
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological activity study of the retro-isomer of RhTx against TRPV1†
Rilei Yu‡
 ac,
Huijie Liu‡b,
Baishi Wang‡a,
Peta J. Harveyd,
Ningning Wei*b and
Yanyan Chu*ac
ac,
Huijie Liu‡b,
Baishi Wang‡a,
Peta J. Harveyd,
Ningning Wei*b and
Yanyan Chu*ac
aKey Laboratory of Marine Drugs, Chinese Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, Qingdao 266003, China. E-mail: chuyanyan@ouc.edu.cn
bDepartment of Pharmacology, School of Pharmacy, Qingdao University, Qingdao 266021, China. E-mail: weiningning@qdu.edu.cn
cLaboratory for Marine Drugs and Bioproducts of Qingdao National Laboratory for Marine Science and Technology, Qingdao 266003, China
dInstitute for Molecular Bioscience, The University of Queensland, Brisbane, QLD, 4072 Australia
First published on 10th January 2020
Abstract
TRPV1 is a ligand-gated ion channel and plays an important role in detecting noxious heat and pain with an unknown mechanism. RhTx from Chinese red-headed centipede activates the TRPV1 channel through the heat activation pathway by binding to the outer pore region, and causes extreme pain. Here, we synthesized RhTx and its retro-isomer RL-RhTx. Their structures were investigated by their circular dichroic spectra and NMR spectra. The effect of RhTx and RL-RhTx on the currents of wild-type and mutants of TRPV1 indicated that RL-RhTx have comparable TRPV1 activation responses to RhTx. A mutagenesis study showed that four TRPV1 residues, including Leu461, Asp602, Tyr632 and Thr634, significantly contributed to the activation effects of RL-RhTx and RhTx, and both peptides probably bind with TRPV1 in similar binding modes. As a novel TRPV1 activator, RL-RhTx provides an essential powerful tool for the investigation of activation mechanisms of TRPV1.
1 Introduction
The transient receptor potential vanilloid 1 (TRPV1), a nonselective cation channel, can be activated by capsaicin.1–3 TRPV1 is a polymodal nociceptor sensitive to different physical and chemical stimuli, including heat and mechanical stimuli.4–6 The channel plays key roles in maintaining stable body temperature in mammals and transduces heat pain.4 Thus, TRPV1 is considered as an essential target for pain therapy.4,8–10 TRPV1 is a homotetramer, each subunit consists of six transmembrane α-helices (Fig. 1A).7,11 The outer pore region contributes to regulation of the mechanisms of channel gating and permeation, and is essential for peptide toxin binding.12 Several types of animal toxins, including DkTx,13,14 RhTx,15,16 APHCs12,17 and HCRG21 (ref. 15 and 18) targeted TRPV1 with high potency. Among these, RhTx is the only toxin that was reported to target TRPV1 through the heat activation pathway.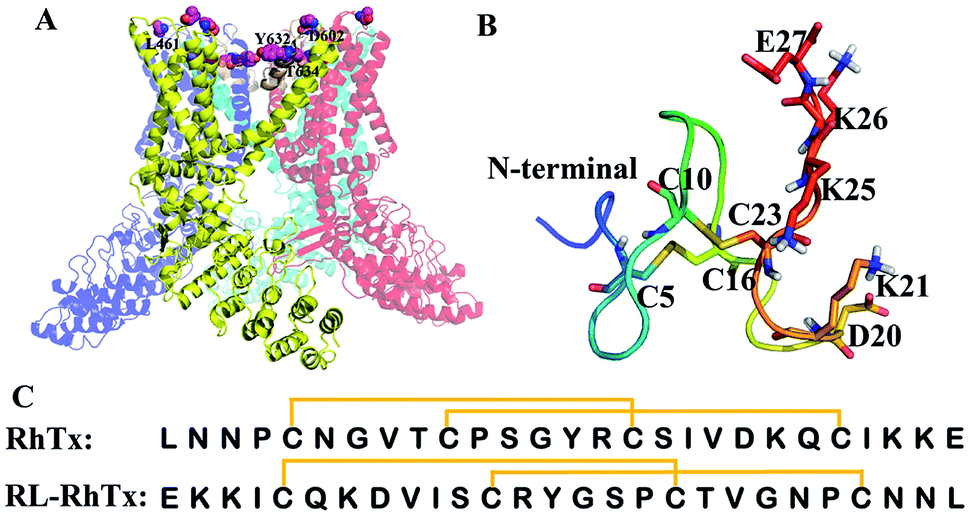 | ||
| Fig. 1 Structures of TRPV1, RhTx and RL-RhTx. (A) The structure of TRPV1 (PDB code: 3J5P7). The constituted four monomers were colored differently. The important pore regions were colored wheat. (B) The structure of RhTx (PDB code: 2MVA). The charged residues of C-terminal were labeled and shown in blue sticks. The cysteines that formed disulfide bonds were in green sticks. (C) The sequences of RhTx and RL-RhTx. Disulfide bonds were indicated by square brackets. | ||
