
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploiting the native inspiratory ability of a mass spectrometer to improve analysis efficiency†
Qian Zhang‡
ab,
Lin Lin‡c,
Quan Yu *a and
Xiaohao Wangab
*a and
Xiaohao Wangab
aDivision of Advanced Manufacturing, Tsinghua Shenzhen International Graduate School, Shenzhen, 518055, China. E-mail: yu.quan@sz.tsinghua.edu.cn
bState Key Laboratory of Precision Measurement Technology and Instruments, Department of Precision Instruments, Tsinghua University, Beijing 100084, China
cMaterials Characterization & Preparation Center, Southern University of Science and Technology, Shenzhen 518055, China
First published on 24th January 2020
Abstract
In this study, a new approach to perform self-aspirating sampling in mass spectrometry (MS) analysis was developed by using the native inspiratory ability of a mass spectrometer. Specifically, the inspiratory channel and sampling inlet of the MS instrument were integrated into a single pathway through a sealed ionization chamber to facilitate analyte delivery and improve sample utilization. Based on this approach, combined with structural simplification and optimization, a versatile electrospray ionization (ESI) source has been constructed and characterized using different mass spectrometers. In addition to the self-aspirating ability, this source configuration can provide sub-ambient pressure (SAP) conditions for ionization, which were conducive to suppressing the background ions generated from some air-involved reactions. Moreover, it can also be used directly for electrospray-driven extraction ionization. With the SAP-ESI source, a conventional mass spectrometer enables rapid analysis of both volatiles and solutions via secondary electrospray ionization and coaxial electrospray ionization, respectively. As the compact gas pathway of the source will promote the efficient transfer and ionization of the sampled substances, the total consumption of the analyte for each analysis can be reduced to subnanogram level and a subppbv limit detection is achieved. Other demonstrated features such as the versatility, easy operation as well as simple assembly will likely contribute to the prevalence of the proposed sampling and ionization strategy.
1. Introduction
Among the many modern analytical methods, mass spectrometry (MS) is undoubtedly one of the most authoritative and versatile chemical identification techniques. Currently, the emergence of a variety of novel sampling and ionization approaches, along with the continuous updating of high-performance instruments, have extended the application of mass spectrometers to various online and on-site analysis fields such as food safety,1,2 environmental monitoring,3 and clinical diagnostics.4 As these areas all have a strong need for rapid time response, the prosperity of ambient ionization5–9 technology can well satisfy such demands by greatly simplifying the sample preparation process to improve the detection efficiency.Advances in ambient ionization are mainly based on the excavation of new sampling and ionization mechanisms. These techniques enable direct ionization of different raw sample types or selective ionization of specific compounds in complex matrices. For instance, as the groundbreaking technique of ambient ionization, desorption electrospray ionization (DESI)10 has greatly shined on the organic and biochemical analysis of sample surfaces. Direct analysis in real time (DART)11 is another pioneer that has been rapidly commercialized and gained reputation for its versatility and high throughput ability. This technique can be used to differentially detect a wide variety of chemical species in vapor, liquid or solid samples. Besides these two representatives, other ambient ionization techniques, such as secondary electrospray ionization (SESI),12–15 extractive electrospray ionization (EESI),16 paper spray (PS)17,18 and dielectric barrier discharge ionization (DBDI),19 are also driving mass spectrometry to conquer more and more application territories.4–9,20 Among these ionization methods, SESI has been proposed for more than two decades, and its recent use in some refreshing applications like breath analysis21–23 and plant metabolism study24,25 is regaining increasing attention to this technique.
In addition to exploring new mechanism, increased efforts in structure optimization are following up to further improve the performance of the ion source or simplify its operation. For example, since ion motion is significantly affected by the state of airflow under non-vacuum conditions, ingenious usage of gas flow can bring many benefits when building an ion source. It allows long-distance ion transportation from the source to the mass spectrometer, as implemented in air flow assisted ionization26 and real-time sampling probe for DESI analysis.27 Collisions between the charged gas flow and the samples can facilitate the liberation and ionization of the analytes in those extractive-electrospray related methods.28–30 For SESI analysis, airflow is especially vital as it is the only carrier of analyte vapor and will also affect charge transfer from the solvent ions to the analyte molecules.23,31,32 Furthermore, some ion sources with special airflow configuration exhibit self-aspirating ability33–35 or high sample utilization36,37, both of which can make the sampling process more efficient.
