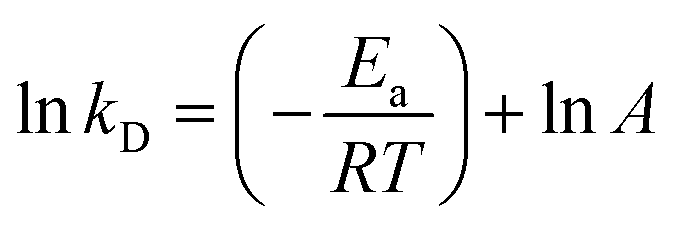DOI:
10.1039/C9RA09067C
(Paper)
RSC Adv., 2020,
10, 2624-2633
Stable immobilization of aldehyde ketone reductase mutants containing nonstandard amino acids on an epoxy resin via strain-promoted alkyne–azide cycloaddition†
Received
3rd November 2019
, Accepted 9th January 2020
First published on 14th January 2020
Abstract
To avoid random chemical linkage and achieve precisely directed immobilization, mutant enzymes were obtained and immobilized using an incorporated reactive nonstandard amino acid (NSAA). For this purpose, aldehyde ketone reductase (AKR) was used as a model enzyme, and 110Y, 114Y, 143Y, 162Q and 189Q were each replaced with p-azido-L-phenylalanine (pAzF). Then, the mutant AKR was coupled to the functionalized support by strain-promoted alkyne–azide cycloaddition (SPAAC). The effects of the incorporation number and site of NSAAs on the loading and thermal stability of the immobilized AKR were examined. The results show that the mutant enzymes presented better specific activity than the wild type, except for AKR-110Y, and AKR-114Y showed 1.16-fold higher activity than the wild type. Moreover, the half-life (t1/2) of the five-point immobilized AKR reached 106 h and 45 h, 13 and 7 times higher than that of the free enzyme at 30 °C and 60 °C, respectively. Comparison of these three types of enzymes shows that multi-point immobilization provides improved loading and thermal stability and facilitates one-step purification. We expect this platform to facilitate a fundamental understanding of precisely oriented and controllable covalent immobilization and enable bio-manufacturing paradigms for fine chemicals and pharmaceuticals.
1. Introduction
In covalent immobilization, random and unwanted chemical linkages are difficult to avoid when crosslinkers are used,1 which could result in enzyme conformation changes and decrease of enzyme activity due to the destroyed and buried active sites.2 In addition, random chemical linking reactions often lead to promiscuous multi-point covalent immobilization. Panova et al.3 have verified that the temperature dependence of the rate constant of irreversible thermal inactivation, kin, of immobilized α-chymotrypsin, is markedly dependent on the number of covalent bonds between the enzyme and support. Nevertheless, the number of covalent bonds between the enzyme and support was estimated from the results of spectrophotometric titration of non-reacted amino groups of the enzyme after the modification step,4 indicating that this number would differ from that of the modification reaction. In addition, it was not possible to control the site where covalent linkage occurs on the enzyme surface. Although multi-point covalent immobilization can enhance the rigidity of the enzyme, consequently improving the thermal stability of the immobilized enzyme, this promiscuous multi-point immobilization also reduced enzyme activity5,6 due to the decrease in the motion of the enzyme.7,8 Thus, uncertainties still exist9,10 as to how to precisely control the exact number of covalent bonds between enzyme and support and sites where the linkages occurs on the enzyme surface with regard to multi-point covalent immobilization. Site-directed mutagenesis seems to be an elegant and versatile way to control the direction of a protein. It can immobilize a protein on a support in different directions and identify areas where it interacts with the support.11 However, the immobilization sites still can't be selected and random covalent linkage would result in burying or destroying the active site of the enzyme.
A linkage base on the tag fused to the enzyme protein12 could efficiently prevent random chemical bonding reactions. Using this tag, enzyme can be covalently and site-specifically attached on the support surface with enhanced activity and thermal stability. Unfortunately, fusion of the tag to the enzyme occurs only at the C-terminus and N-terminus.13–15 However, we were inspired by the nonstandard amino acids (NSAAs) incorporation16–19 for site-specific covalent immobilization. Modification by the incorporation of reactive NSAAs can occur to a specific site to well control enzyme orientation, which resulted from genetic code expansion using an orthogonal aminoacyl-tRNA synthetase (aaRS)/tRNA pair to direct the incorporation of a NSAA into proteins in response to an unassigned codon (commonly the amber stop codon, UAG) introduced at the desired site in a gene of interest.20,21 Because of the pre-designed distal locations from the active site, the enzyme remained active after immobilization. Additionally, multi-point immobilization may be achieved by multi-site mutation.
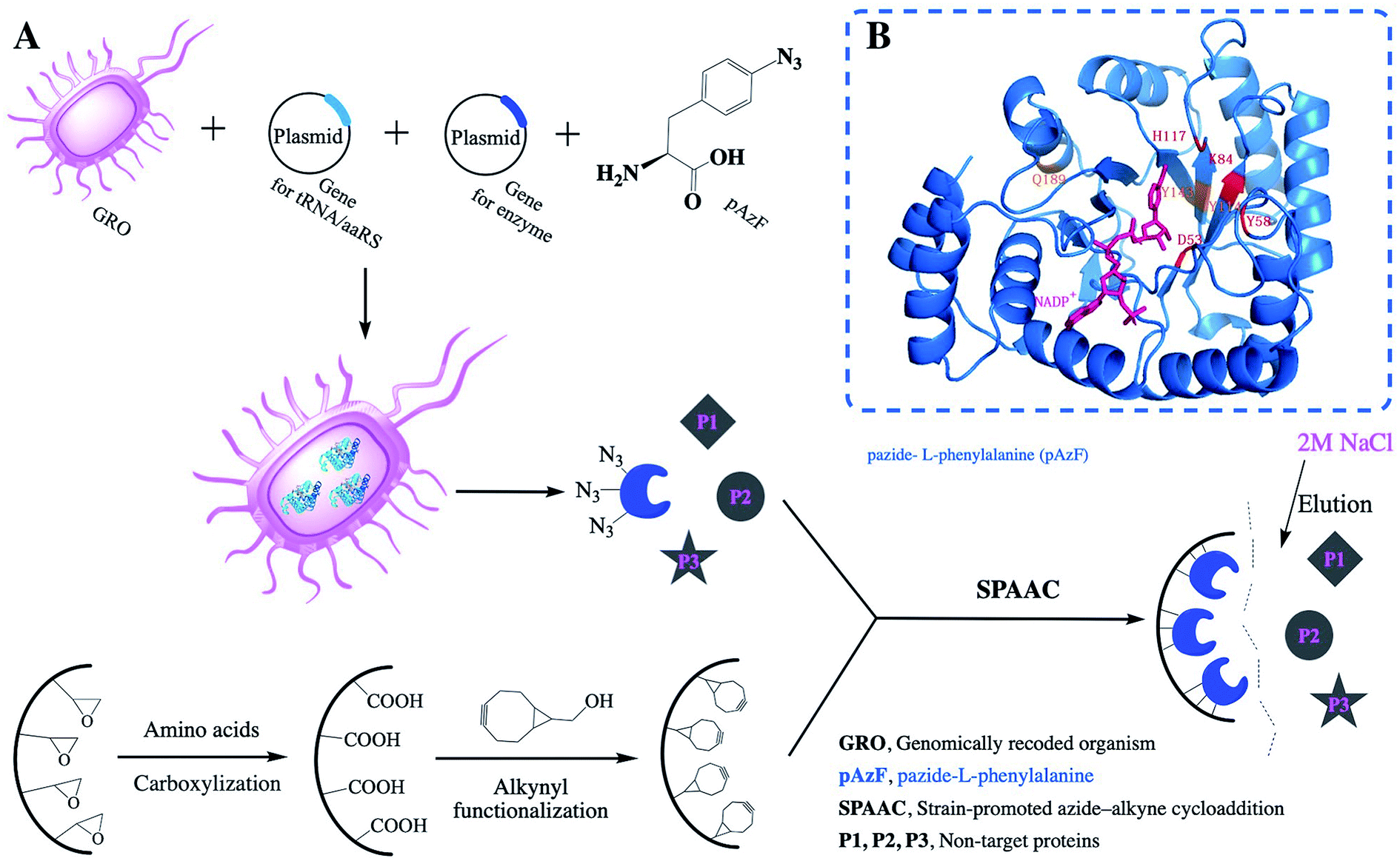
MG1655, a genomically recoded organism (GRO)22 with deletion of release factor 1 and reassignment of the UAG translation function, exhibited improved properties and efficiency for incorporation of NSAAs that expand the chemical diversity of proteins in vivo.23 Even though containing 30 NSAA residues for diverse chemistries, Elastin-like polypeptides could be produced at high yields (∼50 mg L−1) and with high accuracy of incorporation (>95%). NAD(P)H-dependent oxidoreductases are of great interest from an industrial point of view for its critical role in the production of many hard-to-synthesize compounds. This enzyme possess other properties which make them very attractive alternatives to organic chemical synthesis, such as stereospecificity, regiospecificity and the possibility to tailor them to have the appropriate catalytic property and the desired substrate specificity.24 In the present work, we aimed to test the feasibility and reliability of precise multi-point covalent immobilization of aldehyde ketone reductase (AKR) depending on the NSAAs (Fig. 1A). We expect that our proposed approach will successfully address the problems associated with traditional multi-point immobilization. The most stable enzyme derivatives are often produced through multipoint covalent attachment.25