RhTx, isolated from Chinese red-headed centipede Scolopendra subspinipes mutilans, is a 27-residue small peptide with two intramolecular disulfide bonds forming between Cys5–Cys16 and Cys10–Cys23 (Fig. 1B and C). RhTx is rich of charged residues in C-terminal, and the residues contribute to the binding affinity of RhTx with TRPV1.16 By binding to the outer pore of TRPV1, RhTx improves heat-dependent activation of TRPV1 potently.16 However, the interaction mechanism of RhTx remains unclear. More structure and activity investigation benefits to understanding of the TRPV1 heat activation mechanism.
The development of peptide toxins as drugs or biological tools is heavily limited by their poor metabolic stability, low bioavailability and toxicity. Retro-isomerization of a constrained peptide is a strategy in drug design to obtain peptidomimetics lower susceptibility to proteolysis, improved bioavailability,19 and sometimes decreased toxicity.19,20 The peptidomimetics usually possess with comparable activity to the native one.21–26 Partial retro-isomer structure was successfully applied to several families of biological active peptides. For example, retro-isomer of tumor suppressor protein p53 (15–29) had strong binding affinity to MDM2, which is a human E3 ubiquitin ligase that mediates ubiquitination of p53/TP53.21 As expected, the strategy is not universally applicable because the retro-isomer may have different three-dimentional structure from the original one.27 Li et al. reported that stingin emulated the transactivation peptide of p53 and bound with high affinity to MDM2, while its retro isomer completely abolished its MDM2 binding.28 Although, the retro-isomer transformation remains an important peptide modification method and has been widely used for designing novel and proteolytically stable peptides with comparable biological activities by numerous bioorganic chemists.23
In this study, we adopted the retro-isomerization strategy and designed a retro-isomer of RhTx (RL-RhTx), which is expected to harbor increased stability, enhanced TRPV1 activity and low toxicity. RL-RhTx is proved to be a novel TRPV1 agonist. The effects of RhTx and RL-RhTx on TRPV1 currents were evaluated through electrophysiological experiment. The results demonstrated that RL-RhTx and RhTx have equal activation response to TRPV1.
2 Experimental
2.1 Synthesis of RhTx and RL-RhTx
The chlorotrityl chloride resin was swollen in mixture of dichloromethane (DCM) and N,N-dimethylformamide (DMF) (DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMF = 1:1). The first amino acid was coupled to the resin for 24 h in DMF, with a 5-fold excess of amino acid and 10-fold excess of N,N-diisopropylethylamine (DIPEA). Then, 30 mL of MeOH was added to the mixture to cap any remaining reactive groups on the resin. After washing the resin by DCM and DMF (DCM:DMF = 1:1), Fmoc deprotection was carried out with piperidine:DMF (1:4). Subsequent amino acids were coupled using an amino acid:HATU (2-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate):DIPEA (5:5:10). By adding 100 mL of trifluoroacetic acid (TFA):triisopropylsilane (TIPS):H2O (96:2:2), the mixture was stirred for 5 hours. Finally, peptide was cleaved from the resin and all protecting groups were removed. The majority of TFA was removed through vacuum evaporation. Peptide was precipitated with ice-cold diethyl ether and lyophilization.
