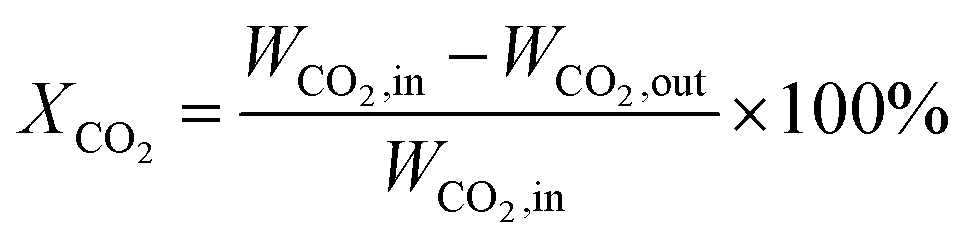DOI:
10.1039/C9RA08967E
(Paper)
RSC Adv., 2020,
10, 2067-2072
Ni nanocatalysts supported on mesoporous Al2O3–CeO2 for CO2 methanation at low temperature†
Received
31st October 2019
, Accepted 2nd December 2019
First published on 10th January 2020
Abstract
The selectivity and activity of a nickel catalyst for the hydrogenation of carbon dioxide to form methane at low temperatures could be enhanced by mesoporous Al2O3–CeO2 synthesized through a one-pot sol–gel method. The performances of the as-prepared Ni/Al2O3–CeO2 catalysts exceeded those of their single Al2O3 counterpart giving a conversion of 78% carbon dioxide with 100% selectivity for methane during 100 h testing, without any deactivation, at the low temperature of 320 °C. The influence of CeO2 doping on the structure of the catalysts, the interactions between the mesoporous support and nickel species, and the reduction behaviors of Ni2+ ions were investigated in detail. In this work, the addition of CeO2 to the composites increased the oxygen vacancies and active metallic nickel sites, and also decreased the size of the nickel particles, thus improving the low temperature catalytic activity and selectivity significantly.
1 Introduction
Natural gas is a potential clean fuel as well as an important feedstock used to produce other key industrial chemicals. The process of carbon dioxide (CO2) hydrogenation to produce methane (CH4) is a promising route for recycling CO2 captured from the combustion of fossil fuels.1–4 CO2 methanation, also known as the Sabatier reaction (4H2 + CO2 → CH4 + 2H2O, ΔH298 K = −165 kJ mol−1), is exothermic and thermodynamically favoured at low temperatures but there are significant kinetic barriers5 and thus it still remains a big challenge to develop a catalyst with both excellent catalytic activity and selectivity at low temperatures. Great efforts have been made to study metal-supported catalysts for the hydrogenation of CO2 to CH4. Compared with expensive noble metals (Rh, Ru, Pd) and other common transition metals (Fe, Co),6–11 Ni-based composites, so far, have been the most extensively used in CO2 hydrogenation to CH4 because of their low cost, excellent catalytic activities and selectivity.12–16 However, the sintering problems of Ni nanoparticles at relatively high reaction temperatures and the deposition of carbon lead to rapid deactivation during the reaction processes.17 Therefore, it is desirable to explore novel nanocatalysts which are highly efficient, mechanically resistant, chemically and physically stable, and resistant to sintering.
Many strategies have been proposed to alleviate the fast deactivation and low selectivity of catalysts for CH4, such as the modification of catalytic supports, the addition of structural or electronic promoters, and adjustments to the preparation routes for the catalysts.18–23 Among these, the modification of supports has drawn much attention because changes to the metal-support interactions affect the reactivity and bonding of chemisorbed molecules as well. For instance, MgO and ZrO2 were investigated for their capacities to improve the catalytic activity and selectivity of heterogeneous catalysts.24–27 In general, CeO2 has acted as an electronic and structural promoter to enhance the performance of Ni-based catalysts by reinforcing the thermal stability, and improving the exchange of oxygen species as well as the uniform distribution of metals over the catalyst.28–30
Here, we describe a Ni-modified catalyst loaded on an Al2O3–CeO2 support through a one-pot sol–gel method, and demonstrate its activity for the hydrogenation of CO2. The increased quantity of active nickel sites combined with the oxygen vacancies of the composite support promoted by CeO2 lead to an excellent performance in the hydrogenation of CO2 to form CH4. Although there are some reports in the literature regarding CeO2-based composites for CO2 methanation, most of these have used CeO2 as a separate carrier or promoter, and very few reports have focused on the Ce species as both a promoter and a carrier at the same time. The low content of Ce and the poor interaction between CeO2 and Al2O3 in composites prepared in previous studies have led to inferior catalytic performance.31–33 In this study, we have developed a mild method to construct Al2O3–CeO2 composites, namely a one-step sol–gel method. These Al2O3–CeO2 composites have high redox activity, high numbers of oxygen vacancies, resistance to sintering and excellent thermal stability. Our Al2O3–CeO2-1.0-supported Ni catalyst exhibited 100% selectivity for CH4 with 78% CO2 conversion in a 100 h test at the low temperature of 320 °C.