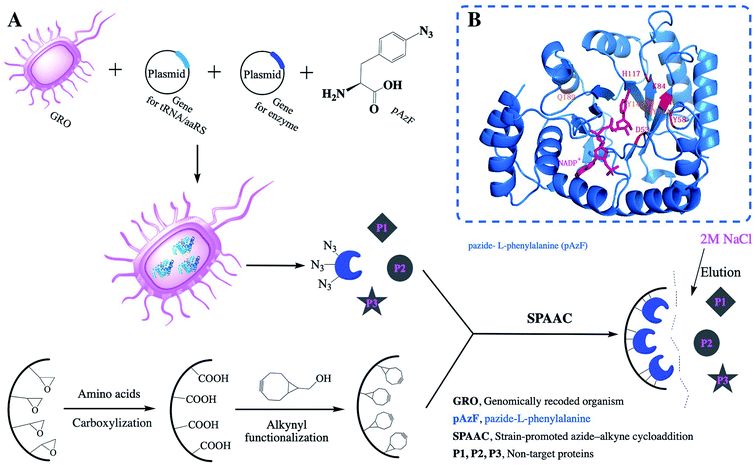 |
| | Fig. 1 (A) Scheme of the precise immobilization and one-step purification of AKR depending on incorporation of pAzF and the relative activities of the immobilized enzyme; (B) three-point AKR mutant. | |
2. Materials and methods
2.1. Materials
The host strains E. coli DH5 and E. coli BL21(DE3) were purchased from Stratagene and Novagen. All standard recombinant DNA products were synthesized or purchased from Shanghai Generay Biotech Co., Ltd. Antibiotics were purchased from Sangon Biotech. p-Azide-L-phenylalanine was purchased from Artis Biotech Co. Ltd. Anhydrotetracycline hydrochloride (aTc) and dihydro-4,4-dimethyl-2,3-furandione were obtained from Sigma-Aldrich. NADPH and NADP+ were purchased from J&K Scientific Ltd. Bicyclo[6.1.0]nonyne (BCN) was purchased from TCI (Japan). The epoxy resin support was obtained from Sunresin New Materials Co. Ltd. Which model is LX-1000EP The protein MARKER is derived from Beijing Solarbio Science & Technology Co., Ltd. All other biological reagents not mentioned above were purchased from Sangon Biotech, while all other chemical reagents were purchased from Sinopharm Chemical Reagent Ltd.
2.2. Site-directed mutagenesis, plasmid construction and bacterial strains
The plasmid pEVOL-pAzF,26 encoding a p-azide-L-phenylalanine (pAzF)-specific engineered tyrosyl-tRNA synthetase/amber suppressor tRNA pair derived from Methanococcus jannaschii (Plasmid ID:31186), was obtained from Addgene (Cambridge, MA) and used without further modification. To construct a bacterial expression vector encoding a C-terminally His×6-tagged recombinant aldehyde ketone reductase (AKR; NCBI gene ID: 897867) originating from Thermotoga maritima MSB8 (strain: MSB8), the coding sequence was amplified from pZE21-GFPaav (plasmid ID: 26643), which was obtained from Addgene. We replaced the GFP gene by digestion with the restriction endonucleases Kpn I and Hind III and cloned the akr gene into pZE21 to obtain pZE21-akr.
By submitting the amino acid sequences of the AKR enzyme to SWISS-MODEL Workspace, we obtained a relatively accurate 3D structure model. The template for AKR modelling was the crystal structure of the novel aldehyde ketone reductase Tm1743 from T. maritima in complex with NADP+ (PDB ID: 5dan.1; resolution, 2.0 Å) with the highest sequence similarity (100%) to AKR. We selected several sites, including 110 (Y), 114 (Y), 143 (Y), 162 (Q) and 189 (Q) (N-terminal), of AKR-pZE21, and site-directed mutagenic PCR was performed with pZE21-akr as a template to replace codons encoding residues 110, 114, 143, 162, and 189 (N-terminal) with the amber codon (TAG) to obtain pZE21-akr-114Y (110Y, 143Y, 162Q, 189Q). Multi-point mutation of the target enzyme gene was carried out on the basis of the successive one-point work. The primers required for construction of AKR-pZE21 and for AKR sites mutations were listed in Table S1 and Primers Table S2,† respectively.
BL21 cells were transformed with pZE21-akr for expression, affording pZE21-akr-BL21 (AKR-WT). As an expression host for pAzF-incorporated akr-114Y (110Y, 143Y, 162Q, 189Q)-pZE21, genomically engineered E. coli C321.ΔA (MG1655), obtained from Addgene (plasmid ID: 48998),27 was used to co-transform pEVOL-pAzF and akr-114Y (110Y, 143Y, 162Q, 189Q)-pZE21 to obtain AKR-114Y (110Y, 143Y, 162Q, 189Q)-pZE21-MG1655.
2.3. Site-specific incorporation of pAzF into AKR-114Y-pZE21-MG1655 and purification of the recombinant enzyme
AKR-114Y-pZE21-MG1655 cells were inoculated into LB medium containing 50 g mL−1 ampicillin, 34 g mL−1 chloramphenicol and 100 g mL−1 kanamycin and were cultured at 34 °C in a shaking incubator (220 rpm). When the OD600 of 0.5 was reached, L-(+)-arabinose was added at a final concentration of 0.2% (w/v). At an OD600 of 0.6, protein expression was induced by adding 30 ng mL−1 aTc (anhydrotetracycline hydrochloride) and pAzF at a final concentration of 1 mmol mL−1. AKR-pZE21-BL21 cells were inoculated into LB medium containing 100 μg mL−1 kanamycin and cultured at 37 °C in a shaking incubator (220 rpm). When the OD600 was 0.6, protein expression was induced by the addition of 30 ng mL−1 aTc. All protein expression was conducted by continuous shaking at 23 °C for 16 h.
Following 16 h of expression, the obtained cells were harvested and pelleted by centrifugation at 8000 rpm for 5 min, resuspended in Tris–HCl buffer (20 mmol L−1, pH 7.4) and lysed by sonication. Soluble and insoluble fractions were also separated by centrifugation. The soluble fractions were incubated with 20 mL of Ni-NTA agarose (Ni Sepharose™ 6 Fast Flow) for 1 hour at 4 °C within a chromatographic column. The chromatographic column was first washed with washing buffer containing Tris–HCl buffer (20 mmol L−1, pH 7.4), 300 mmol L−1 NaCl and 20 mmol L−1 imidazole. Then, the bound protein was eluted by the same buffer with 250 mmol L−1 imidazole. The obtained enzyme was further desalted and concentrated by using an ultrafiltration tube to obtain a pure enzyme solution in phosphate buffer (20 mmol L−1, pH 7.0). The relative molecular mass and expression of the protein were detected by SDS–PAGE followed by staining with Coomassie brilliant blue R250. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) was performed on a Microflex MALDI-TOF mass spectrometer. The instrument was operated in positive ion reflector mode with an accelerating potential of +20 kV. The TOF mass was analyzed using pulsed ion extraction and the spectra were acquired by averaging at least 600 laser shots. Methanol was used as a matrix, and a few protein samples were dissolved in 1 μL of methanol solution. The mixture was then applied onto a clean target plate and air-dried at room temperature. The acquired data were analyzed using Microflex.