Mass spectrometers coupled with atmospheric pressure ionization (API) sources have been well recognized for their low relative detection limit and ultratrace analysis38 capability. In reality, when ionization is operated in an open environment, waste of samples is inevitable that only a small fraction of the produced ions is sampled into these API-MS instruments. Sample utilization efficiency is generally not concerned in routine MS analysis because of the abundant sample amount. From another perspective, microanalysis38 or low absolute detection limit is required when the available sample volume is small. To meet such requirement, minimizing the sampling amount along with maximizing ionization efficiency are two vital elements. Pulsed ionization methodology is a good choice in this case and it can even realize ultra-microanalysis of solution and solid samples.39–41 Another practical way to reduce sample consumption in routine MS analysis is self-aspirating sampling, namely, gaseous or liquid samples are directly drawn to the ion source with minimal residual during the sampling process.33–35,42–44 This approach can endue the mass spectrometer with sniffing ability similar to the olfaction of animals to detect volatile mixtures,44 but additional pump or assistant gas flow are commonly needed to create the inspiration channel to drive the sample introduction.
So far, the family of ambient ionization technology is still expanding, but in practice only a few members become widely dispersed in analytical field. Those rapidly popularized sources, such as DESI and PS, are generally featured with simple structures and convenient operations. These characteristics should be important design concepts for the development of practical ion sources. Herein, a simplified sampling and ionization strategy with inherent aspirating ability, which is provided by the vacuum system of a mass spectrometer, is introduced and used for rapid detection of gas and solution samples, aiming to maximize the sample utilization and reduce the sample consumption for routine MS analysis. The proposed method can be implemented with a commercial ion source, but using a tailored device with a low-pressure ionization chamber can present superior performance.
2. Experimental
Two commercial MS platforms from Thermo Fisher Scientific, including an LCQ Fleet ion trap mass spectrometer and a Q-Exactive Orbitrap mass spectrometer, were used in this study. Both instruments were originally equipped with the same ionization system consisting of an Ion Max ESI source and an atmospheric interface. By using a new simple sampling strategy (described in Fig. S1†), the standardized ion source can be directly applied for routine SESI analysis without structural modification. In addition, another simplified and compact device, which was called sub-ambient pressure ESI (SAP-ESI, see Fig. 1) source, has been constructed to perform more efficient SESI ionization. It comprised a sample inlet module that was introduced earlier45,46 and a sealed spray chamber (ID 20 mm and 35 mm in length) connected to the MS inlet. The outlet hole of the chamber was matched with the MS inlet tube that the connection of the entire module to the MS interface can be manually assembled and meet the airtightness requirement. | ||
| Fig. 1 Schematic structure of the developed SAP-ESI sampling and ionization system. | ||
During the operation, the spray solvent was syringed into the spray chamber through a fused-silica capillary (OD 360 μm, ID 50 μm) at a rate of 3 μL min−1, and a high voltage of +6 kV was applied onto the solvent to generate a stable electrospray. Simultaneously, the gas or liquid samples were placed near the sampling inlet and continuously drawn into the MS instrument along with the airflow, and then the SESI process could occur in the spray chamber. Standard 1/16 inch OD PEEK tubing with different internal diameters was used as the sample inlet to obtain variable sampling rates and chamber pressures.
The experiments were mostly carried out on the Q-Exactive Orbitrap instrument unless specially indicated. The scan parameters for MS analysis were as follows: scan range, 50 to 750 m/z; resolution, 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000; microscans, 1; AGC target, 1e6; maximum inject time, 30 ms. The capillary temperature was set at 320 °C, with a S-lens RF level of 50. No sheath or auxiliary gas was used.
000; microscans, 1; AGC target, 1e6; maximum inject time, 30 ms. The capillary temperature was set at 320 °C, with a S-lens RF level of 50. No sheath or auxiliary gas was used.
Most of the chemical analytes were purchased from Aladdin Reagent (Shanghai, China), and the reagents were purchased from Anpel Company (Shanghai). The dried rosemary leaves were purchased from an ordinary supermarket. Methanol with 0.01% hydrochloric acid was used as the spray solvent when performing SESI analysis.