2 Experimental section
2.1 Preparation of catalysts
Al(NO3)3·9H2O (99.99%), Ce(NO3)3·6H2O (99.99%) and Ni(NO3)3·6H2O (99.99%) were purchased and used without further purification. A series of Ni-supported mesoporous Al2O3–CeO2 composites with different Al/Ce ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 10:1, 5:1, 2.5:1 and 1:1) were prepared via a one-pot sol–gel route.34 Briefly, Ce(NO3)3·6H2O (7.5 g, 0.02 mol) and Al(NO3)3·9H2O (8.68 g, 0.02 mol) were dissolved in 50 mL EtOH, and a moderate amount of Ni(NO3)3·6H2O (2.46 g, 8 mmol) was added to the solution with vigorous stirring. Then 1,2-epoxypropane (25 mL) was added dropwise to the above solution with stirring until gels formed. After being aged at room temperature for 48 h, the gels were five times solvent exchanged with EtOH to remove impurities, and dried at 80 °C for two days. The white product Ni/Al2O3–CeO2-1.0 (Al/Ce mole ratio = 1) was obtained by calcining the powder at 500 °C for 7 h. In the composites, the sum mole ratio of Al and Ce was fixed at 0.04 mol, and the Ni content was maintained at 10 wt%. The resulting porous 10Ni/Al2O3–CeO2 catalysts were named Ni/Al2O3–ZrO2-x (x = 10, 5.0, 2.5, and 1.0), where x represents the Al/Ce ratio. Single Ni/Al2O3 catalysts were prepared for comparison using the same process but without adding the Ce source.
:0, 10:1, 5:1, 2.5:1 and 1:1) were prepared via a one-pot sol–gel route.34 Briefly, Ce(NO3)3·6H2O (7.5 g, 0.02 mol) and Al(NO3)3·9H2O (8.68 g, 0.02 mol) were dissolved in 50 mL EtOH, and a moderate amount of Ni(NO3)3·6H2O (2.46 g, 8 mmol) was added to the solution with vigorous stirring. Then 1,2-epoxypropane (25 mL) was added dropwise to the above solution with stirring until gels formed. After being aged at room temperature for 48 h, the gels were five times solvent exchanged with EtOH to remove impurities, and dried at 80 °C for two days. The white product Ni/Al2O3–CeO2-1.0 (Al/Ce mole ratio = 1) was obtained by calcining the powder at 500 °C for 7 h. In the composites, the sum mole ratio of Al and Ce was fixed at 0.04 mol, and the Ni content was maintained at 10 wt%. The resulting porous 10Ni/Al2O3–CeO2 catalysts were named Ni/Al2O3–ZrO2-x (x = 10, 5.0, 2.5, and 1.0), where x represents the Al/Ce ratio. Single Ni/Al2O3 catalysts were prepared for comparison using the same process but without adding the Ce source.
2.2 Material characterizations
X-ray Photoelectron Spectroscopy (XPS) was conducted on an ESCALAB250Xi XPS spectrometer (Thermo Fisher Scientific), and the binding energies of all photoelectron peaks were calibrated using C 1s spectra (binding energy at 284.8 eV). Powder X-ray Diffraction (PXRD) characterization was performed on a Smartlab diffractometer (Rigaku) with filtered Cu Kα radiation (λ = 1.5405 Å). N2 adsorption and desorption isotherms were performed on an Autosorb iQ2 analyzer (Quantachrome) in a liquid nitrogen bath at 77 K. H2-temperature programmed reduction (H2-TPR) was conducted using an Altamira AMI 200-R-HP unit with a thermal conductivity detector (TCD). Thermogravimetric analysis (TGA) was performed on a DTG-60 thermal gravimetric analyser (Shimadzu) in an air atmosphere. All prepared catalysts were stored in an inert glovebox (O2 < 0.1 ppm, H2O < 0.1 ppm, Mikrouna) before use and characterization.
2.3 Catalytic tests
CO2 methanation was performed in a continuous fixed-bed reactor in a stainless steel tube with a length of 330 mm and an inner diameter of 12 mm at normal pressure and various temperatures. Briefly, 0.5 g catalyst was mixed with an equivalent weight of quartz sand (40–70 mesh) and reduced in situ under pure H2 with a gas hourly space velocity (GHSV) of 2000 mL g−1 h−1 at 500 °C for 9 hours before the catalytic test. Then the instrument was cooled to 160 °C and a mixed stream of CO2 and H2 (volumetric ratio of H2/CO2 = 4) was introduced into the gas circuit as the feedstock. The gases in the outflow were analysed using an online gas chromatograph (Fuli 9790II). CH4 (SCH4) selectivity and CO2 (XCO2) conversion were determined by the following equations:| |
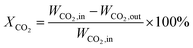 | (1) |
| |
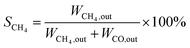 | (2) |
where, WCO2,in denotes the moles of CO2 in the feedstock, and WCO2,out, WCO,out, and WCH4,out denote the carbon moles of CO2, CO and CH4 at the outflow reactor, respectively.
3 Results and discussion
3.1 Characterization of the catalysts
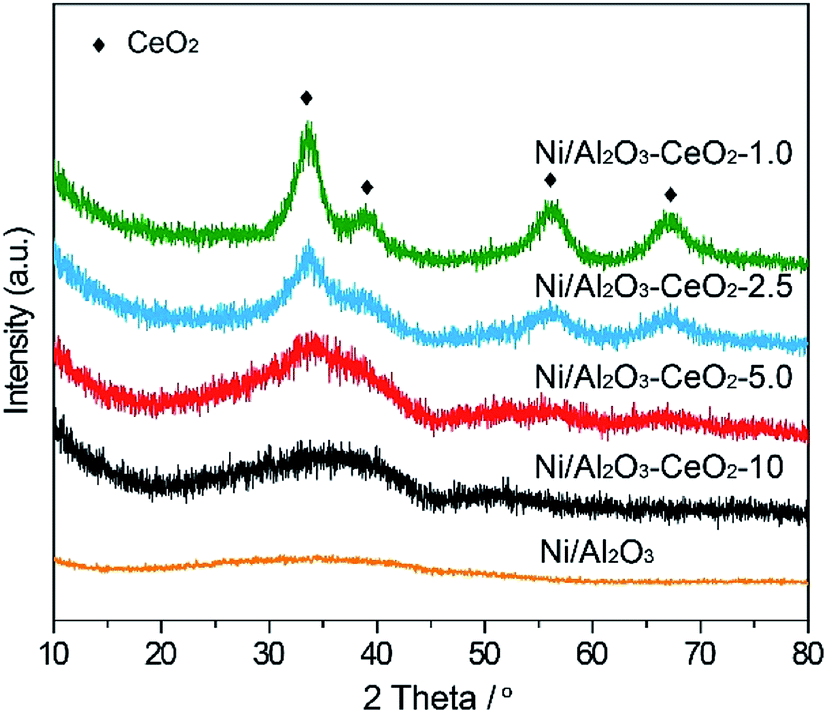
The X-ray powder diffraction (PXRD) patterns of fresh Ni/Al2O3–CeO2-x (x = 10, 5.0, 2.5, and 1.0) samples are shown in Fig. 1. In the pattern of the Ni/Al2O3 catalyst, we could observe a broad peak from 29.9 to 45.9°, which was assigned to the amorphous phase of Al2O3. Four new diffraction peaks at 33.3°, 38.6°, 55.8° and 66.5° appeared after the introduction of the Ce species, which corresponded to the characteristic peaks of CeO2. In general, a higher Ce loading in the composite resulted in an increased degree of crystallization of Al2O3, as confirmed by the gradual narrowing of the broad peak (Fig. 1). Meanwhile, no obvious characteristic diffraction peaks of bulk NiO could be detected in the Ni/Al2O3–CeO2-x composites, suggesting an amorphous form and an even distribution of Ni-based materials on the surface of the Al2O3–CeO2 composites, as expected.
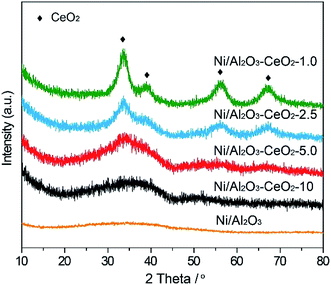 |
| | Fig. 1 PXRD patterns of the fresh Ni/Al2O3–CeO2-x catalysts. | |
The influence of the presence of cerium and nickel on the Brunauer–Emmett–Teller (BET) pore size distributions and specific surface areas of the Ni/Al2O3–CeO2 composites with different molar ratios of Al/Ce were evaluated by N2 sorption isotherms at 77 K (Fig. 2). Similar to the single Ni–Al2O3 sample, all the Ni/Al2O3–CeO2 composites displayed type IV isotherms with big hysteresis loops, demonstrating the existence of mesopores in these composite supports. The pristine mesoporous Al2O3-modified Ni catalyst had a high specific surface area of 291 m2 g−1 with an average pore volume of 0.35 cm3 g−1. As the Ce loading in the Ni/Al2O3–CeO2-x composites was increased, the region of the hysteresis loops tended to decrease (Fig. 2A); at the same time, the average pore diameters widened gradually in the range 3.66 to 4.35 nm (Fig. 2B), indicating that the Ce and Ni species might occupy the small pores of Al2O3 or cause partial collapse of pristine structures. The basic data on the catalysts are listed in Table S1.† The prepared Ni/Al2O3–CeO2-x samples exhibited obvious decreases in pore volumes and specific surface areas, but an increase in pore diameters in comparison to the Ni/Al2O3 catalyst. These results may be ascribed to the covering of the Al2O3 surface by Ce or Ni species, or the aggregation of NiO species.
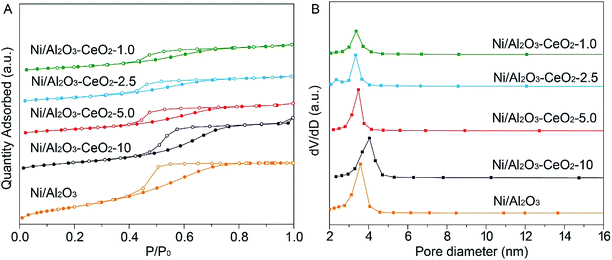 |
| | Fig. 2 (A) N2 adsorption–desorption isotherms and (B) BJH pore size distributions of the fresh Ni/Al2O3–CeO2-x catalysts. | |
3.2 Effect of Ce content
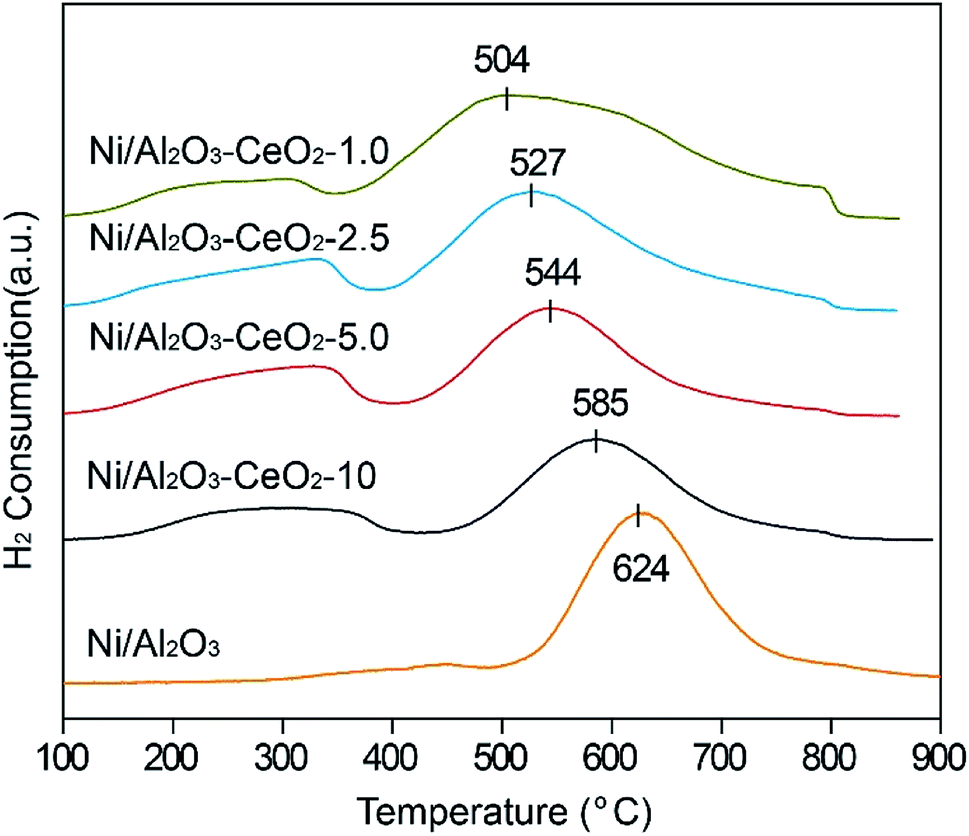
H2-TPR was used to test the reduction behaviour of the Ni/Al2O3–CeO2-x catalysts (Fig. 3), and the Ni/Al2O3 catalyst was also measured for comparison. The Ce content had a remarkable effect on the reduction of Ni2+ ions in the composites. With increasing amounts of Ce in the Ni/Al2O3–CeO2 composites, the reduction peaks for the NiO species shifted gradually to lower temperatures (624 °C for Ni/Al2O3 to 504 °C for Ni/Al2O3–CeO2-1.0). This indicated that the doping of cerium into the Ni/Al2O3–CeO2 supports broke the reciprocities between Al2O3 and NiO due to the formation of the Al2O3–CeO2 composite, which was beneficial for the reduction of NiO. The reduction peak at around 624 °C could be observed for the Ni/Al2O3 sample; the high temperature could be ascribed to the intense interactions between the Al2O3 support and NiO species. For Ce-doped samples, the reduction peaks for Ni/Al2O3–CeO2-x shifted slowly to lower temperature with increasing Ce loading, and the consumption of H2 also increased, which could be attributed to the Ce3+/Ce4+ couple, which could create both bulk and surface oxygen vacancies.35 The inferior reduction behaviour of the Ni-based species on mesoporous Al2O3 led to inadequate numbers of active metallic Ni sites in comparison with those in the Ni-supported Al2O3–CeO2 composites. The best reduction behaviour of NiO species in this study was obtained for the catalyst with the Al/Ce molar ratio of 1.0.
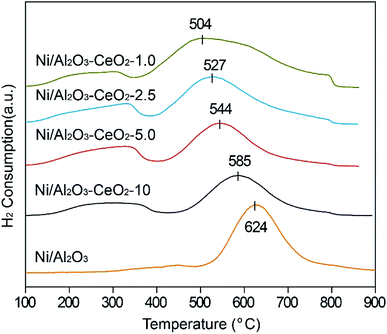 |
| | Fig. 3 H2-TPR profiles of the fresh Ni/Al2O3–CeO2-x catalysts. | |
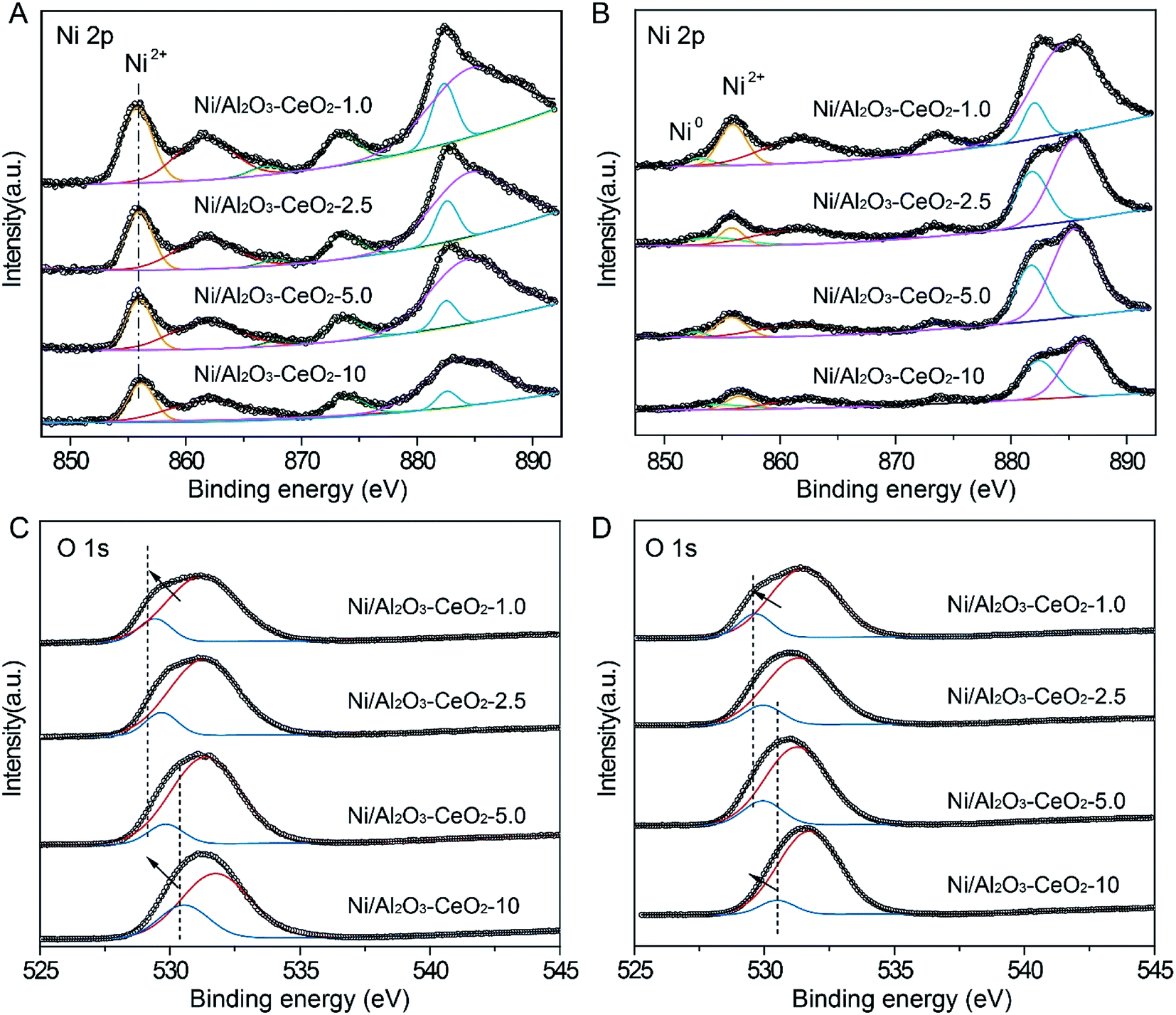
The valence state of Ni, the interactions between NiO species and the Al2O3–CeO2 support, and the surface chemical environment of fresh Ni/Al2O3–CeO2-x catalysts were further revealed by XPS (Fig. 4). The binding energy at 855.9 eV of Ni 2p3/2 is the characteristic peak of Ni2+, and no obvious peak shift could be detected for Ni/Al2O3–CeO2-x regardless of the Al/Ce ratio (Fig. 4A), which demonstrated that the Ce content did not change the chemical environment of the NiO species dispersed on the surface of the composites. From this observation in combination with the PXRD analysis, we could infer that the nickel existed on the surface of the catalyst mainly as highly dispersed NiO species. However, the intensity of the Ni2+ peak strengthened with increasing Ce content, indicating the generation of vast numbers of active sites on the support surface in the reduction process, which could be further confirmed by the XPS of spent catalysts (Fig. 4B). The XPS peak at around 529.9 eV corresponded to the lattice oxygen (OL) on the surface of CeO2 or Al2O3 (Fig. 4C and D), and another peak at around 530.9 eV was assigned to adsorbed oxygen (OA) on the surface.36 The detailed information about the OA to OL ratio for the fresh composites on the basis of the OL and OA area percentages is summarized in Table S2.† The OA and OL peaks for the Ni-supported Al2O3–CeO2 catalysts were located in the ranges 531.2–531.9 eV and 529.6–531.0 eV, respectively. With increasing Ce content, the peaks shifted slowly to lower binding energies, which may be ascribed to the ever-increasing numbers of oxygen vacancies on the surface of the Al2O3–CeO2 catalysts at higher Ce content, thus contributing to the adsorption and conversion of CO2 by the catalysts.37
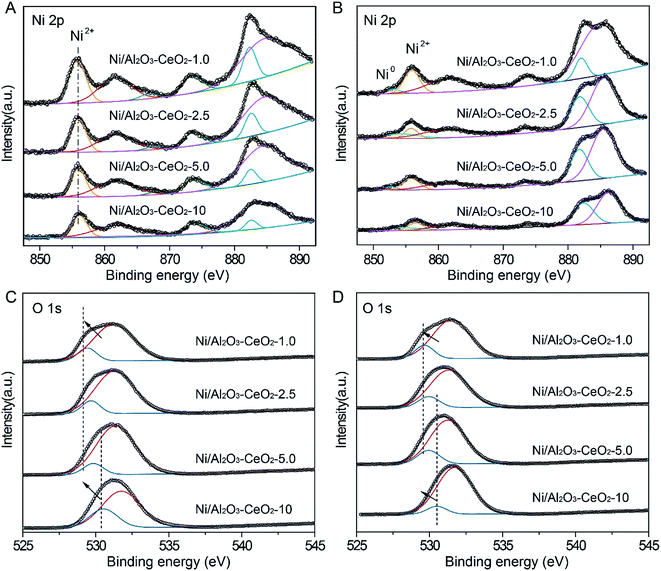 |
| | Fig. 4 XPS spectra for (A) fresh and (B) spent Ni/Al2O3–CeO2-x catalysts in the Ni 2p region, and (C) fresh and (D) spent Ni/Al2O3–CeO2-x catalysts in the O 1s region. | |
3.3 Catalytic performance
The catalytic hydrogenation of CO2 to CH4 with the Ni/Al2O3–CeO2-x catalysts was performed in a fixed-bed reactor (GHSV of 6000 mL g−1 h−1, H2/CO2 = 4.0, atmospheric pressure, temperature varied from 150 to 450 °C). As the reaction temperature was progressively increased, the CO2 conversion first increased for all catalysts, but it then reached a peak value at an optimum reaction temperature and started to decrease (Fig. 5A). The catalytic activity declined when the temperature was further increased to 350 °C, which could be ascribed to the endothermic reverse reaction. The CeO2-modified catalysts displayed an obviously higher CH4 selectivity compared with the single Ni/Al2O3 sample (Fig. 5B). The amount of CeO2 in the Al2O3–CeO2 composite had a critical impact on the catalytic performance, especially at lower temperature. The Ni/Al2O3 catalyst without CeO2 displayed a low CO2 conversion of only 9.8% at the low reaction temperature of 250 °C, but when we introduced trace amounts of Ce species into the catalyst (Ni/Al2O3–CeO2-10), the conversion of CO2 increased sharply to the value of 42.9%. It was noteworthy that the CO2 conversion increased step-by-step when the CeO2 loading was increased in the Ni-modified Al2O3–CeO2 samples. Apparently, with an increase of CeO2 content, a lower temperature was sufficient to reach the same CO2 conversion level; the excellent catalytic performance of 78% CO2 conversion with 100% CH4 selectivity was obtained for the Ni/Al2O3–CeO2-1.0 composite catalyst at the relatively low temperature of 320 °C.
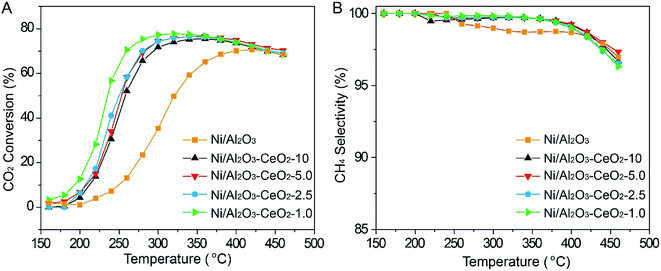 |
| | Fig. 5 (A) CO2 conversion and (B) CH4 selectivity of the Ni/Al2O3–CeO2-x catalysts. GHSV = 6000 mL g−1 h−1, P = 0.1 MPa, H2/CO2 = 4. | |
In order to investigate the effect of Ce species on the long-term durability of the catalyst, measurements were performed over a time period of 100 h for the Ni/Al2O3–CeO2-1.0 catalyst (Fig. 6). Indeed, the loading of CeO2 in the composite had a significant influence on the catalytic performance of CO2 methanation. The Ni/Al2O3–CeO2-1.0 sample exhibited 78% CO2 conversion with almost 100% CH4 selectivity during the 100 h test, demonstrating excellent long-term stability and selectivity, and showing that CeO2 doping significantly improved the long-term stability of Ni/Al2O3 catalysts. Obviously, the introduction of CeO2 could promote catalytic stability and activity at the same time, which may be attributed to the generation of oxygen vacancies on the surface of the support and the increased metallic nickel surface area, as evidenced by XPS (Fig. 4C and D). On the one hand, Ni species provided active sites for activating molecular CO2 and could facilitate the formation of atomic hydrogen by dissociating H2 from the Ni-based catalyst. On the other hand, the surface oxygen vacancies resulted in the formation of carbon species, which could react with the atomic hydrogen on the surface of the catalyst to form CH4. The structural stability of the Ni/Al2O3–CeO2-1.0 catalyst was further confirmed by PXRD after the long-term reaction; no obvious change could be observed when it was compared with the fresh catalyst (Fig. S1†).
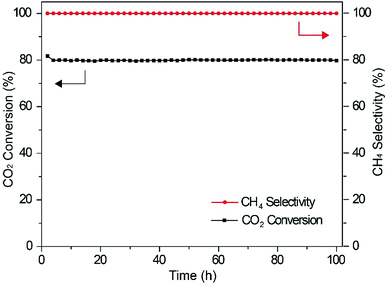 |
| | Fig. 6 Stability tests on the Ni/Al2O3–CeO2-1.0 catalyst at 320 °C, GHSV = 6000 mL g−1 h−1, P = 0.1 MPa, H2/CO2 = 4. | |
4 Conclusions
To sum up, Ni-modified mesoporous Al2O3–CeO2 composite catalysts containing various amounts of CeO2 were synthesized through a one-pot sol–gel route and used for CO2 conversion to CH4 at low reaction temperatures. The mesoporous Ni/Al2O3–CeO2 catalysts displayed excellent CH4 selectivity and CO2 conversion in comparison to the single Ni-modified Al2O3 catalyst. The uniform distribution of Ni species combined with the improved surface oxygen vacancies resulting from CeO2 loading on the support made the excellent catalytic activity and CH4 selectivity possible at lower temperatures. The Ni/Al2O3–CeO2-1.0 catalyst displayed impressive catalytic properties of 78% CO2 conversion with 100% CH4 selectivity at 320 °C; this performance was retained without any decay during 100 h of testing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for the support from the National Natural Science Foundation of China (51861145102, 21978225), the Science and Technology program of Shenzhen (JCYJ20180302153928437), the Foundation of State Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering (2019-KF-06) and the Fundamental Research Fund for the Central Universities (2042019kf0221).
References
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- M. Gassner and F. Marechal, Energy Environ. Sci., 2012, 5, 5768–5789 RSC.
- C. Janke, M. S. Duyar, M. Hoskins and R. Farrauto, Appl. Catal. B Environ., 2014, 152–153, 184–191 CrossRef CAS.
- I. Dimitriou, P. García-Gutiérrez, R. H. Elder, R. M. Cuéllar-Franca, A. Azapagic and R. W. K. Allen, Energy Environ. Sci., 2015, 8, 1775–1789 RSC.
- K. Mueller, M. Staedter, F. Rachow, D. Hoffmannbeck and D. Schmeisser, Environ. Earth Sci., 2013, 70, 3771–3778 CrossRef CAS.
- M. Younas, L. Loong Kong, M. J. K. Bashir, H. Nadeem, A. Shehzad and S. Sethupathi, Energy Fuels, 2016, 30, 8815–8831 CrossRef CAS.
- P. Frontera, A. Macario, M. Ferraro and P. Antonucci, Catalysts, 2017, 7, 59 CrossRef.
- J. Gao, Q. Liu, F. Gu, B. Liu, Z. Zhong and F. Su, RSC Adv., 2015, 5, 22759–22776 RSC.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and A. Ahma, Green Chem., 2015, 17, 2647–2663 RSC.
- Y. Zhu, S. Zhang, Y. Ye, X. Zhang, L. Wang, W. Zhu, F. Chang and F. Tao, ACS Catal., 2012, 2, 2403–2408 CrossRef CAS.
- A. Karelovic and P. Ruiz, ACS Catal., 2013, 3, 2799–2812 CrossRef CAS.
- M. Younas, L. L. Loong, M. J. K. Bashir, H. Nadeem, A. Shehzad and S. Sethupathi, Energy Fuels, 2016, 30, 8815–8831 CrossRef CAS.
- S. He, C. Li, H. Chen, D. Su, B. Zhang, X. Cao, B. Wang, M. Wei, D. G. Evans and X. Duan, Chem. Mater., 2013, 25, 1040–1046 CrossRef CAS.
- G. A. Du, S. Y. Lim, Y. H. Yang, C. Wang, L. Pfefferle and G. L. Haller, J. Catal., 2007, 249, 370–379 CrossRef CAS.
- D. Wierzbicki, R. Debek, M. Motak, T. Grzybek, M. E. Gálvez and P. Da Costa, Catal. Commun., 2016, 83, 5–8 CrossRef CAS.
- S. Rahmani, M. Rezaei and F. Meshkani, J. Ind. Eng. Chem., 2014, 20, 346–352 Search PubMed.
- D. Wierzbicki, R. Baran, R. Dębek, M. Motak, M. E. Gálvez, T. Grzybek, P. Da Costa and P. Glatzel, Appl. Catal. B Environ., 2018, 232, 409–419 CrossRef CAS.
- X. Zou, X. Wang, L. Li, K. Shen, X. Lu and W. Ding, Int. J. Hydrogen Energy, 2010, 35, 13191–13200 Search PubMed.
- W. Wang and J. Gong, Front. Chem. Sci. Eng., 2011, 5, 2–10 CrossRef CAS.
- C. Cheng, D. Shen, R. Xiao and C. Wu, Fuel, 2017, 189, 419–427 CrossRef CAS.
- T. A. Le, M. S. Kim, S. H. Lee, T. W. Kim and E. D. Park, Catal. Today, 2017, 293–294, 89–96 CrossRef CAS.
- G. Zhou, H. Liu, K. Cui, H. Xie, Z. Jiao, G. Zhang, K. Xiong and X. Zheng, Int. J. Hydrogen Energy, 2017, 42, 16108–16117 CrossRef CAS.
- J. Lin, C. Ma, J. Luo, X. Kong, Y. Xu, G. Ma, J. Wang, C. Zhang, Z. Li and M. Ding, RSC Adv., 2019, 9, 8684–8694 RSC.
- Y. Yan, Y. Dai, H. He, Y. Yu and Y. Yang, Appl. Catal. B Environ., 2016, 196, 108–116 CrossRef CAS.
- K. M. Lee and W. Y. Lee, Catal. Lett., 2002, 83, 65–70 CrossRef CAS.
- M. Romero-Sáez, A. B. Dongil, N. Benito, R. Espinoza-González, N. Escalona and F. Gracia, Appl. Catal. B Environ., 2018, 237, 817–825 CrossRef.
- J. Lin, C. Ma, Q. Wang, F. Xu, G. Ma, J. Wang, H. Wang, C. Dong, C. Zhang and M. Ding, Appl. Catal. B Environ., 2019, 243, 262–272 CrossRef CAS.
- Y. Lu, S. Li and L. Guo, Fuel, 2013, 103, 193–199 CrossRef CAS.
- L. Zhou, Q. Wang, L. Ma, J. Chen, J. Ma and Z. Zi, Catal. Lett., 2015, 145, 612–619 CrossRef CAS.
- M. Ding, J. Tu, Q. Zhang, M. Wang, N. Tsubaki, T. Wang and L. Ma, Biomass Bioenergy, 2016, 85, 12–17 CrossRef CAS.
- J. Chen, R. Wang, J. Zhang, F. He and S. Han, J. Mol. Catal. A: Chem., 2005, 235, 302–310 CrossRef CAS.
- M. M. Barroso-Quiroga and A. E. Castro-Luna, Int. J. Hydrogen Energy, 2010, 35, 6052–6056 CrossRef CAS.
- L. Zhou, Q. Wang, L. Ma, J. Chen, J. Ma and Z. Zi, Catal. Lett., 2015, 145, 612–619 CrossRef CAS.
- D. Chen, D. He, J. Lu, L. Zhong, F. Liu, J. Liu, J. Yu, G. Wan, S. He and Y. Luo, Appl. Catal. B Environ., 2017, 218, 249–259 CrossRef CAS.
- H. Liu, X. Zou, X. Wang, X. Lu and W. Ding, J. Nat. Gas Chem., 2012, 21, 703–707 CrossRef CAS.
- W. Zheng, J. Zhang, Q. Ge, H. Xu and W. Li, Appl. Catal. B Environ., 2008, 80, 98–105 CrossRef CAS.
- W. Nie, X. Zou, X. Shang, X. Wang, W. Ding and X. Lu, Fuel, 2017, 202, 135–143 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08967e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Jianghui Lina,
Guangyuan Maa,
Yanfei Xua,
Jianli Zhangb,
Chanatip Samartc and
Mingyue Ding
a,
Jianghui Lina,
Guangyuan Maa,
Yanfei Xua,
Jianli Zhangb,
Chanatip Samartc and
Mingyue Ding