2.4. Functionalization and characterization of porous resin support
2.4.1. Preparation of amino-activated epoxy resin support. Epoxy resin polymer (120 mol epoxy resin, 1 g) was added to the aqueous reaction solution (3 mL). Then, 2 eq. of lysine (240 mol, 15 mg) was added (glycine and lysine react in parallel), and the pH of the solution was adjusted to 10 by adding 1 mol L−1 NaOH aqueous. The reaction was carried out at room temperature overnight. Then, the reaction was removed and centrifuged, the supernatant was drained with a syringe, and the reaction was washed 3 times with water and dried in a vacuum oven at 50 °C.
2.4.2. Functionalization of the resin support using BCN. BCN (100 mol), amino-activated epoxy resin support intermediate (100 mol), DCC (N,N′-dicyclohexylcarbodiimide) (120 mol) and DMAP (4-dimethylaminopyridine) (50 mol) were added to 2 mL of DCM (dichloromethane) and reacted overnight at room temperature in the dark. After the reaction, the resin support was washed 4 times with an aqueous solution to remove excess DCC and DMAP. Then, the BCN-functionalized resin was dried in a vacuum drying oven at 50 °C. After drying, the infrared test was performed.
2.4.3. Characterization of the functionalized resin support. 0.5–1 mg of unmodified epoxy resin, lysine, BCN and BCN-modified epoxy resin were mixed with KBr in an agate mortar and ground for 1 min separately to obtain the test sample for tableting, respectively. The FT-IR spectra of the samples were recorded using a Thermo Nicolet iS5 FT-IR spectrophotometer from 4000 to 500 cm−1.
2.5. Precise multi-point immobilization and one-step purification of the AKR mutants
BCN-functionalized resin support (0.5 g) and 3 mL of pZE21-AKR114 crude enzyme supernatants of the cell lysates of the harvested cultured strains were added to 1 mL of phosphate buffer (0.02 M, pH 7.0) and incubated at 20 °C with shaking at 160 rpm for 18 h. Then, the immobilized enzyme was washed twice with 2 M NaCl solution and 0.02 M phosphate buffer (pH 7.0). The azide-targeted protein was coupled to the functionalized support by a promoted azide–alkyne cycloaddition and Cu-free click reaction. The heteroprotein without pAzF could only be adsorbed into the pores of the support wall and was easily eluted with 2 M NaCl solution, which resulted in one-step purification and simultaneous immobilization. The immobilized enzyme was separated by centrifugation and washed with phosphate buffer (0.01 mol L−1, pH 7.0) until no protein was detected in the supernatant by Coomassie brilliant blue R250 staining. The amount of enzyme remaining in the supernatant was examined and measured by a Bradford protein assay kit (Quick Start™, Bio-Rad, USA)28 to detect the loading of the immobilized enzyme. The method of measuring the enzyme concentration is as follows: the protein concentration was determined using a modified Bradford method protein concentration determination kit. That is, the absorbance value was measured at 595 nm and the protein concentration standard curve was used to calculate the protein concentration value. The loading efficiency was investigated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE, 12% w/v acrylamide), and the relative intensity of each SDS–PAGE band was calculated by ImageJ software.29
2.6. Enzymatic activity assay of AKR preparations
AKR activity was determined based on a previous report30 as follows: 100 L of NADPH solution (2.5 mg mL−1) and 100 L of dihydro-4,4-dimethyl-2,3-furandione (5 mg mL−1) were transferred into 0.7 mL of potassium phosphate buffer (KPB; 0.1 mol L−1, pH 7.0). This mixture was first pre-warmed at 30 °C for 3 min with shaking at 150 rpm. Then, AKR was added to the mixture and incubated for 1 min. The whole reaction was monitored by using the decrease in absorbance at 340 nm as a measure of the conversion of the cofactor NADPH to NADP+. One unit of AKR activity is defined as the amount of enzyme required for conversion of 1 μmol of dihydro-4,4-dimethyl-2,3-furandione per minute. The method of measuring enzyme activity is as follows: the reduced coenzyme NADPH has a special absorption peak at 340 nm, and no other substance has a significant absorption peak at 340 nm during the synthesis of the catalytic substrate. Using the UV spectrophotometer, we can judge the consumption and reduction of NADPH by measuring the change in absorbance.
2.7. Thermal stability of enzyme preparations
To evaluate the thermal stability of AKR, the free and immobilized enzymes were transferred to a reaction medium and incubated at 30 °C, 40 °C, 50 °C, and 60 °C for different periods of time. Then, the suspension sample was taken out periodically, and the remaining activity was determined as described above. Thermal stability is reported to be the ratio of the residual activity to the initial activity of immobilized derivatives. The initial activity of the enzyme preparation was set to 100%. For the apparent dynamics of thermal stability, the nonlinear decay model proposed by Sadana and Henley31 was used to calculate the deactivation rate constant (kD, t−1) and the half-life of the enzyme (t1/2, h) (eqn (1) and (2)). The activation energy (Ea, kJ mol−1) for the thermal denaturation of AKR was also determined by a plot of ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kD versus 1/T as previously shown by the linearized Arrhenius equation (eqn (3)) during the immobilization process. The values were calculated from the data fit to a first-order exponential decay equation as follows:
kD versus 1/T as previously shown by the linearized Arrhenius equation (eqn (3)) during the immobilization process. The values were calculated from the data fit to a first-order exponential decay equation as follows:| |
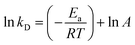 | (3) |
In the activation energy calculation equation, A (t−1) is the Arrhenius frequency of the collision factor, R is the general gas constant (8.314 × 10−3 kJ mol−1 K−1), and T (K) is the absolute temperature.
3. Results and discussion
3.1. Expression and purification of the recombinant AKR containing p-azide-L-phenylalanine (pAzF)
Generally, amino acid mutation sites are selected away from the active center and preferably used for covalent immobilization, in order to protect the protein structure from burying or blocking the active site of the enzyme.19 The corresponding bases for the chosen amino acid residue were mutated to the TAG codon by site-directed mutagenesis. Then, we generated plasmids pZE21-akr-114Y (110Y, 143Y, 162Q, 189Q), pZE21-akr-114Y-189Q, pZE21-akr-114Y-143Y-189Q, and pZE21-akr-110Y-114Y-143Y-162Q-189Q. These plasmids were co-transformed with pEVOL-pAzF into the fully recoded C321.ΔA strain (MG1655)32,33 and inserted azido-phenylalanine to generate different full-length AKR mutants (Fig. 1A), respectively. The simulated structures of the three-point AKR mutant is shown in Fig. 1B and the one-point, two-point, and five-point mutants are shown in the ESI (Fig. S1–S3†). The results in Table S3† show that the highest expression level and activity of the purified mutant protein (AKR-114Y) were 182 mg L−1 and 1.08 U mg−1, similar with those of wild-type AKR (AKR-WT). According to the study, the substrate specificity of the orthogonal MjTyrRS/tRNA is directed to tyrosine among the natural amino acids.34 We found three tyrosine sites away from the active center, namely, AKR110, AKR114, and AKR143, through 3D structural analysis, and thus, tyrosine at these sites was replaced with the desired tyrosine analogue, pAzF. Unfortunately, as the introduction of this NSAA in the non-tyrosine site and increase 25 of the mutation site number, a slight decrease occurs to the expression level and activity of enzyme mutants. The insertion of multiple NSAAs may disturb the native protein structure to some extent, leading to a slight decrease in enzyme activity. Further optimization for mutant sites is necessary and expected to weaken these negative effects.
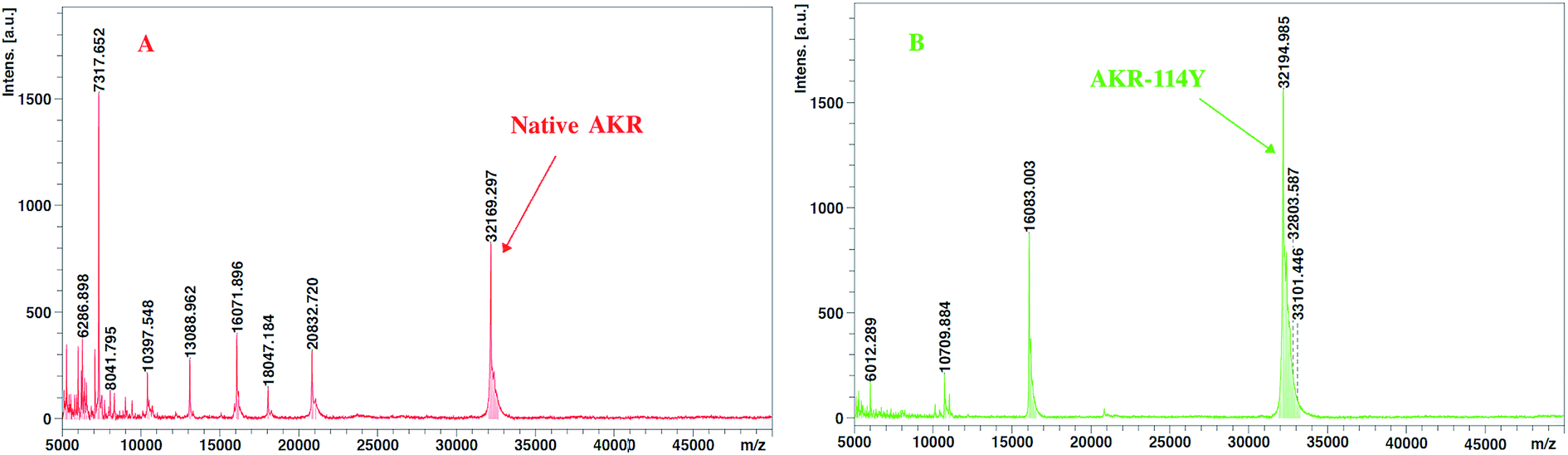
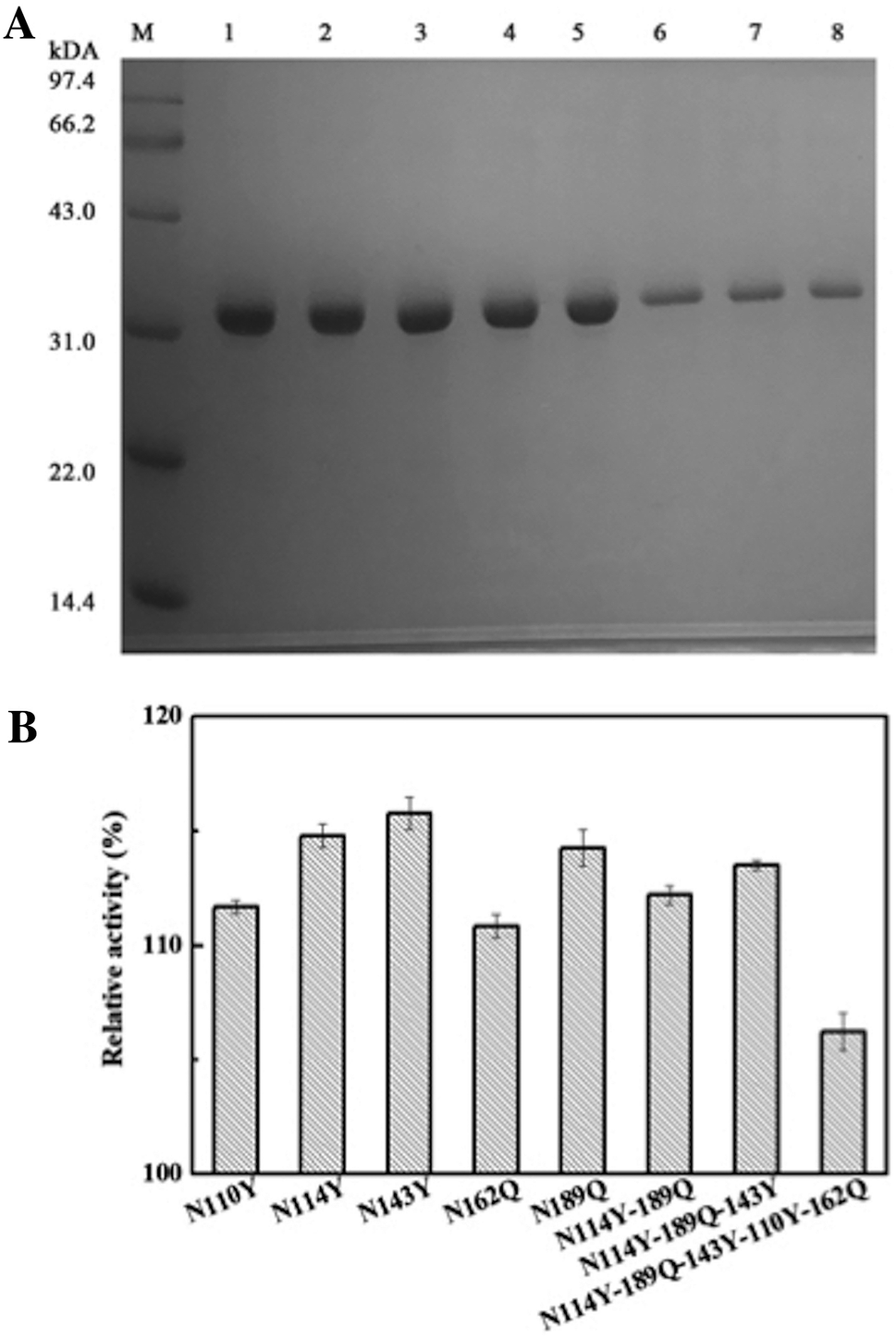
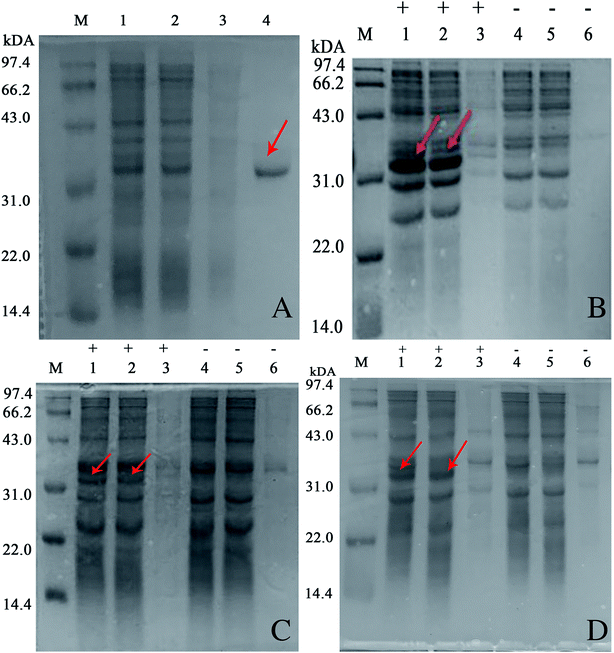
All the obtained purified proteins were analyzed using SDS–PAGE, and the apparent molecular weight of native AKR was approximately 32190 kDa (Fig. 2A). SDS–PAGE analysis and enzyme activity assays also showed the high expression of recombinant proteins and the induction temperature was set based on our previous work.35 The protein bands for one -point AKR mutants, such as AKR-114Y, AKR-110Y, AKR-143Y, AKR-162Q, and AKR-189Q, were all detected at approximately 32 kDa (Fig. 2B). Similarly, bands for multi-point mutant enzymes were also observed at this location (Fig. 2C and D). Control expression experiments to examine the NSAAs incorporation, were carried out without pAzF and no band at approximately 32 kDa was detected. In addition, the results in the MALDI-TOF/MS analysis spectrum (Fig. 3) show that the molecular weight of the AKR-114Y mutant is 25 Da more than that of native AKR. The value was just equal to the molecular weight difference between pAzF and tyrosine, which further confirmed that pAzF molecules were successfully incorporated into the AKR enzyme protein.
 |
| | Fig. 2 (A) SDS–PAGE analysis of wild-type AKR (lane M, protein marker; lane 1, cell lysate; lane 2, cellular soluble fraction; lane 3, cellular insoluble fraction; lane 4, purified AKR); (B–D) SDS–PAGE analysis of AKR containing pAzF (lane M, protein marker; lanes 1 and 4, cell lysate; lanes 2 and 5, cellular soluble fraction; lanes 3 and 6, cellular insoluble fraction), the arrow points to the expressed AKR mutant. | |
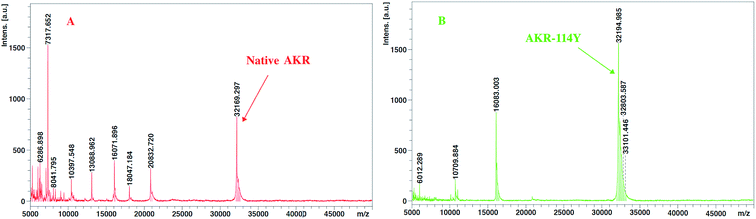 |
| | Fig. 3 Intact protein MALDI-TOF/MS spectrum of the affinity purified native AKR (A) and AKR-N114Ymutant (B). Inset, major components of 32169.297 Da and 32194.985 Da were shown in the (A) and (B), respectively. | |
3.2. Effect of mutation site of AKR on the precise immobilization
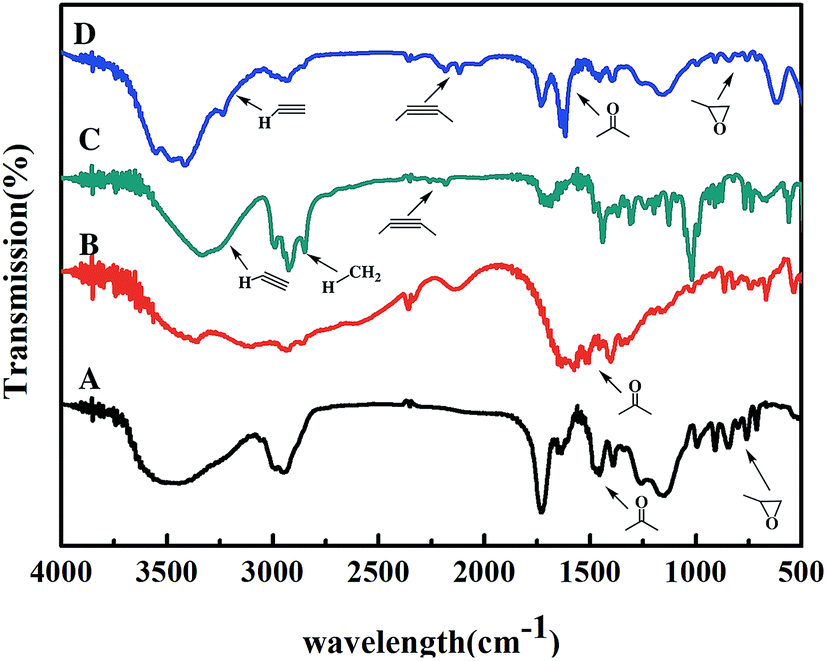
In the covalent immobilization of AKR mutants, supernatant from cells lysate were used to achieve one-step purification because of the biorthogonal strain-promoted alkyne–azide cycloaddition (SPAAC) click reaction (Fig. 1A). We have purchased BCN for infrared testing, such as the (C) curve in Fig. 4. A sharp vibrational peak at ∼2150 cm−1 was observed, which we judge to be the characteristic absorption peak of cyclooctyne. At the same time, in the modified epoxy resin, we were observed at a sharp vibrational peak at ∼2150 cm−1. The amino acid in curve (B) in Fig. 4 is characteristic infrared absorption of lysine. For the support characterization, a sharp vibrational peak at ∼2150 cm−1 was observed (Fig. 4), this peak is ascribable to the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– stretch, indicating the exposure of the cyclooctyne group on porous support after alkynyl-functionalization using BCN.
C– stretch, indicating the exposure of the cyclooctyne group on porous support after alkynyl-functionalization using BCN.
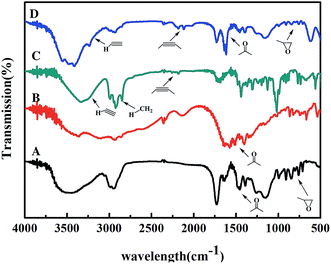 |
| | Fig. 4 FT-IR spectra of support and small molecules ((A) the original epoxy resin; (B) lysine; (C) BCN; (D) the alkynylated epoxy resin). | |
This bioorthogonal SPAAC,36,37 was used as chemical coupling reaction for site-specific immobilization was also biocompatible for use in imaging studies in living organisms.38 No cytotoxic Cu(I) salts were used in this click coupling reaction, which improved the biocompatibility and bioorthogonality of the azide–alkyne cycloaddition. Using structural analysis of AKR for the first time, we chose a mutant site in the other side of active site to stabilize the enzyme structure and activity. After the precise immobilization, the full activity of the immobilized AKR mutants remained close to or even higher than that of the wild type, and the specific activity of immobilized AKR114 relative to free AKR114 is 116% (Fig. 5B). As shown in Fig. 5B, no decrease in activity was observed after the precise covalent immobilization, indicating that the chosen mutation sites were appropriate and efficient, and the structure of enzyme was clearly undisturbed. Furthermore, to test the activity difference between multi-point and one-point immobilization, the enzyme mutants, ranging from one-point to five-point mutants, were covalently attached to support. The results in Fig. 4B show that no marked fluctuation was observed in the relative activities of the immobilized enzyme mutants as the mutation site number increased, which indicated the feasibility of the choice of mutant sites and the efficiency of the precise multi-point covalent immobilization depending on the incorporation of NSAAs.
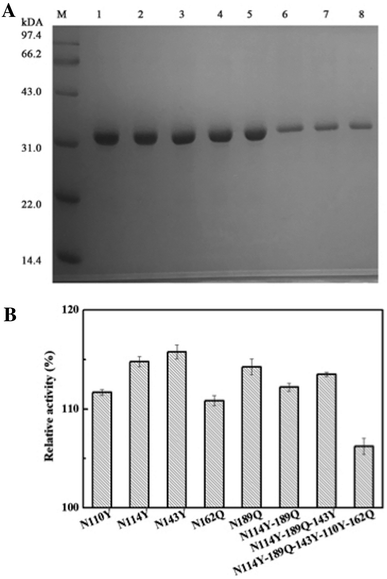 |
| | Fig. 5 (A) SDS–PAGE photograph of the purified AKR mutants (lane M, protein marker; lane 1, purified AKR-110Y; lane 2, purified AKR-114Y; lane 3, purified AKR-143Y; lane 4, purified AKR-162Q, lane 5, purified AKR-189Q, lane 6, purified AKR-N114Y-189Q, lane 7, purified AKR-N114Y-189Q-143Y, lane 8, purified AKR-N114Y-189Q-143Y-110Y-162Q); (B) the relative activities of the immobilized enzyme mutants. | |
3.3. One-step purification and immobilization of AKR using cell lysate
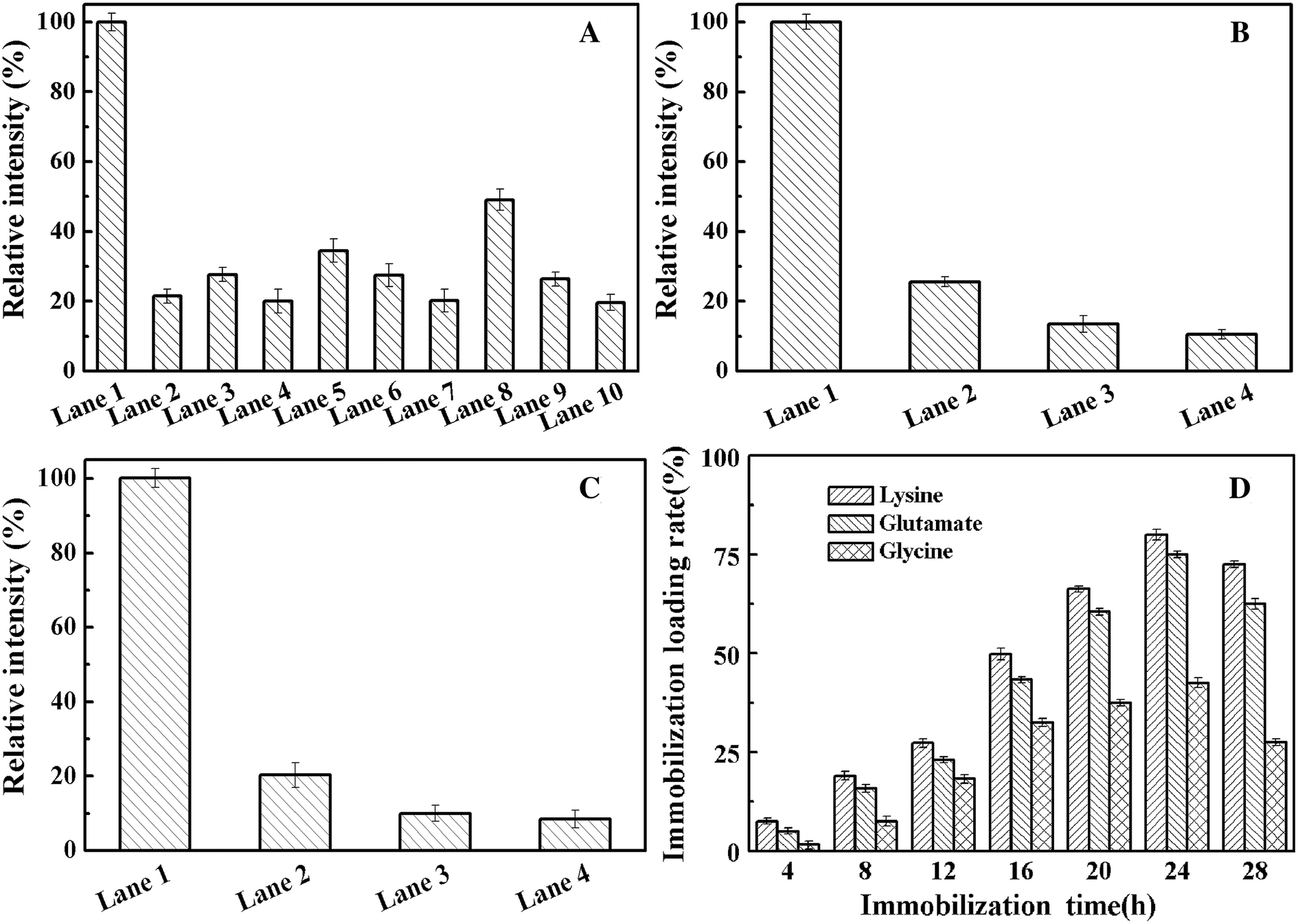
To examine whether the amino acid used to modify the resin support affects enzyme loading during immobilization, SDS–PAGE of the supernatant and elution from the whole process was carried out to analyze the protein content by using ImageJ.39,40 We use the SDS–PAGE gray analysis method to detect the protein concentration. We use a known protein concentration as a standard for a relative emission intensity of 100%. The value ratio is the relative emission intensity of the protein concentration of the other bands. According to the software-based analysis, nearly 30% pure enzyme recovery was achieved by one-point immobilization (Fig. 6A and S4†). At the same time, highly efficient enzyme immobilization was achieved by the lysine-modified resin. Using this support, the multi-point immobilization (Fig. 6B, three-point; Fig. 6C, five-point) of enzyme mutant presents a purification yield of about 50% and 60%, respectively (Fig. 6, S5 and S6†). For the goal of the ideal choice for multi-point coupling of a biocatalyst onto a support surface,41 we precisely controlled the number of chemical bonds between enzyme and support first time. This accurate and reasonable immobilization protects the active site of enzyme from undesired covalent linkage and can be combined with one-step purification by site-specific Cu-free click reaction to effectively simplify the tedious protein purification.
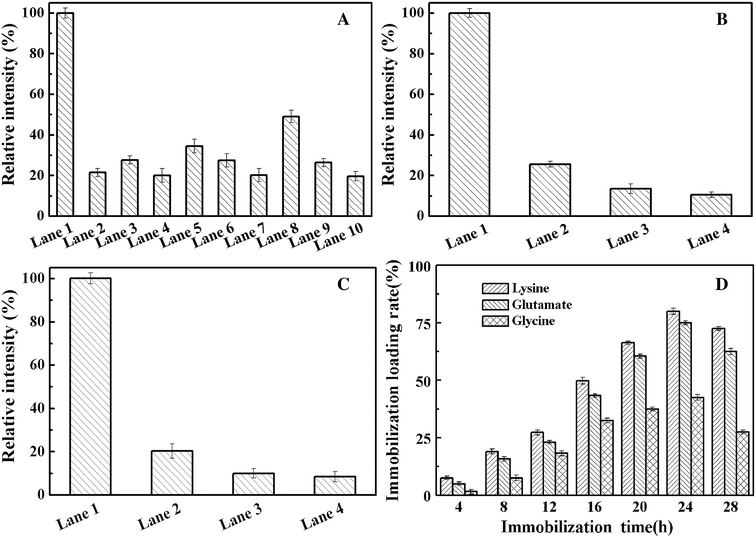 |
| | Fig. 6 The relative emission intensity of each fraction and effect of immobilization time on the immobilization loading rate. The relative emission intensity was calculated using ImageJ software ((A) one-point immobilization; (B) three-point immobilization; (C) five-point immobilization; (A–C) lane 1, pre-immobilization crude enzyme; lane 2, supernatant from immobilization; lane 3, elution of first time from the immobilized enzyme using 2 M NaCl aqueous; lane 4, elution of second time from the immobilized enzyme using 2 M NaCl aqueous. Three groups, lanes 2–4; 5–7; 8–10; were for the resin modified using lysine, glutamate, glycine, respectively). (D) Effect of click reaction time on the immobilization loading rate (%) of immobilized AKR mutants. | |
Fig. 6D shows the effect of the click reaction time on the immobilization loading rate (%) of the immobilized AKR mutants. The results show that the covalent bonds of the epoxy resin reached saturation after 24 h, and slight cleavage of the covalent bonds was observed after 24 h. At the same time, through modification of the resin using different amino acids, we found that the effect of lysine on enzyme immobilization is better than that of glycine and glutamic acid. The isoelectric points of the three amino acids, namely, lysine, glycine and glutamic acid, were 9.74, 5.97 and 3.22, respectively. Under alkaline conditions, lysine is more difficult to protonate than glycine and glutamic acid, which implies that amino groups are readily exposed to the support and easily react with the epoxy groups. Upon reacting with the epoxy group, the bare amino group can open the epoxy ring under basic conditions to complete the group modification.
3.4. Thermal stability of the AKR preparations
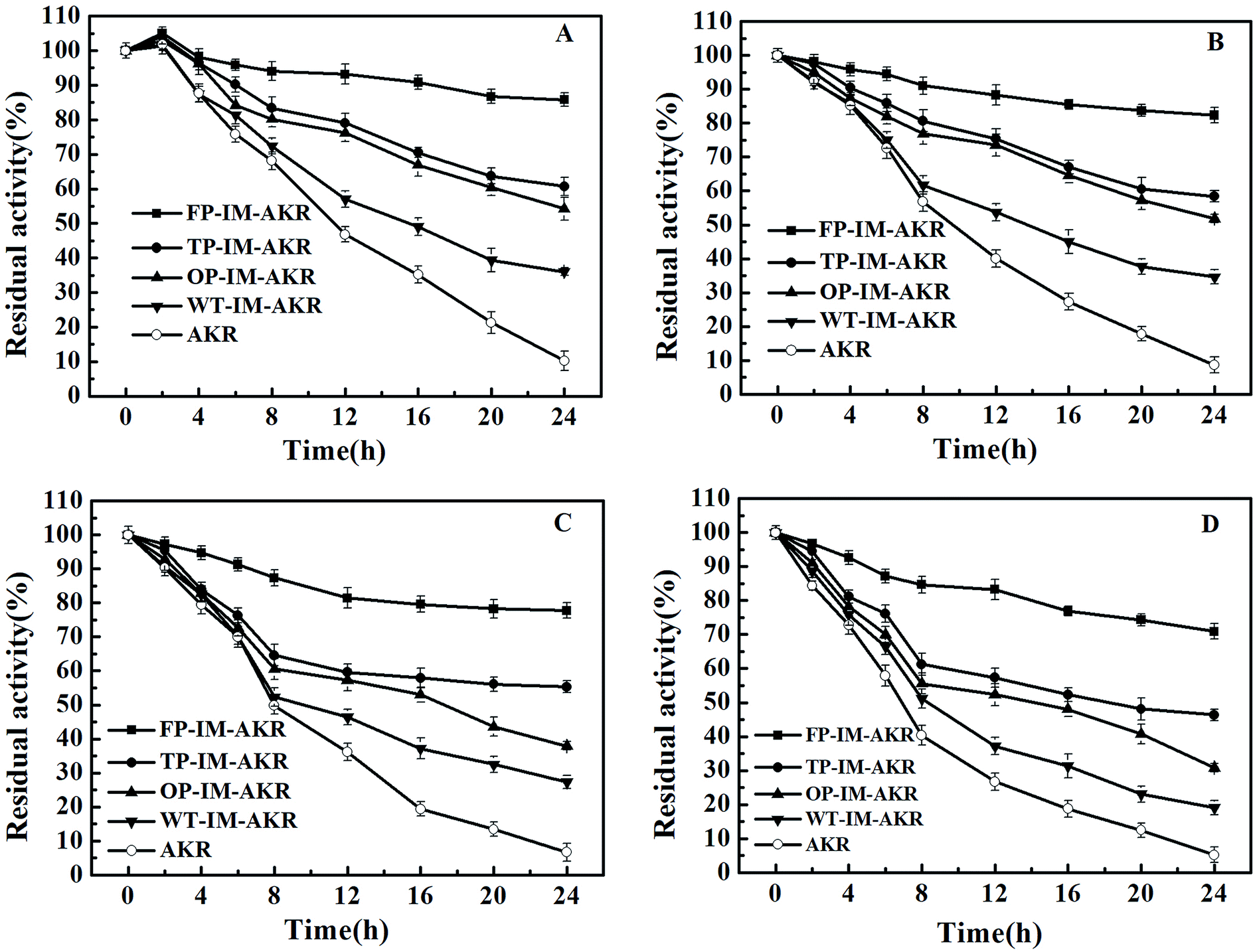
The thermal stability of AKR preparations surges as the increase of the number of mutation sites and chemical bonds between enzyme and support (Fig. 7). In five-point immobilization, more than 70% of the initial activity remained even after 24 h at 70 °C, however, free AKR lost almost of its activity after the same time period. The thermal stability of the enzyme preparations was consistent with their deactivation rate constant and half-life (t1/2) values. The half-life (t1/2) of the five-point immobilized enzyme reached 106 and 45 h, 13 and 7 times higher than that of the free enzyme at 30 °C and 60 °C, respectively (ESI, Table S4†). A long half-life increased the possibility of industrial application at that temperature.42 On the one hand, as the number of immobilization sites increased, the half-life of the enzyme activity increased gradually, and the denaturation rate constant decreased, which further proves that the stability of the immobilized enzyme is related to the number of covalent bonds between the enzyme and support. During the heating process, the activation energy of denaturation (Ea) is the energy required to denature the enzyme preparation (irreversible conformational change); thus, a high value of this parameter is a strong indication of high thermal stability of the enzyme preparation.43 The results (ESI, Table S5†) indicate that as the number of mutation and immobilization sites increased, the activation energy of the denaturation gradually increased, which may be attributed to the high structural rigidity. The insertion of multiple pAzF residues can increase the collision probability of azides and alkynes, leading to improved covalent binding of the enzyme to the support which would restrict the motion of the enzyme and increase the rigidity.
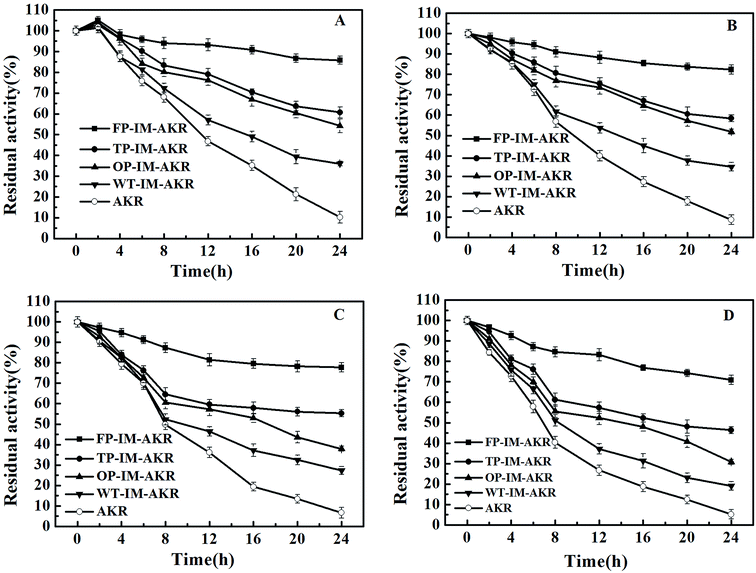 |
| | Fig. 7 Thermal stability of AKR enzyme preparations ((A) 30 °C; (B) 40 °C; (C) 50 °C; (D) 60 °C). FP-IM-AKR refers to five-point immobilized AKR, TP-IM-AKR refers to three-point immobilized AKR, OP-IM-AKR refers to one-point immobilized AKR, and WT-IM-AKR refers to the wild-type immobilized AKR. | |
4. Conclusions
In summary, we have developed a precise multi-point immobilization and one-step purification method by using a bioorthogonal copper-free click reaction for covalent enzyme immobilization based on NSAA insertion. By this method, we can effectively control the orientation for immobilization and tune the number of covalent bonds between the enzyme and support based on protein structure analysis and site mutation. Additionally, the enzyme mutants did not lose their activities, and the five-point immobilized enzyme presented heavily enhanced thermal stability due to accurate chemical linking and protection of the active site during covalent immobilization. This method can potentially be applied to a variety of enzymes, and the range of bioorthogonal reactions can be further expanded to prepare the efficient biocatalyst and perform catalytic applications under various conditions.
List of abbreviations
| AKR | Aldehyde ketone reductase |
| pAzF | p-Azido-L-phenylalanine |
| SPAAC | Strain-promoted alkyne–azide cycloaddition |
| NSAAs | Nonstandard amino acids |
| BCN | Bicyclo[6.1.0]nonyne |
| MG1655 | C321.ΔA |
| aTc | Anhydrotetracycline hydrochloride |
| DCC | N,N′-Dicyclohexylcarbodiimide |
| DMAP | 4-Dimethylaminopyridine |
| DCM | Dichloromethane |
| KPB | Potassium phosphate buffer |
| FP-IM-AKR | Five-point immobilized AKR |
| TP-IM-AKR | Three-point immobilized AKR |
| OP-IM-AKR | One-point immobilized AKR |
| WT-IM-AKR | Wild-type immobilized AKR |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to Dr Yinjie Tang (Professor) in the McKelvey School of Engineering at Washington University in St. Louis for advices in the experimental design and manuscript preparation. This study was supported by the National Natural Science Foundation of China (21576062), the Natural Science Foundation of Zhejiang Province (LY18B060009, LY15B060011), the Technology Research and Development Program of Hangzhou (20191203B09), the Key Projects of National Natural Science Foundation of China (81730108), the Research Plan for Sprout Talents in University in Zhejiang Province (2019R426081), the National Innovation and Entrepreneurship Training Program for Undergraduate (201810346008), the “Star and light” Project for Talent Students in Hangzhou Normal University (2019).
References
- L. Cao, Carrier-bound Immobilized Enzymes: Principles, Application and Design, 2006 Search PubMed.
- O. Barbosa, C. Ortiz, A. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, RSC Adv., 2013, 4, 1583–1600 RSC.
- A. A. Panova, V. Y. Levitsky, A. V. Levashov and V. V. Mozhaev, Biotechnol. Tech., 1995, 9, 13–18 CrossRef CAS.
- K. Martinek, A. M. Klibanov, V. S. Goldmacher, A. V. Tchernysheva, V. V. Mozhaev, I. V. Berezin and B. O. Glotov, Biochim. Biophys. Acta, 1977, 485, 13–28 CrossRef CAS.
- C. W. Rivero, E. C. De Benedetti, F. L. Gallego, B. C. Pessela, J. M. Guisan and J. A. Trelles, J. Biotechnol., 2017, 249, 34–41 CrossRef CAS PubMed.
- K. Martinek, A. M. Klibanov, V. S. Goldmacher and I. V. Berezin, Biochim. Biophys. Acta, Enzymol., 1977, 485, 1–12 CrossRef CAS.
- T. Saleh and C. G. Kalodimos, Science, 2017, 355, 247–248 CrossRef CAS PubMed.
- S. R. Tzeng and C. G. Kalodimos, Nature, 2012, 488, 236–240 CrossRef CAS PubMed.
- A. H. Orrego, M. Romero-Fernandez, M. D. Millan-Linares, M. D. Yust, J. M. Guisan and J. Rocha-Martin, Catalysts, 2018, 8, 333–347 CrossRef.
- C. Bernal, A. Illanes and L. Wilson, Langmuir, 2014, 30, 3557–3566 CrossRef CAS PubMed.
- K. Hernandez and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2011, 48, 107–122 CrossRef CAS PubMed.
- A. Wang, F. Du, F. Wang, Y. Shen, W. Gao and P. Zhang, Biochem. Eng. J., 2013, 73, 86–92 CrossRef CAS.
- D. Rabuka, J. S. Rush, G. W. deHart, P. Wu and C. R. Bertozzi, Nat. Protoc., 2012, 7, 1052–1067 CrossRef CAS PubMed.
- I. S. Carrico, B. L. Carlson and C. R. Bertozzi, Nat. Chem. Biol., 2007, 3, 321–322 CrossRef CAS PubMed.
- S. Lim, G. A. Jung, R. J. Muckom, D. J. Glover and D. S. Clark, Chem. Commun., 2019, 55, 806–809 RSC.
- M. T. Smith, J. C. Wu, C. T. Varner and B. C. Bundy, Biotechnol. Prog., 2013, 29, 247–254 CrossRef CAS PubMed.
- B. K. Raliski, C. A. Howard and D. D. Young, Bioconjugate Chem., 2014, 25, 1916–1920 CrossRef CAS PubMed.
- J. C. Y. Wu, C. H. Hutchings, M. J. Lindsay, C. J. Werner and B. C. Bundy, J. Biotechnol., 2015, 193, 83–90 CrossRef CAS PubMed.
- A. Wang, F. Du, X. Pei, C. Chen, S. G. Wu and Y. Zheng, J. Mol. Catal. B: Enzym., 2016, 132, 54–60 CrossRef CAS.
- J. W. Chin, in Annual Review of Biochemistry, ed. R. D. Kornberg, 2014, vol. 83, pp. 379–408 Search PubMed.
- C. C. Liu and P. G. Schultz, in Annual Review of Biochemistry, ed. R. D. Kornberg, C. R. H. Raetz, J. E. Rothman and J. W. Thorner, 2010, vol. 79, pp. 413–444 Search PubMed.
- H. H. Wang, F. J. Isaacs, P. A. Carr, Z. Z. Sun, G. Xu, C. R. Forest and G. M. Church, Nature, 2009, 460, 894–898 CrossRef CAS PubMed.
- M. J. Lajoie, A. J. Rovner, D. B. Goodman, H.-R. Aerni, A. D. Haimovich, G. Kuznetsov, J. A. Mercer, H. H. Wang, P. A. Carr, J. A. Mosberg, N. Rohland, P. G. Schultz, J. M. Jacobson, J. Rinehart, G. M. Church and F. J. Isaacs, Science, 2013, 342, 357–360 CrossRef CAS PubMed.
- C. M. Nealon, M. M. Musa, J. M. Patel and R. S. Phillips, ACS Catal., 2015, 5, 2100–2114 CrossRef CAS.
- C. A. Godoy, B. de las Rivas and J. M. Guisan, Process Biochem., 2014, 49, 1324–1331 CrossRef CAS.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS PubMed.
- M. J. Lajoie, A. J. Rovner, D. B. Goodman, H.-R. Aerni, A. D. Haimovich, G. Kuznetsov, J. A. Mercer, H. H. Wang, P. A. Carr and J. A. Mosberg, science, 2013, 342, 357–360 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671 CrossRef CAS PubMed.
- Z. Magomedova, A. Grecu, C. W. Sensen, H. Schwab and P. Heidinger, J. Biotechnol., 2016, 221, 78–90 CrossRef CAS PubMed.
- A. Sadana and J. P. Henley, Biotechnol. Bioeng., 1987, 30, 717–723 CrossRef CAS PubMed.
- F. J. Isaacs, P. A. Carr, H. H. Wang, M. J. Lajoie, B. Sterling, L. Kraal, A. C. Tolonen, T. A. Gianoulis, D. B. Goodman, N. B. Reppas, C. J. Emig, D. Bang, S. J. Hwang, M. C. Jewett, J. M. Jacobson and G. M. Church, Science, 2011, 333, 348–353 CrossRef CAS PubMed.
- M. Amiram, A. D. Haimovich, C. Fan, Y.-S. Wang, H.-R. Aerni, I. Ntai, D. W. Moonan, N. J. Ma, A. J. Rovner, S. H. Hong, N. L. Kelleher, A. L. Goodman, M. C. Jewett, D. Soell, J. Rinehart and F. J. Isaacs, Nat. Biotechnol., 2015, 33, 1272–1279 CrossRef CAS PubMed.
- Q. Wang, A. R. Parrish and L. Wang, Chem. Biol., 2009, 16, 323–336 CrossRef CAS PubMed.
- X. Chen, L. Xu, A. Wang, H. Li, C. Wang, X. Pei, P. Zhang and S. G. Wu, J. Chem. Technol. Biotechnol., 2019, 94, 236–243 CrossRef CAS.
- C. M. Madl, L. M. Katz and S. C. Heilshorn, Adv. Funct. Mater., 2016, 26, 3612–3620 CrossRef CAS PubMed.
- X. Ren, A. H. El-Sagheer and T. Brown, Nucleic Acids Res., 2016, 44, e79 CrossRef PubMed.
- Q.-Y. Hu, F. Berti and R. Adamo, Chem. Soc. Rev., 2016, 45, 1691–1719 RSC.
- T. Shibata, K. Maki, J. Hadano, T. Fujikawa, K. Kitazaki, T. Koshiba and S. Kawabata, PLoS Pathog., 2016, 12, e1005670 CrossRef PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- A. G. Marangoni, Enzyme kinetics: a modern approach, John Wiley & Sons, 2003 Search PubMed.
- A. A. N. Saqib, M. Hassan, N. F. Khan and S. Baig, Process Biochem., 2010, 45, 641–646 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09067c |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Ningning Lia,
Ru Wangb,
Jing Zhanga,
Xinxin Chena,
Xiaolin Pei
*a,
Ningning Lia,
Ru Wangb,
Jing Zhanga,
Xinxin Chena,
Xiaolin Pei