:DMF = 1:1). The first amino acid was coupled to the resin for 24 h in DMF, with a 5-fold excess of amino acid and 10-fold excess of N,N-diisopropylethylamine (DIPEA). Then, 30 mL of MeOH was added to the mixture to cap any remaining reactive groups on the resin. After washing the resin by DCM and DMF (DCM:DMF = 1:1), Fmoc deprotection was carried out with piperidine:DMF (1:4). Subsequent amino acids were coupled using an amino acid:HATU (2-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate):DIPEA (5:5:10). By adding 100 mL of trifluoroacetic acid (TFA):triisopropylsilane (TIPS):H2O (96:2:2), the mixture was stirred for 5 hours. Finally, peptide was cleaved from the resin and all protecting groups were removed. The majority of TFA was removed through vacuum evaporation. Peptide was precipitated with ice-cold diethyl ether and lyophilization.
The two disulfide bonds were formed selectively using Trt and Acm protecting groups, Fmoc-Cys(Trt)-OH and Fmoc-Cys(Acm)-OH. The first disulfide bond was generated by oxidization in a solution of 0.1 M NH4HCO3 (pH 8.3). The second disulfide bond was formed by oxidization in a solution of 0.1 M I2 in acetonitrile (ACN). The products were purified by 2-step semi-preparative reversed-phase HPLC. First, the peptides were dissolved in solvent A (0.05% TFA in 90% H2O, 10% ACN), filtered through a 0.22 μm filter, and loaded into 20.5 mm C18 column. Then, the peptides were purified using a linear gradient from 10% to 50% solvent B (0.05% TFA, 10% H2O, 90% ACN) for 40 min, at 6 mL min−1 flow rate, and detected at 214 nm. The HPLC fractions were collected and analyzed by ESI-MS (Fig. S2 and S4†). Finally, the fraction containing desired products was lyophilized and its purity was verified using the analytical HPLC (Fig. S1 and S3†).
2.2 Circular dichroic (CD) spectra assay
CD spectra of the synthetic peptides were obtained at room temperature using a Jasco J-815 CD spectrometer with 0.5 cm path length. The data was collected between 195–260 nm with a scan speed of 100 nm min−1, a response time of 1 s, and a bandwidth of 1 nm. Peptide samples with a final concentration of 5 mM were prepared containing 50% 2,2,2-trifluoroethanol (TFE). The baseline scan was acquired by measuring the buffer alone.2.3 NMR analysis
RL-RhTx was prepared as a 1 mM sample in H2O/D2O (90:10 v/v) at pH 3.0. All spectra were acquired on a Bruker Avance III 600 MHz spectrometer equipped with a TCI cryoprobe at 298 K. Two-dimensional spectra (TOCSY and NOESY) were recorded at mixing times of 80 and 200 ms, respectively, and referenced to internal 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) at 0 ppm. Spectral analysis was performed with CCPNMR (Version 2.4.4).