3. Results and discussion
Mass spectrometer with an atmospheric pressure interface will continuously draw air into the instrument during the operation, and this airflow can also be used for sample delivery. Using the inherent vacuum of a mass spectrometer as the self-aspirating force for gas or liquid sampling will simplify both the structure and operation and implement high sample utilization.45–47 Currently, SESI-MS has become an increasingly powerful technique for online analysis, especially when coupled to a high-resolution (HR) instrument,22,23,48 and the simplification in sampling operation can make it more convenient to use. A routine SESI-MS analysis experiment was first carried out in this study by using a conventional Q-Exactive Orbitrap mass spectro-meter coupled with an Ion Max source. Using the sampling method described in Fig. S1† allows the MS instrument to analyze the aroma components in some plants directly. When placing rosemary leaves near the sample inlet, the corresponding SESI-MS spectrum could be rapidly acquired, as shown in Fig. 2. The high mass accuracy of the Q-Exactive instrument (generally less than 3 ppm) enabled direct assignment of the molecular formulas based on the m/z values, especially for those small ions. Then the main components of the volatiles could be roughly identified by comparing with the previous ref. 49 and were also given in the spectrum. The above experiment preliminarily verified the feasibility of using the native inspiratory flow of a mass spectrometer for sample transmission, and the subsequent sampling mode can turn a standard laboratory MS device into an electronic nose with the ability of sniffing44 and rapid identification, especially when coupled with an HR instrument.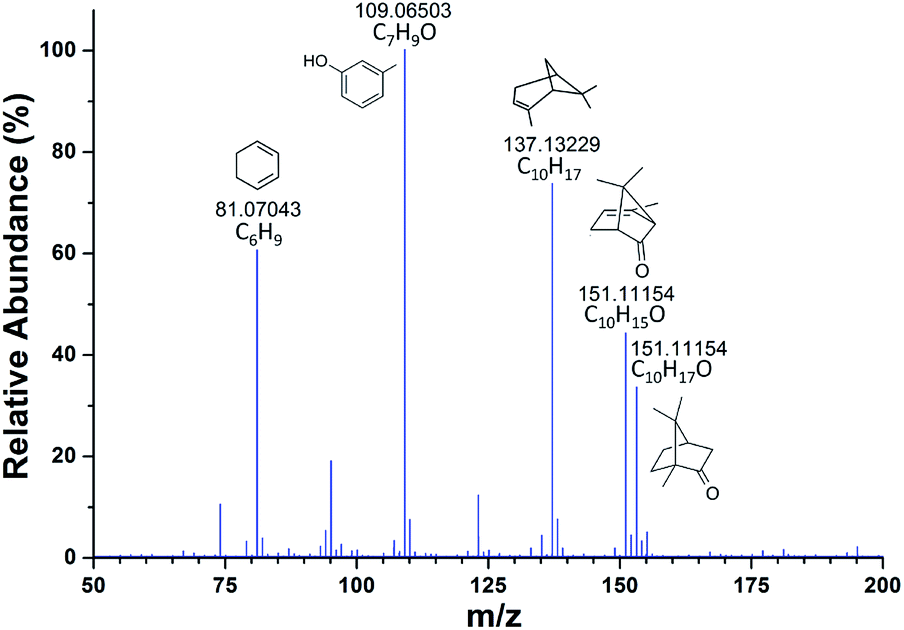 | ||
| Fig. 2 High-resolution SESI-MS spectrum of rosemary leaves by using the Ion Max source. | ||
Since the Ion Max source is not designed for SESI analysis, its large ionization chamber and diagonal gas path configuration will reduce detection efficiency. Then efforts were devoted to improve the overall performance of the self-aspirating SESI-MS analysis. The ionization efficiency of SESI is affected by many factors, such as the yield of the charged spray, the charge transfer rate and the airflow field condition.31,50 In addition, enhancing ion transmission efficiency during the atmospheric sampling process should also be critical for high detection sensitivity. Based on the above considerations, a small and simplified SAP-ESI device was designed to favor the ionization and sampling process. It evolved from the previously described vacuum electrospray source that was adopted in a miniature mass spectrometer.45 The pulsed pinch valve became unnecessary and was removed in the current device because the lab-scale MS instruments were capable of continuous atmospheric sampling. Furthermore, an enclosed spray chamber was added to create a variable pressure environment for different ionization processes. In this structure, almost all samples and sprays in gas or aerosol phase could be drawn into the mass spectrometer. Besides, the coaxial design of the ESI capillary and sample tube outlet was expected to produce straight and full impacts between the analytes and the charged drops to achieve a high ionization efficiency.50–52
During the experiment, the pressure in the spray chamber of the SAP-ESI source was determined by the pumping speed of the MS instrument and the intake rate of the sample inlet. The former remained constant while the latter could be adjusted by changing the sampling tubing. The normal fore vacuum pressure of the MS system with or without the Ion Max source was about 1.8 mbar. When the SAP-ESI source was mounted, the pressure dropped as the tubing ID decreased. Using an inlet tube with an ID greater than 1 mm will not change the pressure, while using a 250 μm tube will reduce the pressure to 0.4 mbar. To meet the operational requirement of the commercial MS instrument in terms of fore vacuum pressure (higher than 0.65 mbar), most of the SESI experiments were carried out using a 500 μm tube that led to a pressure of about 0.8 mbar. Under this condition, the actual pressure in the spray chamber was about 0.55 bar, indicating that a sub-ambient pressure environment was formed.
ESI is widely renowned and used as an atmospheric ionization technique, but in fact, it has a certain tolerance to the operating pressure. Prior to our recently developed vacuum ESI source,45 it has been reported that performing ESI at SAP condition was conducive to both the reduction of the solvent background peaks and the improvement in ionization efficiency and ion transmission.53–56 Current HR-MS can well identify the analyte ions from massive background species, but those instruments with low resolution often encounter severe peak interference issues. The influence of the dropped chamber pressure on the ionization process and the MS analysis was roughly evaluated at first by coupling the ion source to a common LCQ ion trap mass spectrometer. In this experiment, switching between ambient and sub-ambient conditions inside the spray chamber was simply implemented by successively removing and installing the sampling tubing of the SAP-ESI source. A mixed methanol solution containing 1 ppm 9,9-dioxopromethazine (DPZ) and 2,4-dimethylaniline was prepared as the electrospray solvent to perform routine ESI-MS analysis. Mass spectra acquired under AP and SAP conditions were shown and compared in Fig. 3a. It was found that severe interference of background ions appeared mainly in low mass range for AP-ESI analysis that almost submerged the protonated 2,4-dimethylaniline ion peak (m/z 122). In addition, the extracted ion chromatograms of the protonated DPZ (m/z 317) and one of the background ion species (m/z 81) were given in Fig. 3b, indicating a close correlation between the ion formation and the source pressure. It was speculated that the generation of the background ions during the AP-ESI process was probably related to the air-involved reaction products rather than the solvent clusters. Hence, the background ions could be effectively suppressed under SAP condition due to less air participation during the ionization, while the analyte ion current was slightly increased probably owning to the reduced competition effect among different ions. Although the developed sub-ambient source was aimed at facilitating SESI analysis, its capability in suppressing ESI background interferences was also remarkable, and this can be quite conducive to the analysis of small volatile compounds.
 | ||
| Fig. 3 (a) ESI mass spectra and (b) extracted ion chromatograms acquired under sub-ambient pressure and ambient pressure (AP) condition. | ||
The usage of an enclosed spray chamber can prevent the diffusion of gas samples and the influence of external airflow turbulence during ionization, and the structure of the chamber will also affect the ionization efficiency. For example, gaseous sample will easily remain in the large ionization chamber of the Ion Max source and thus cause residual effect and lower the sample utilization. To improve the ionization efficiency, the SAP-ESI source was carefully designed to eliminate dead volume in its injection pathway. Direct SESI analysis of an aqueous solution of 10 ppm acetone was then carried out using the two sources for comparison. Although acetone has low sensitivity in SESI-MS analysis probably due to its low mass and low protonated ionization efficiency, its strong volatility can reduce residues in transmission and make the results more representative. During the experiment, the solution was quickly moved away after being placed under the sample inlets, then the volatilized analytes can be drawn into the spray chamber and ionized. The chromatogram profiles of protonated acetone ion acquired using the two sources were shown in Fig. 4. As the self-aspirating airflow can accelerate the sampling process, both sources had fairly fast response time, creating sharp rising edges of the signal profiles. However, the ion signal acquired by the commercial source exhibited a long-term tailing due to sample retention in the chamber, which also caused dilution of the gas analytes and reduction of signal intensity. In contrast, a shorter and stronger signal response can be obtained using the SAP-ESI source, because the compact gas path enabled a more efficient ionization process and therefore a higher detection sensitivity in terms of convenient SESI analysis.
 | ||
| Fig. 4 Comparison of the SESI ion signal responses acquired using the Ion Max source and SAP-ESI source. The accurate time of sample injection was difficult to determine and therefore not marked on the chromatogram. | ||
To evaluate the quantitative ability of the SAP-ESI source in SESI analysis, a series of aqueous solutions of the moderately volatile N,N-dimethylaniline (DMA) with concentrations ranging from 100 ppm to 1.7 ppb were prepared and directly analyzed. According to the Henry's law,57 the actual content of the evaporated DMA in the gaseous state was estimated to be about 0.47 times the solution concentration (i.e. 0.47 ppbv in gaseous phase corresponds to 1 ppb in solution, the calculation is shown in the ESI†). The centrifuge tubes containing these solutions were successively placed under the sample inlet for a few tens of seconds to make the solution volatile to be sampled and detected. Then the corresponding SESI-MS signals could be rapidly obtained, as shown in Fig. 5. By processing this result, a calibration curve was plotted and given in Fig. 5b, indicating a linear response between ion intensity and analyte concentration in a relatively wide range. For detection of 1.7 ppb DMA solution, a signal to noise ratio of about 17 was obtained, then the detection limit was estimated to be less than 1 ppbv for DMA vapor. It is worth mentioning that natural volatilization and diffusion of the analyte from the solution to the open lab environment can result in an uncontrolled enhancement of DMA ion background, which will affect the detection of low-content samples and reduce detection sensitivity. In addition, analysis of high-concentration samples (higher than 3.7 ppm) will cause signal saturation and a certain degree of residual effect, hence it was not suitable for current experimental platform. Nevertheless, the proposed device and sampling strategy still provided a simple method, despite some flaws, for rapid quantitative analysis of volatiles in liquids.
 | ||
| Fig. 5 (a) EIC chromatogram and (b) calibration curve acquired from successive SESI-MS detections of N,N-dimethylaniline vapors above different solutions. | ||
With the analytical protocol described above, the SAP-ESI source can provide a convenient way for rapid and sensitive detection of gaseous compounds or solution volatiles. Since the electrospray cloud has indeed been well served as a charge source to ionize different forms of analytes through charge transfer reactions, the application of the developed source could be further extended. Efforts have been invested in performing extractive analysis of liquid using the SAP-ESI source by directly drawing solution through the sample inlet. It was found that continuous injection of liquid would block the gas path, leading to a sharp decrease in the fore pressure of the MS instrument and causing system error notification. Then a pulsed introduction procedure was applied to avoid such error in direct liquid analysis. A mixed aqueous solution containing 100 ppb 2,4-lutidine (v/v), 100 ppb DMA (v/v), 10 ppb arginine (w/w) and 10 ppb DPZ (w/w) was prepared and analyzed following a two-step procedure. The first step was to sample 3 μL solution with a 10 μL pipette, followed by a sample transfer step that the tip was removed from the pipette and quickly connected to the inlet tubing of the source. Then, the micro-volume samples can be rapidly sucked into the source without causing pressure fluctuations. The total consumption of each compound in a single analysis was at sub-nanogram level, and it was sufficient to produce ion signals with a certain time width, as shown in Fig. 6.
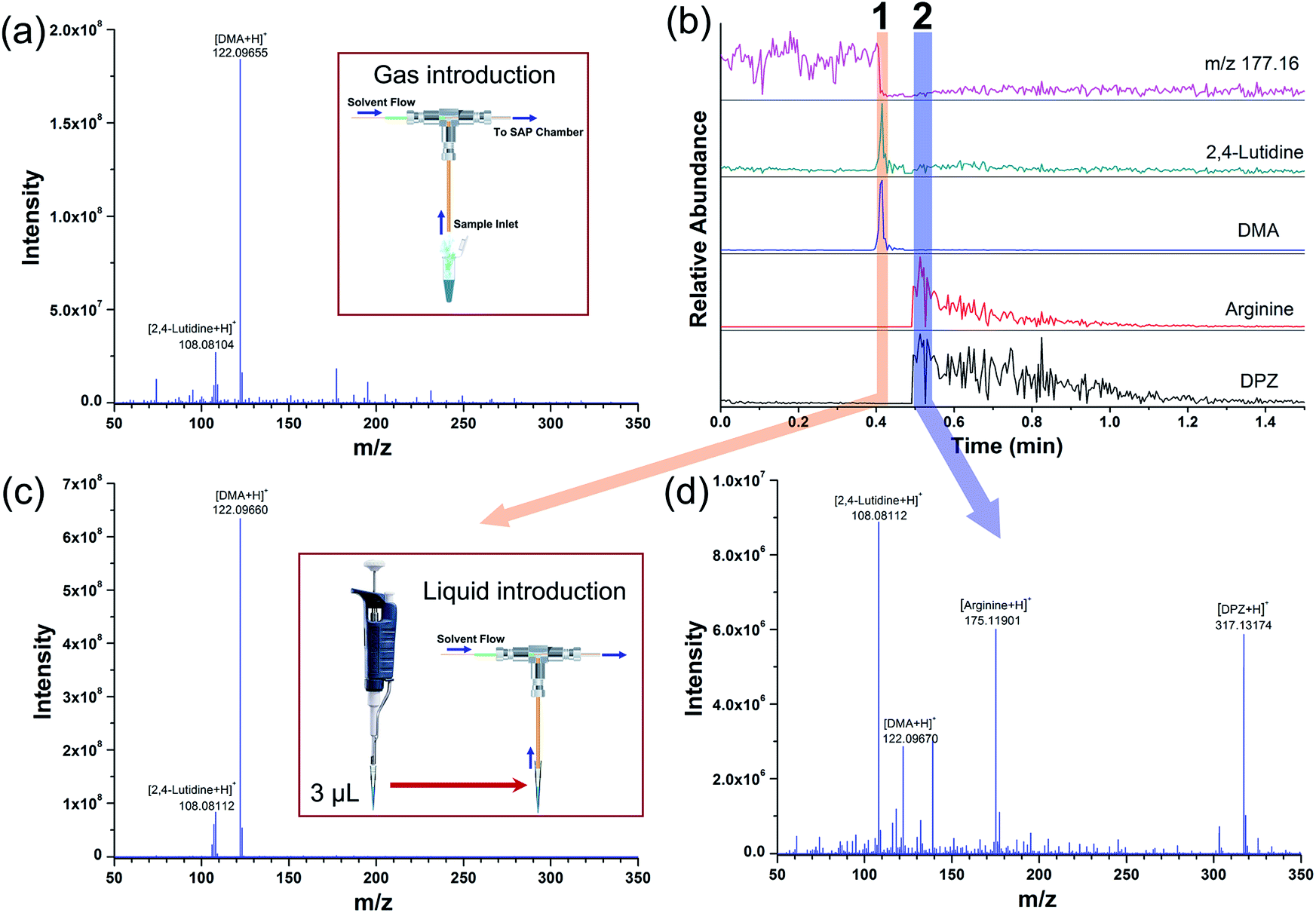 | ||
| Fig. 6 (a) A typical SAP-ESI mass spectrum of the mixed aqueous solution acquired using gas introduction; (b) EICs for different compounds and a background ion (m/z 177.16) acquired after directly injecting 3 μL mixed solution into the SAP-ESI source; average mass spectra recorded in (c) interval 1 and (d) interval 2. | ||
In this experiment, the SAP-ESI device behaved similar to a coaxial ESI source51 but with a pulsed operating mode. Protonated ions of all analytes, both volatile and non-volatile, can be ionized and appear in the mass spectra. The EICs for the standard solutes and a background ion (m/z 177.16) were also obtained and shown in Fig. 6b, which depicted the time profiles of different ion components during a typical detection process. The background ion signal descended rapidly after sample injection and then remained substantially stable, while the analyte signals exhibited synchronous responses with a time width of a few seconds. However, the ion signals of volatile and non-volatile compounds showed different response characteristics that two distinct signal intervals can be observed. The volatile compounds (2,4-lutidine and DMA) mainly appeared in the mass spectrum of the former interval (as seen in Fig. 6c). The acquired ion signals were higher than that in Fig. 6a, indicating a better sample utilization in the pulse liquid introduction analysis. Such operation can achieve high injection rate for a short period to maximize sampling efficiency. After a few seconds, the ion peaks of the non-volatile components (arginine and DPZ) could be observed in the second interval along with some residual ion signals of the volatile analytes. Based on this approach, MS detection of peptide samples can also be realized (see Fig. S2†). The generation of two signal intervals in Fig. 6 may due to the volatilization of the analytes during the transport and the higher transmission rate of gas in the tube than that of liquid. Even so, the ability to easily perform microanalysis on both gaseous and liquid samples potentializes the SAP-ESI technique to be a common and versatile ionization method, considering that all these analyses can be implemented on the same device.
4. Conclusions
The renowned ESI technology and its many derivatives, including DESI, EESI and SESI, have brought us too many surprises through their magical and superb capability in charging various compounds. Until now, its inexhaustible potential along with related applications are still being excavated. For SESI process, the charge transfer reactions between the electrospray cloud and its surrounding substances makes ionization of some gas-phase analytes exceptionally easy to achieve. A simplified SAP-ESI device is then designed to further facilitate the SESI operation and has been successfully employed to detect gas or volatile samples. In particular, it also allows direct analysis of both volatile and non-volatile compounds in liquid by combining pulse injection method. MS instrument coupled with this SAP-ESI source presents some impressive merits, such as convenient operation, fast response, high sensitivity, and minimal sample consumption. Although surrounding condition is a crucial factor for electrospray process, it is usually ignored in traditional applications. The small sealed chamber in the SAP-ESI device can provide a variable pressure environment for ESI and SESI, both of which are found to function well in low-pressure atmosphere with the background interferences being evidently suppressed. The SAP-ESI source has a compact and smooth gas pathway that enables the analytes to be efficiently ionized and introduced into the mass spectrometer. Besides, its ability to directly aspirate samples for MS analysis, like a human nose but with more powerful identification capability, can simplify the operation and minimize sample waste.Some analytical protocols demonstrated in this study can be further extended. For instance, creating a low-pressure ionization condition to reduce interference should be effective for other conventional API techniques. Furthermore, using the native aspiration capability of a mass spectrometer to implement efficient sample acquisition and transmission will simplify both the assembly and operation of the MS instrument.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is was financially supported by National Natural Science Foundation of China (Grant No. 21775085 and 21605076) and the Fundamental Research Program of Shenzhen (Grant No. JCYJ20180508152013054 and JSGG20180508153006719).Notes and references
- M. E. Monge, G. A. Harris, P. Dwivedi and F. M. Fernández, Chem. Rev., 2013, 113, 2269–2308 CrossRef CAS PubMed.
- C. Black, O. P. Chevallier and C. T. Elliott, Trends Anal. Chem., 2016, 82, 268–278 CrossRef CAS.
- A. T. Lebedev, Annu. Rev. Anal. Chem., 2013, 6, 163–189 CrossRef CAS.
- W. P. Zhang, X. Wang, Y. Xia and Z. Ouyang, Theranostics, 2017, 7, 2968–2981 CrossRef CAS.
- R. G. Cooks, Z. Ouyang, Z. Takats and J. M. Wiseman, Science, 2006, 311, 1566–1570 CrossRef CAS PubMed.
- A. Venter, M. Nefliu and R. Graham Cooks, Trends Anal. Chem., 2008, 27, 284–290 CrossRef CAS.
- M.-Z. Huang, C.-H. Yuan, S.-C. Cheng, Y.-T. Cho and J. Shiea, Annu. Rev. Anal. Chem., 2010, 3, 43–65 CrossRef CAS PubMed.
- G. A. Harris, A. S. Galhena and F. M. Fernandez, Anal. Chem., 2011, 83, 4508–4538 CrossRef CAS PubMed.
- P. Nemes and A. Vertes, Trends Anal. Chem., 2012, 34, 22–34 CrossRef CAS.
- Z. Takats, J. M. Wiseman, B. Gologan and R. G. Cooks, Science, 2004, 306, 471–473 CrossRef CAS PubMed.
- R. B. Cody, J. A. Laramée and H. D. Durst, Anal. Chem., 2005, 77, 2297–2302 CrossRef CAS PubMed.
- Y. H. Chen, H. H. Hill and D. P. Wittmer, J. Microcolumn Sep., 1994, 6, 515–524 CrossRef CAS.
- C. Wu, W. F. Siems and H. H. Hill, Anal. Chem., 2000, 72, 396–403 CrossRef CAS PubMed.
- P. Martínez-Lozano, J. Rus, G. Fernández de la Mora, M. Hernández and J. Fernández de la Mora, J. Am. Soc. Mass Spectrom., 2009, 20, 287–294 CrossRef PubMed.
- J.-C. Wolf, M. Schaer, P. Siegenthaler and R. Zenobi, Anal. Chem., 2015, 87, 723–729 CrossRef CAS PubMed.
- H. Chen, A. Venter and R. G. Cooks, Chem. Commun., 2006, 2042–2044 RSC.
- J. Liu, H. Wang, N. E. Manicke, J.-M. Lin, R. G. Cooks and Z. Ouyang, Anal. Chem., 2010, 82, 2463–2471 CrossRef CAS PubMed.
- H. Wang, J. Liu, R. G. Cooks and Z. Ouyang, Angew. Chem., 2010, 122, 889–892 CrossRef.
- N. Na, C. Zhang, M. Zhao, S. Zhang, C. Yang, X. Fang and X. Zhang, J. Mass Spectrom., 2007, 42, 1079–1085 CrossRef CAS PubMed.
- Z.-P. Yao, Mass Spectrom. Rev., 2011, 31, 437–447 CrossRef PubMed.
- J. Zhu, H. D. Bean, Y.-M. Kuo and J. E. Hill, J. Clin. Microbiol., 2010, 48, 4426–4431 CrossRef CAS PubMed.
- D. Garcia-Gomez, T. Gaisl, L. Bregy, P. Martinez-Lozano Sinues, M. Kohler and R. Zenobi, Chem. Commun., 2016, 52, 8526–8528 RSC.
- A. Tejero Rioseras, K. D. Singh, N. Nowak, M. T. Gaugg, T. Bruderer, R. Zenobi and P. M. L. Sinues, Anal. Chem., 2018, 90, 6453–6460 CrossRef CAS PubMed.
- C. Barrios-Collado, D. García-Gómez, R. Zenobi, G. Vidal-de-Miguel, A. J. Ibáñez and P. Martinez-Lozano Sinues, Anal. Chem., 2016, 88, 2406–2412 CrossRef CAS PubMed.
- R. R. Farrell, J. Fahrentrapp, D. García-Gómez, P. Martinez-Lozano Sinues and R. Zenobi, Food Control, 2017, 81, 107–112 CrossRef CAS.
- J. He, F. Tang, Z. Luo, Y. Chen, J. Xu, R. Zhang, X. Wang and Z. Abliz, Rapid Commun. Mass Spectrom., 2011, 25, 843–850 CrossRef CAS PubMed.
- C.-H. Chen, Z. Lin, R. Tian, R. Shi, R. G. Cooks and Z. Ouyang, Anal. Chem., 2015, 87, 8867–8873 CrossRef CAS PubMed.
- W. S. Law, R. Wang, B. Hu, C. Berchtold, L. Meier, H. Chen and R. Zenobi, Anal. Chem., 2010, 82, 4494–4500 CrossRef CAS PubMed.
- X. Li, B. Hu, J. Ding and H. Chen, Nat. Protoc., 2011, 6, 1010–1025 CrossRef CAS PubMed.
- A. K. Badu-Tawiah, L. S. Eberlin, Z. Ouyang and R. G. Cooks, Annu. Rev. Phys. Chem., 2013, 64, 481–505 CrossRef CAS PubMed.
- C. Barrios-Collado, G. Vidal-de-Miguel and P. Martinez-Lozano Sinues, Sens. Actuators, B, 2016, 223, 217–225 CrossRef CAS.
- L. A. Dillon, V. N. Stone, L. A. Croasdell, P. R. Fielden, N. J. Goddard and C. L. Paul Thomas, Analyst, 2010, 135, 306–314 RSC.
- Q. Yu, J. Zhang, K. Ni, X. Qian and X. Wang, J. Mass Spectrom., 2017, 52, 109–115 CrossRef CAS PubMed.
- N. V. Schwab, A. M. Porcari, M. B. Coelho, E. M. Schmidt, J. L. Jara, J. V. Visentainer and M. N. Eberlin, Analyst, 2012, 137, 2537–2540 RSC.
- C. L. Yu, X. Qian, Y. Chen, Q. Yu, K. Ni and X. H. Wang, RSC Adv., 2016, 6, 50180–50189 RSC.
- G. Vidal-de-Miguel, M. Macía, P. Pinacho and J. Blanco, Anal. Chem., 2012, 84, 8475–8479 CrossRef CAS PubMed.
- R. G. Ewing and B. R. Valenzuela, Anal. Chem., 2018, 90, 7583–7590 CrossRef CAS PubMed.
- N. Omenetto, G. A. Petrucci, P. Cavalli and J. D. Winefordner, Fresenius. J. Anal. Chem., 1996, 355, 878–882 CAS.
- Z. Liang, S. Zhang, X. Li, T. Wang, Y. Huang, W. Hang, Z. Yang, J. Li and Z. Tian, Sci. Adv., 2017, 3(12), eaaq1059 CrossRef PubMed.
- Z. Wei, X. Xiong, C. Guo, X. Si, Y. Zhao, M. He, C. Yang, W. Xu, F. Tang, X. Fang, S. Zhang and X. Zhang, Anal. Chem., 2015, 87, 11242–11248 CrossRef CAS PubMed.
- Z. Yin, X. Cheng, R. Liu, X. Li, L. Hang, W. Hang, J. Xu, X. Yan, J. Li and Z. Tian, Angew. Chem., Int. Ed., 2019, 58, 4541–4546 CrossRef CAS PubMed.
- K. G. Asano, M. J. Ford, B. A. Tomkins and G. J. Van Berkel, Rapid Commun. Mass Spectrom., 2005, 19, 2305–2312 CrossRef CAS PubMed.
- P. Martínez-Lozano and J. F. de la Mora, J. Am. Soc. Mass Spectrom., 2009, 20, 1060–1063 CrossRef PubMed.
- M.-M. Chen, H.-F. Su, Y. Xie, L.-F. He, S.-C. Lin, M.-L. Zhang, C. Wang, S.-Y. Xie, R.-B. Huang and L.-S. Zheng, Sci. Bull., 2018, 30, 1351–1357 CrossRef.
- Q. Yu, Q. Zhang, X. Lu, X. Qian, K. Ni and X. Wang, Anal. Chem., 2017, 89, 12938–12944 CrossRef CAS PubMed.
- X. Lu, Q. Yu, Q. Zhang, K. Ni, X. Qian, F. Tang and X. Wang, J. Am. Soc. Mass Spectrom., 2017, 28, 1702–1708 CrossRef CAS PubMed.
- W. Shi, X. Lu, J. Zhang, Q. Yu and X. Wang, Rapid Commun. Mass Spectrom., 2018, 32, 2159–2165 CrossRef CAS PubMed.
- D. García-Gómez, P. Martínez-Lozano Sinues, C. Barrios-Collado, G. Vidal-de-Miguel, M. Gaugg and R. Zenobi, Anal. Chem., 2015, 87, 3087–3093 CrossRef PubMed.
- C. Tschiggerl and F. Bucar, Sci. Pharm., 2010, 78, 483–492 CrossRef CAS PubMed.
- P. Martinez-Lozano Sinues, E. Criado and G. Vidal, Int. J. Mass Spectrom., 2012, 313, 21–29 CrossRef CAS.
- B. N. Sundberg and A. F. Lagalante, J. Am. Soc. Mass Spectrom., 2018, 29, 2023–2029 CrossRef CAS PubMed.
- K. D. Swanson, A. L. Worth and G. L. Glish, Anal. Methods, 2017, 9, 4997–5002 RSC.
- I. Marginean, J. S. Page, A. V. Tolmachev, K. Tang and R. D. Smith, Anal. Chem., 2010, 82, 9344–9349 CrossRef CAS PubMed.
- J. S. Page, K. Tang, R. T. Kelly and R. D. Smith, Anal. Chem., 2008, 80, 1800–1805 CrossRef CAS PubMed.
- K. Tang, J. S. Page, I. Marginean, R. T. Kelly and R. D. Smith, J. Am. Soc. Mass Spectrom., 2011, 22, 1318–1325 CrossRef CAS PubMed.
- J. T. Cox, I. Marginean, R. T. Kelly, R. D. Smith and K. Tang, J. Am. Soc. Mass Spectrom., 2014, 25, 2028–2037 CrossRef CAS PubMed.
- R. Sander, Atmos. Chem. Phys., 2015, 15, 4399–4981 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1: using Ion Max source to perform SESI-MS analysis; calculation of DMA vapour concentration based on Henry's law. See DOI: 10.1039/c9ra09104a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |