii) sodium hydroxide (NaOH), methanol, 80 °C, 5 h. | |
 |
| | Scheme 5 Synthetic route to target compounds: (ix) DMTMM, N-methylmorpholine, amine, methanol, rt, 15 h. | |
Consecutive reactions of intermediate compound 3 with hydrazine in AcOH at room temperature gave the corresponding hydrazones 4a–4f in 74–91% yields respectively.4 Their 13C NMR spectra showed characteristic carbon signals at δ 147.3–157.9, attributed to C-20. Derivative 4b was subjected to purification by recrystallization in CH2Cl2/CH3OH/n-hexane (1:0.1:3, v/v/v) at 4 °C, which afforded the product as yellow crystals. The absolute configuration of 4b was determined by single-crystal X-ray diffraction, and spatial configurations of the parent nuclei of derivatives were determined. By measuring the single crystal diffraction of 4b, we speculated and determined the spatial configuration of compounds 4a–4f.
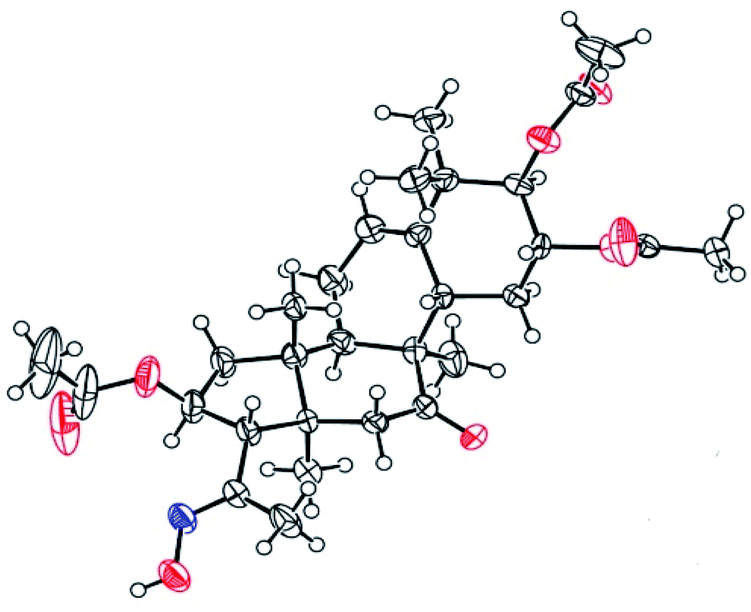
Compounds 3 were mixed with hydroxylamine hydrochloride in pyridine at 50 °C to give compounds 5a and 5b in 55% and 42% yields, respectively. The structures of compounds 5a and 5b were confirmed by their respective 13C NMR spectra, which showed characteristic C-21 carbon signals at δ 15.6 and 19.8, respectively. To confirm the significant differences between 5a and 5b, derivative 5a was subjected to purification by recrystallization in CHCl2/CH3OH/n-hexane (1:0.1:3, v/v/v) at 4 °C, which afforded the product as white crystals. The absolute configuration of 5a was determined by single-crystal X-ray diffraction, and the spatial configurations of the parent nuclei of derivatives were determined. The differences in chirality between 5a and 5b were also determined (Fig. 3).16
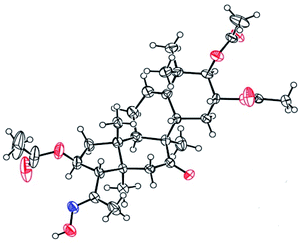 |
| | Fig. 3 Molecular structure of derivative 5a. | |
Compounds 5a and 5b were treated with sodium hydroxide in methanol at 50 °C to afford compounds 6a and 6b in 93% and 91% yields, respectively. The 13C NMR spectra of compounds 6a and 6b showed carbon signals characteristic of C-21 at δ 15.9 and 19.9, respectively.
Consecutive reactions of intermediate 3 with sodium hypochlorite in methanol at room temperature gave the corresponding methyl ester (7) in 93% yield. Compound 7 was hydrolyzed with sodium hydroxide in methanol at 80 °C to obtain corresponding carboxylic acid 8 in 78% yield.
Consecutive reactions of intermediate compound 8 with DMTMM, N-methylmorpholine, and amine in methanol at room temperature gave corresponding amides 9a–9h in 75–93% yields respectively. Their 13C NMR spectra showed characteristic carbon signals at δ 170.7–172.4 attributed to C-20.
Cytotoxic activity
The sulforhodamine B (SRB) assay is a method for detecting cell proliferation. SRB is a pink anionic dye that is readily soluble in water and specifically binds to basic amino acids of proteins in cells under acidic conditions, producing an absorption peak at 540 nm, and absorbing light linearly with cell mass. SRB can also be used as a quantitative test for the number of cells. The anticancer activities of the synthesized compounds were studied by SRB assay using the following human cancer cell lines in cell culture:20 SKOV3 (an ovarian cancer cell line), LOVO (a colon cancer cell line), HT-29 (a colon cancer cell line), MCF-7 (a breast cancer cell line), and HepG2 (a liver cancer cell line). Cisplatin and cucurbitacin IIa were used as reference standards in this study. We first measured the inhibitory rate of cucurbitacin IIa derivatives at a concentration of 10 μM, with the obtained suppression ratio values shown in Table 1. The IC50 values of cells were then determined for derivatives with suppression ratio values of more than 50%. The obtained IC50 values are shown in Table 2. Three cucurbitacin IIa derivatives showed dose-dependent (1.2–13.7 μM) anticancer activities against the five cancer cells, with lower toxicity against human normal HEK293 cells (human embryonic kidney cell line). Among the tested compounds, compounds 3 and 7 without branched chains were not cytotoxic, and acetylated compound 2 still showed good activity against SKOV3 cells, indicating that the branched chain was an important pharmacophore of cucurbitacin IIa. The IC50 values of 2,4,6-trichlorophenylhydrazine derivative 4a and 2-hydrazinopyridine derivative 4d against LOVO cells were 3.9 ± 0.15 μM and 2.2 ± 0.20 μM, respectively, and their cytotoxicity toward human normal cells HEK293 was significantly reduced, indicating that changing the branched chain of cucurbitacin IIa to hydrazone derivatives is an important strategy.
Table 1 Inhibition of derivatives on SKOV3, HT29, HEPG2, MCF-7, and LOVO cell lines at 10 μM
| Compound |
% inhibitiona (10 μM) |
| SKOV3 |
HT29 |
HEPG2 |
MCF-7 |
LOVO |
| Inhibition values were calculated from two independent experiments using the SRB assay after 72 h treatment. Cisplatin is a positive control anticancer drug. |
| 1 |
95 |
97 |
95 |
98 |
91 |
| 2 |
61 |
4 |
4 |
3 |
3 |
| 3 |
7 |
4 |
5 |
2 |
0 |
| 4a |
95 |
93 |
84 |
61 |
64 |
| 4b |
8 |
3 |
10 |
0 |
0 |
| 4c |
4 |
12 |
9 |
1 |
1 |
| 4d |
58 |
62 |
6 |
7 |
71 |
| 4e |
7 |
8 |
8 |
5 |
9 |
| 4f |
4 |
5 |
4 |
13 |
0 |
| 5a |
7 |
5 |
10 |
1 |
1 |
| 5b |
8 |
8 |
9 |
2 |
7 |
| 6a |
1 |
12 |
8 |
2 |
4 |
| 6b |
6 |
7 |
7 |
5 |
6 |
| 7 |
6 |
3 |
11 |
2 |
2 |
| 9a |
2 |
1 |
3 |
6 |
18 |
| 9b |
1 |
0 |
1 |
0 |
0 |
| 9c |
1 |
1 |
2 |
0 |
1 |
| 9d |
1 |
0 |
1 |
2 |
0 |
| 9e |
0 |
3 |
0 |
0 |
3 |
| 9f |
0 |
1 |
6 |
0 |
1 |
| 9g |
4 |
2 |
7 |
1 |
4 |
| 9h |
5 |
1 |
5 |
0 |
8 |
| Cisplatinb |
0.57 ± 0.13 |
2.19 ± 0.06 |
2.06 ± 0.11 |
1.71 ± 0.09 |
0.95 ± 0.13 |
Table 2 In vitro cytotoxic activity of compounds 1, 2, 4a, and 4d against HEK293, SKOV3, LOVO, HT29, MCF-7, and HEPG2 cell lines
| Compound |
IC50a (μM) |
| HEK293 |
SKOV3 |
LOVO |
HT29 |
MCF-7 |
HEPG2 |
| IC50 values were calculated from two independent experiments. The values were calculated using Graphpad Prism 5 and reported as the mean ± SD. Cisplatin is a positive control anticancer drug. |
| 1 |
0.51 ± 0.02 |
0.14 ± 0.01 |
0.14 ± 0.01 |
0.25 ± 0.08 |
0.26 ± 0.02 |
0.52 ± 0.04 |
| 2 |
7.4 ± 1.15 |
1.22 ± 0.12 |
— |
— |
— |
— |
| 4a |
24.61 ± 1.47 |
6.22 ± 0.05 |
3.95 ± 0.38 |
11.88 ± 0.05 |
7.2 ± 1.47 |
13.70 ± 0.66 |
| 4d |
11.07 ± 0.97 |
2.23 ± 0.05 |
2.19 ± 0.38 |
9.00 ± 1.22 |
— |
— |
| Cisplatinb |
2.12 ± 0.13 |
0.57 ± 0.13 |
0.95 ± 0.13 |
2.19 ± 0.06 |
1.71 ± 0.09 |
2.06 ± 0.11 |
Effect of derivative 4a on SKOV3 cell cycle
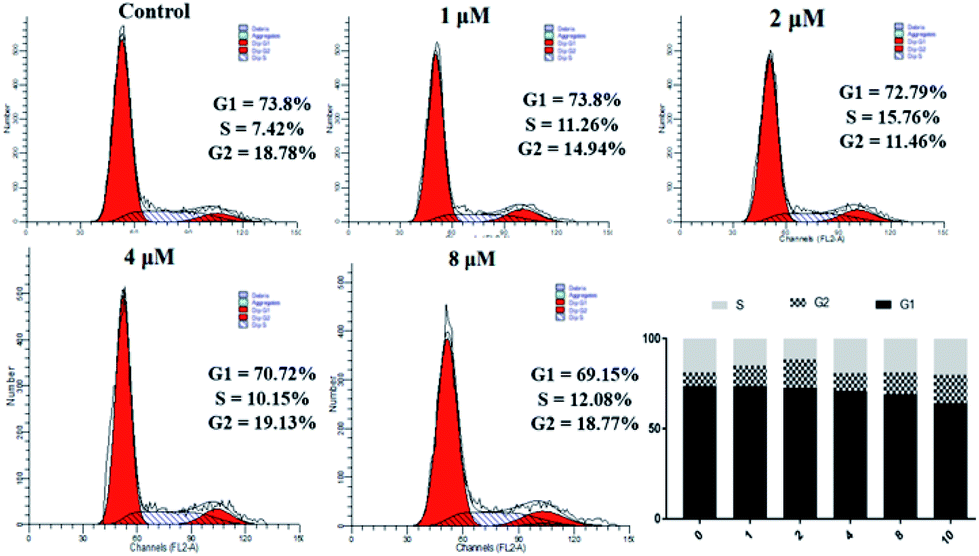
The data in Table 2 shows that 4a has significant antiproliferative activity against SKOV3 cells. Therefore, we continued to study the biological effects of 4a on SKOV3 cells. A PI staining kit was used to investigate the effect of 4a on cell cycle distribution. SKOV3 cells were stained with PI and analyzed using a flow cytometer, after treating with 4a (1, 2, 4, and 8 μM) or the control for 48 h. As shown in Fig. 4, with increasing concentration of 4a, cells in the G1 phase accumulated less compared with the control group (73.8%), ranging from 69.15% (0.1 μM) to 73.8% (3 μM), exhibiting a concentration-dependent change. However, cells in the G2 and S phases showed little change. From the above results, the cytotoxicity mechanism of 4a might not involve influencing the cell cycle.
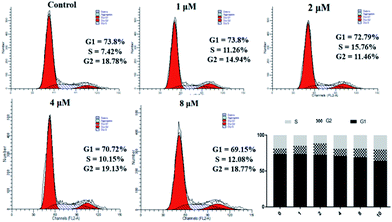 |
| | Fig. 4 Effect of derivative 4a on SKOV3 cell cycle. | |
SKOV3 cell apoptosis induced by compound 4a
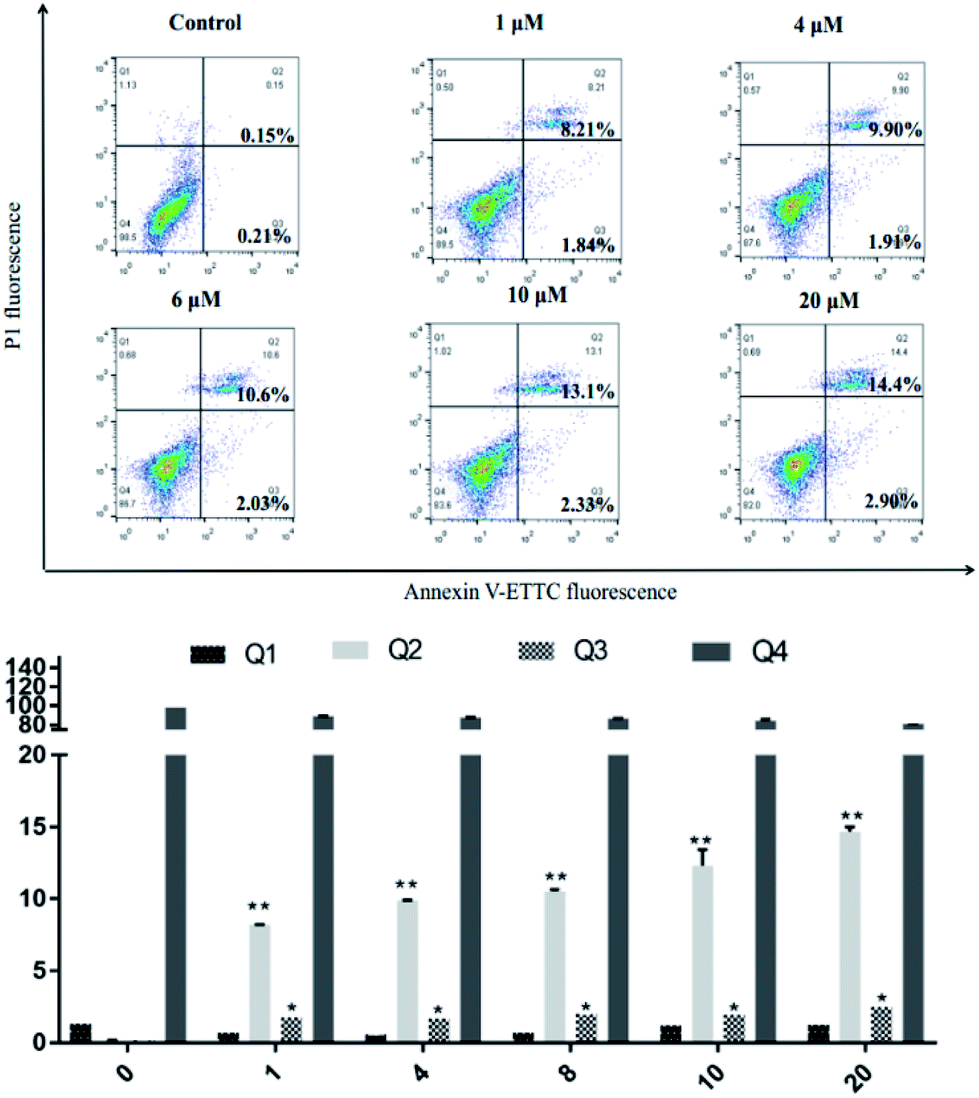
SKOV3 cells were examined using an Annexin V-FITC/PI FACS assay after treating with 4a (1, 4, 6, 10, 20 μM) for 48 h, with the aim to explore the cell death mechanism. As shown in Fig. 5, with increasing concentration of 4a, the percentage apoptotic population in SKOV3 cells was significantly increased, ranging from 10.05% to 17.3% after 48 h. The apoptotic rate of SKOV3 cells was positively correlated with the concentration of 4a in a dose-dependent manner.
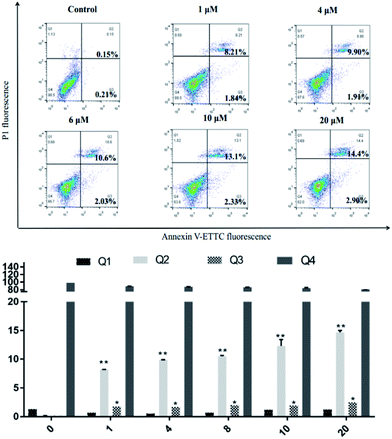 |
| | Fig. 5 SKOV3 cell apoptosis induced by compound 4a. | |
Determination of kinase inhibitory activity
To explore structure–activity relationships (SAR), cucurbitacin IIa and its derivatives (2, 4a, and 4d) were evaluated for inhibition of CDK1/cyclinB(h), AMPKα2(h), EGFR(h), GSK3α(h), JAK2(h), MAPK1(h), mTOR(h), PKBα(h), and PI3K p110α(E542K)/p85α by conducting preliminary tests.20–22 As shown in Tables 2 and 3, the selectivity of the antiproliferative activity of compound 2 was better than that of cucurbitacin IIa. Furthermore, the CDK1/cyclinB (H) inhibition by 2 was better than that by cucurbitacin IIa, which did not inhibit cell proliferation in this fashion. Furthermore, cucurbitacin IIa and its derivatives showed no inhibitory activity against kinase JAK2.11 The tetracyclic structure is not changed from 1 to 4d, which both show inhibitory activity against CDK1/cyclinB, indicating that the tetracyclic structure might be a matching site for CDK1/cyclinB, and that this activity is of great significance to the later modification and mechanism of cucurbitacin IIa.
Table 3 Comparison of cucurbitacin IIa and its analogues on the inhibition of kinases
| compound |
Activity (% control)a |
| CDK1/cyclinB |
AMPKα2 |
EGFR |
GSK3α |
JAK2 |
MAPK1 |
mTOR |
PKBα |
PI3K p110α(E542K)/p85α |
| Activity% values were obtained from twice independent experiments, and activity% = {(countC − countB)/(countP − countB)} × 100%. CountC: the counts of testing compounds. CountP: the counts of positive control. CountB: the counts of blank. |
| 1 (1 μM) |
26 |
81 |
89 |
104 |
114 |
103 |
71 |
90 |
87 |
| 2 (1 μM) |
11 |
82 |
85 |
96 |
115 |
93 |
78 |
92 |
104 |
| 4a (1 μM) |
33 |
77 |
92 |
104 |
105 |
95 |
85 |
108 |
103 |
| 4d (1 μM) |
27 |
92 |
94 |
100 |
100 |
89 |
82 |
113 |
92 |
Conclusions
Cucurbitacin IIa is abundant in Hemsleya. Although it can inhibit cancer cell activity, it cannot be used as an anticancer drug owing to its high toxicity. The toxicity of imine derivatives obtained from cucurbitacin IIa as precursor toward HEK293 cells was significantly reduced, and the cancer cell inhibition activity was maintained, indicating that the imine derivatives showed improved selectivity. After the cucurbitacin IIa side chain was removed, its anticancer activity decreased, proving that the branched chain of cucurbitacin IIa is an important pharmacodynamic group. Protecting the 2-, 3-, and 16-hydroxyl groups of cucurbitacin IIa resulted in only inhibition of human ovarian cancer (SKOV3) cells, and the synthesized derivatives no longer showed broad-spectrum activity, which indicated that its anticancer mechanism had changed. Presently, the modification of natural products is still a focus of drug research and development. This experiment mainly involved changing the structure of cucurbitacin-type tetracyclic triterpenoids, and provided a novel tetracyclic framework derived from a natural product. Through studying its pharmacological activity, cucurbitacin IIa can be concluded to have advantages of good activity, well-defined chirality, and easy access. The late-stage modification of cucurbitane-type tetracyclic triterpenoids provides a useful reference for future research.
Conflicts of interest
The authors confirm that the content of this article has no conflicts of interest.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 81903473) and the Innovation Project of the Shandong Academy of Medical Sciences.
Notes and references
- R. K. Tiwari, J. Geliebter, V. P. S. Garikapaty, S. P. K. Yedavelli and A. Mittelman, Int. J. Oncol., 1999, 14, 713–719 CAS.
- Z. Darzynkiewicz, F. Traganos, J. M. Wu and S. Chen, Int. J. Oncol., 2000, 17, 729–736 CAS.
- D. V. H. S. Jean-Paul, S. Delignat, M. F. Bloch, M. D. Kazatchkine and S. V. Kaveri, Chemotherapy, 2001, 47, 366–376 CrossRef PubMed.
- H. Zhang, Y. Mu, F. Wang, L. Song, J. Sun and Y. Liu, R. Soc. Open Sci., 2018, 5, 171510 CrossRef PubMed.
- X. B. Chen, G. Y. Chen, J. H. Liu, M. Lei, Y. H. Meng and D. A. Guo, Fitoterapia, 2014, 94, 88–93 CrossRef CAS PubMed.
- Z. Biao-Yi, Y. Yu and Y. Zeng-Liang, J. Ethnopharmacol., 2008, 116, 89–95 CrossRef PubMed.
- R. R. Tian, J. C. Chen, G. H. Zhang, M. H. Qiu, Y. H. Wang, L. Du, X. Shen, N. F. Liu and Y. T. Zheng, Chin. J. Nat. Med., 2008, 6, 214–218 CrossRef CAS.
- Y. Li, W. X. Wang, Z. F. Zheng, Y. Y. Mu, Y. J. Liu, H. Y. Wang and Q. Q. Yao, J. Asian Nat. Prod. Res., 2017, 20, 36–48 CrossRef PubMed.
- J. Wu, Y. Wu and B. B. Yang, Life Sci., 2002, 71, 2161–2170 CrossRef CAS PubMed.
- Y. Li, Z. F. Zheng, L. Zhou, Y. J. Liu, H. Y. Wang and Q. Q. Yao, Phytochem. Lett., 2015, 14, 239–244 CrossRef CAS.
- C. Boykin, G. Zhang, Y. H. Chen, R. W. Zhang, X. E. Fan and W. M. Yang, Br. J. Cancer, 2011, 104, 781–789 CrossRef CAS PubMed.
- W. W. Wang, H. R. Yang, Y. Li, Z. F. Zheng, Y. J. Liu, H. Y. Wang, Y. L. Mu and Q. Q. Yao, R. Soc. Open Sci., 2018, 5, 1–16 CrossRef PubMed.
- J. Ren, X. Shi, X. N. Li, L. W. Li and Q. S. Zhao, Org. Lett., 2016, 18, 3948–3951 CrossRef CAS PubMed.
- M. Ihara, T. Taniguchi, Y. Tokunaga and K. Fukumoto, J. Org. Chem., 1994, 59, 8092–8100 CrossRef CAS.
- A. Regueiro-Ren, J. J. Swidorski, Z. Liu and Y. Chen, J. Org. Chem., 2018, 61, 7289–7313 CAS.
- X. Zhang, X. Ma, T. Zhang, B. Li, S. Jiang and G. Zhang, RSC Adv., 2018, 8, 42262–42268 RSC.
- D. C. Braddock, P. D. Lickiss, B. C. Rowley, D. Pugh, T. Purnomo and G. Santhakumar, Org. Lett., 2018, 20, 950–953 CrossRef CAS PubMed.
- W. Meng, J. Wang, W. Xue and A. Wu, Dyes Pigm., 2013, 97, 475–480 CrossRef.
- H. Quan, H. Liu, C. Li and L. Lou, J. Pharmacol. Exp. Ther., 2009, 330, 326–333 CrossRef CAS PubMed.
- M. T. Bengoechea-Alonso and J. Ericsson, Cell Cycle, 2006, 5, 1708–1718 CrossRef CAS PubMed.
- R. Chu, D.
T. Terrano and T. C. Chambers, Biochem. Pharmacol., 2012, 83, 199–206 CrossRef CAS PubMed.
- J. Kundu, D. H. Kim, J. K Kundu and K. S. Chun, Food Chem. Toxicol., 2014, 65, 18–26 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1951946 and 1951947. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra09113k |
| ‡ These authors contributed equally to the work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Qingqiang Yao*b
*b and
Qingqiang Yao*b