2.4 Cell culture and transient transfection
Human embryonic kidney (HEK) 293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum at 37 °C with 5% CO2. The HEK-293 cells were passaged at 2 day intervals and plated on glass coverslips. Transient transfection was performed by using Lipofectamine 2000 (Invitrogen) and wild type human TRPV1 cDNAs (accession number NM_080704.3) and mutants of mouse TRPV1 were generously given by Yang's Lab.7 Patch-clamp recordings were performed between 18 and 36 h after transfection.2.5 Electrophysiology
Whole-cell recordings were performed by a HEKA EPC10 amplifier with PatchMaster software. Patch pipettes were prepared from borosilicate glass and fire-polished to resistance about ∼4.0 MΩ. Both bath and pipette solutions contained 130 mM NaCl, 0.2 mM EDTA, and 3 mM HEPES, pH 7.2–7.4, adjusted with NaOH. Membrane potential was held at 0 mV, and currents were elicited by a protocol consisting of a 300 ms step to +80 mV followed by a 300 ms step to −80 mV at 1 s intervals. One-way ANOVA was used to determine statistical significance for electrophysiology experiments between wild type group and different mutants groups.3 Results and discussion
3.1 Synthesis of RhTx and RL-RhTx
Both RhTx and RL-RhTx were synthesized by solid phase peptide synthesis method using Fmoc chemistry (Fmoc SPPS).29–31 Fmoc SPPS allows milder reaction conditions and low cost for peptide synthesis.32 The versatility of the method was proved.29 Synthesis of the two disulfide bonds was based on orthogonal protection strategy. The trityl (Trt) and acetamidomethyl (Acm) protection groups were used for the four cysteines to control the disulfide bond connectivity. In this way, the two disulfide bonds formed selectively (Fig. S1 and S2†). Finally, the two peptides were purified by RP-HPLC (Fig. 2). | ||
| Fig. 2 RP-HPLC chromatography of RhTx (A) and RL-RhTx (B). | ||
3.2 Structural characterization of RhTx and RL-RhTx
To characterize the conformations of RhTx and RL-RhTx, the circular dichroic (CD) spectra and NMR spectra were used (Fig. 3 and S3†). The CD spectra for the native RhTx was consistent with previous results.16 Despite the sequence reversal, the CD spectra of RL-RhTx remained similar to that of RhTx. Thus, considerable structural similarity existed between RL-RhTx and RhTx despite of significantly conformation perturbation.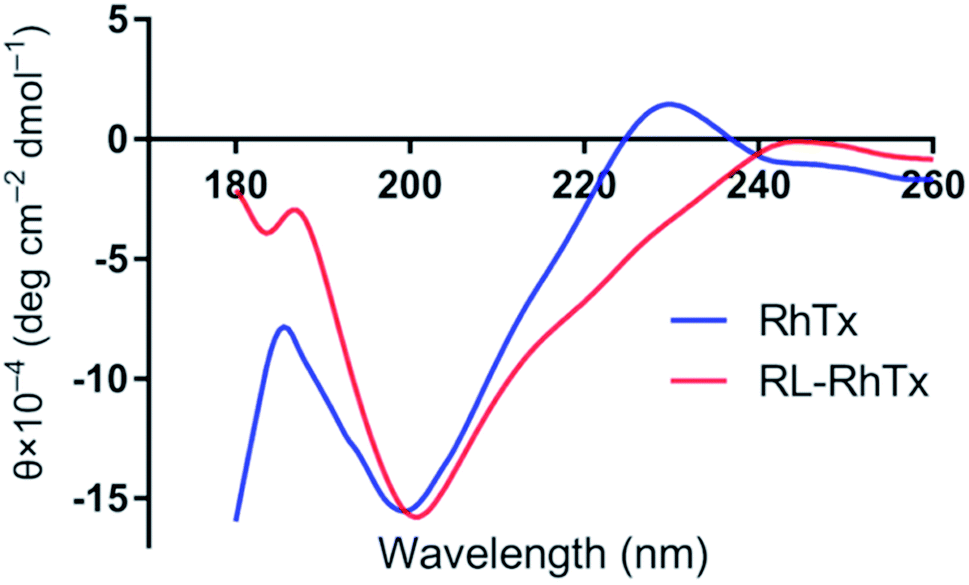 | ||
| Fig. 3 The CD spectra of RhTx and RL-RhTx. | ||
The 1D proton NMR spectrum of RL-RhTx shows very little dispersion in the amide region and a large amount of peak overlap (Fig. S3†). Two conformers of the peptide were observed in the 2D spectra; however, due to the limited dispersion and overlap, full assignment was not possible. Addition of a co-solvent (acetonitrile) along with temperature and pH changes did not improve the resolution nor altered the equilibrium of possible conformational exchange.
There was no exchange of the cis–trans proline bonds in either of the two conformers since NOEs were clearly observed between the Hαi-1–HδPi protons of Pro17 and Pro24 and their preceding residues. There are no defined secondary structural elements in either of the two conformers. Actually, RhTx has no defined secondary structure elements either (Fig. 1B). Due to the restraint from the disulfide bonds, it is likely that RL-RhTx form a compact globular structure similar to that described for RhTx.16
3.3 Interaction mode of RhTx and RL-RhTx with TRPV1
Using HEK293 cells overexpressing TRPV1, we tested the currents of 50 μM RL-RhTx and 50 μM RhTx on TRPV1. TRPV1 agonist capsaicin in 10 μM was used as control. Similar to RhTx and capsaicin, RL-RhTx is an activator for TRPV1 (Fig. 4A). And RL-RhTx has comparable TRPV1 activation response to RhTx at the concentration of 50 μM.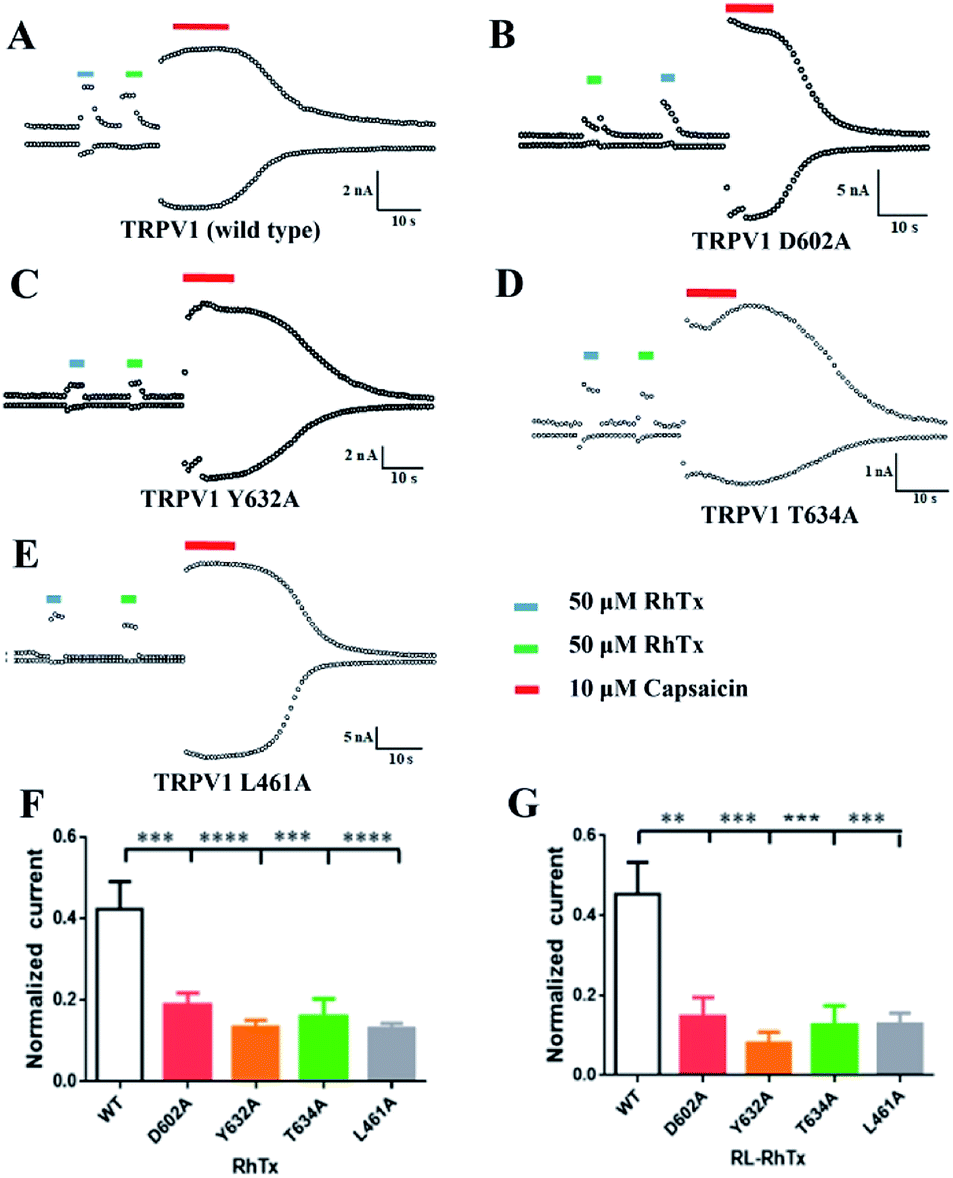 | ||
| Fig. 4 RhTx and RL-RhTx target the TRPV1 outer pore. (A–E) The whole-cell current response of wild type and point mutants induced by RhTx, RL-RhTx and capsaicin. (F) and (G) The normalized current of wild type, L461A, D602A, Y632A and T634A induced by RhTx or RL-RhTx. n = 5, data are the means ± SEM, **p < 0.01, ***p < 0.001, ****p < 0.0001 vs. wild type group. | ||
To compare the interaction modes of RhTx and RL-RhTx, we carried out site-specific mutagenesis. It was previously reported that four TRPV1 residues Leu461 in the S1–S2 linker, Thr634 in the pore helix, Asp602 and Tyr632 in the tunnel were critical for RhTx activation activity (Fig. 4).16 Therefore, we mutated each of these residues to alanine. And the currents of 50 μM RL-RhTx and 50 μM RhTx on wild type and mutants of TRPV1 were investigated. Again, capsaicin in 10 μM was used as control. The normalized current was calculated by the peptide-induced current divided by capsaicin-induced current (Fig. 4). The results showed that comparing with wild type TRPV1, normalized currents of the four mutants, including L461A, Y632A, D602A and T634A, were substantially reduced after applying the RhTx, which is consistent with previous reported results.16 For the RL-RhTx treatment group, the normalized currents of the four mutants were also significantly lower than that of the wild type. The same trend of the normalized current was found when RhTx and RL-RhTx were applied on TRPV1 and its mutants (Fig. 4). Thus, we concluded that the four mutated residues contributed to the interaction of RL-RhTx and RhTx with TRPV1, and may directly participate in the binding of the peptide with TRPV1. Both RhTx and RL-RhTx may bind in a similar mode. Since all of the four residues are located in the extracellular region, and Asp602, Tyr632 and Thr634 are close to the outer pore, RhTx and RL-RhTx may affect the entry of permeable ions into the pore. Since the two peptides are TRPV1 activator, RhTx and RL-RhTx may induce the conformational change after binding to the TRPV1 (Fig. 5).
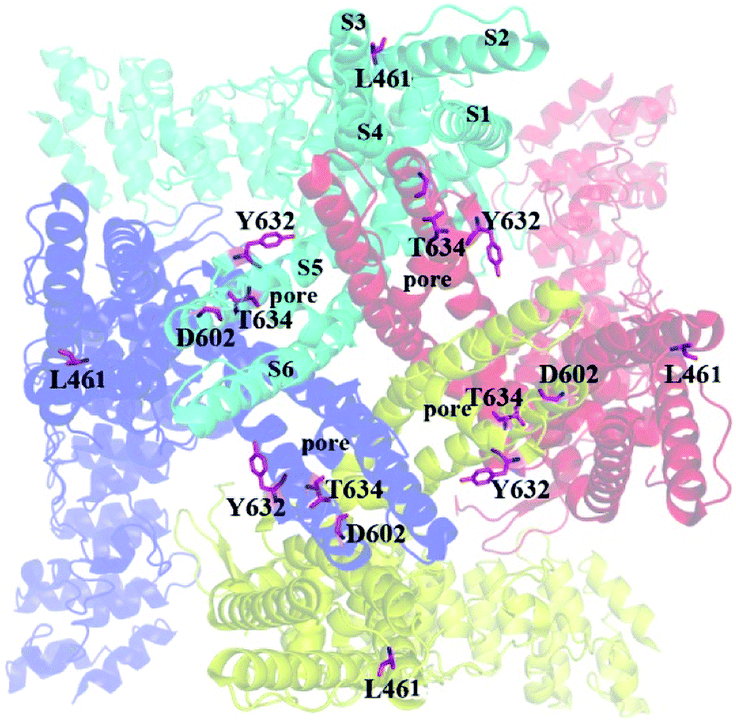 | ||
| Fig. 5 The location of key residues mutated in this study. The residues were shown in pink sticks. The view is along the pore from the extracellular sides. | ||
Based on the current study, RL-RhTx and RhTx have similar structural characteristics, and possess the same or similar binding sites on TRPV1. We assume that RL-RhTx may act on TRPV1 in the same mechanisms as RhTx. They may induce the conformation change and activate the ion channel by directly binding in the outer pore of TRPV1. Overall, the RL-RhTx provides a tool for the investigation on the structure–activity relationship of TRPV1 agonist, and the interaction mechanisms of TRPV1.
4 Conclusions
In summary, we obtained a novel TRPV1 activator, named RL-RhTx, by means of retro-isomerization strategy. RL-RhTx is a retro-isomer of centipede toxin RhTx. It was obtained by solid phase peptide synthesis. The CD and NMR spectrum were used to characterize the structure of the peptide. The results showed that the conformation of RL-RhTx is somehow similar to RhTx. Moreover, there are no defined secondary structural elements for RL-RhTx according to NMR spectra analysis, similar to RhTx. The activity evaluation shows that RL-RhTx and RhTx have comparable activation response to TRPV1. And four TRPV1 residues, Leu461, ASP602, Tyr632 and Thr634, have the same effect on the binding affinity of RhTx and RL-RhTx. Thus, RhTx and RL-RhTx probably adopt similar binding modes with TRPV1. The new peptide, RL-RhTx, provides a tool for the investigation of the structure–activity relationship of TRPV1 agonist. Besides, the results also indicate that the retro-isomerization strategy is suitable for designing RhTx derivatives as TRPV1 agonists.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the National Laboratory Director Fund (QNLM201709), the Scientific and Technological Innovation Project financially supported by the Qingdao National Laboratory for Marine Science and Technology (2018SDKJ0402), the National Natural Science Foundation (NSFC81703579), Natural Science Foundation of Shandong Province (ZR2017BH082), the Key Program of National Natural Science Foundation of China (NSFC41830535), the Fundamental Research Funds for the Central Universities (201762011), a grant from the National Natural Science Foundation of China (NSFC81502977) and the Taishan Scholars Program of Shandong, China.Notes and references
- F. Yang, X. Xiao, B. H. Lee, S. Vu, W. Yang, V. Yarov-Yarovoy and J. Zheng, Nat. Commun., 2018, 9, 2879 CrossRef PubMed.
- L. Darre and C. Domene, Mol. Pharm., 2015, 12, 4454–4465 CrossRef CAS PubMed.
- M. J. Caterina, M. A. Schumacher, M. Tominaga, T. A. Rosen, J. D. Levine and D. Julius, Nature, 1997, 389, 816–824 CrossRef CAS PubMed.
- I. Jardin, J. J. Lopez, R. Diez, J. Sanchez-Collado, C. Cantonero, L. Albarran, G. E. Woodard, P. C. Redondo, G. M. Salido, T. Smani and J. A. Rosado, Front. Physiol., 2017, 8, 392 CrossRef PubMed.
- I. S. Ramsey, M. Delling and D. E. Clapham, Annu. Rev. Physiol., 2006, 68, 619–647 CrossRef CAS PubMed.
- J. Zheng, Compr. Physiol., 2013, 3, 221–242 Search PubMed.
- M. Liao, E. Cao, D. Julius and Y. Cheng, Nature, 2013, 504, 107–112 CrossRef CAS PubMed.
- A. M. Maite, G. R. Sara and F. M. Antonio, Front. Mol. Biosci., 2018, 5, 73 CrossRef PubMed.
- L. Basso and C. Altier, Curr. Opin. Pharmacol., 2017, 32, 9–15 CrossRef CAS PubMed.
- A. D. Mickle, A. J. Shepherd and D. P. Mohapatra, Pharmaceuticals, 2016, 9, 72 CrossRef PubMed.
- Y. Gao, E. Cao, D. Julius and Y. Cheng, Nature, 2016, 534, 347–351 CrossRef CAS PubMed.
- M. Geron, R. Kumar, W. Zhou, J. D. Faraldo-Gomez, V. Vasquez and A. Priel, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, e11837–e11846 CrossRef CAS PubMed.
- C. J. Bohlen, A. Priel, S. Zhou, D. King, J. Siemens and D. Julius, Cell, 2010, 141, 834–845 CrossRef CAS PubMed.
- C. Bae, C. Anselmi, J. Kalia, A. Jara-Oseguera, C. D. Schwieters, D. Krepkiy, C. Won Lee, E. H. Kim, J. I. Kim, J. D. Faraldo-Gomez and K. J. Swartz, eLife, 2016, 5, e11273 CrossRef PubMed.
- M. Geron, A. Hazan and A. Priel, Toxins, 2017, 9, 326 CrossRef PubMed.
- S. Yang, F. Yang, N. Wei, J. Hong, B. Li, L. Luo, M. Rong, V. Yarov-Yarovoy, J. Zheng, K. Wang and R. Lai, Nat. Commun., 2015, 6, 8297 CrossRef CAS PubMed.
- Y. A. Andreev, S. A. Kozlov, S. G. Koshelev, E. A. Ivanova, M. M. Monastyrnaya, E. P. Kozlovskaya and E. V. Grishin, J. Biol. Chem., 2008, 283, 23914–23921 CrossRef CAS PubMed.
- M. Monastyrnaya, S. Peigneur, E. Zelepuga, O. Sintsova, I. Gladkikh, E. Leychenko, M. Isaeva, J. Tytgat and E. Kozlovskaya, Mar. Drugs, 2016, 14, e229 CrossRef PubMed.
- C. Adessi and C. Soto, Curr. Med. Chem., 2002, 9, 963–978 CrossRef CAS PubMed.
- P. Juvvadi, S. Vunnam and R. B. Merrifield, J. Am. Chem. Soc., 1996, 118, 8989–8997 CrossRef CAS.
- A. Atzori, A. E. Baker, M. Chiu, R. A. Bryce and P. Bonnet, PLoS One, 2013, 8, e68723 CrossRef CAS PubMed.
- C. Li, M. Pazgier, J. Li, C. Li, M. Liu, G. Zou, Z. Li, J. Chen, S. G. Tarasov, W. Y. Lu and W. Lu, J. Biol. Chem., 2010, 285, 19572–19581 CrossRef CAS PubMed.
- M. Chorev and M. Goodman, Acc. Chem. Res., 1993, 26, 266–273 CrossRef CAS.
- M. Goodman and M. Chorev, Acc. Chem. Res., 1979, 12, 173–179 CrossRef.
- J. Rai, Chem. Biol. Drug Des., 2007, 70, 552–556 CrossRef CAS PubMed.
- D. M. Vadukul, O. Gbajumo, K. E. Marshall and L. C. Serpell, FEBS Lett., 2017, 591, 822–830 CrossRef CAS PubMed.
- G. H. Zerze, F. H. Stillinger and P. G. Debenedetti, FEBS Lett., 2019, 13558 Search PubMed.
- C. Li, C. Zhan, L. Zhao, X. Chen, W. Y. Lu and W. Lu, Bioorg. Med. Chem., 2013, 21, 4045–4450 CrossRef CAS PubMed.
- O. Cheneval, C. I. Schroeder, T. Durek, P. Walsh, Y. H. Huang, S. Liras, D. A. Price and D. J. Craik, J. Org. Chem., 2014, 79, 5538–5544 CrossRef CAS PubMed.
- R. Yu, S. N. Kompella, D. J. Adams, D. J. Craik and Q. Kaas, J. Med. Chem., 2013, 56, 3557–3567 CrossRef CAS PubMed.
- R. Yu, H. S. Tae, N. Tabassum, J. Shi, T. Jiang and D. J. Adams, J. Med. Chem., 2018, 61, 4628–4634 CrossRef CAS PubMed.
- R. Behrendt, P. White and J. Offer, J. Pept. Sci., 2016, 22, 4–27 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1: High resolution mass spectrometry of RL-RhTx. Fig. S2: High resolution mass spectrometry of RhTx. Fig. S3: 1H NMR spectrum of RL-RhTx, 600 MHz, detail of amide region. See DOI: 10.1039/c9ra08829f |
| ‡ The authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |