
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanisms that control the adsorption–desorption behavior of phosphate on magnetite nanoparticles: the role of particle size and surface chemistry characteristics
Lei Hou a,
Qibin Lianga and
Fang Wang*b
a,
Qibin Lianga and
Fang Wang*b
aCollege of Ecology and Environment, Southwest Forestry University, Kunming, 650024, China
bTianjin Key Laboratory of Water Resources and Environment, Tianjin Normal University, Tianjin 300387, China. E-mail: wangfang@tjnu.edu.cn
First published on 13th January 2020
Abstract
Eutrophication caused by excessive phosphate discharge into surface water has raised wide concern, and the efficient removal of phosphates from wastewater using sorption methods is very important. In our study, magnetite particles with two different sizes and different surface characteristics were chosen as the sorbents to examine their adsorption and desorption behavior toward phosphate. Scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS) and N2 adsorption–desorption methods were used to characterize the morphological and surface chemical properties of the two differently sized magnetite particles. Adsorption kinetics and isotherm models (including the pseudo-first-order, Freundlich, Langmuir and Temkin models) were used to fit the experimental data, and to help with the mechanistic discussions. It was found that the nanometer-sized magnetite (nFe3O4) has a much higher surface area, larger pore volume, higher amounts of surface functional groups, and a lower point of zero charge (pHPZC) value than the micrometer-sized magnetite (Fe3O4). The adsorption kinetics show that reaching adsorption equilibrium in the case of nFe3O4 is much slower, and the particle size or surface characteristics of the magnetite may become the main factor determining the adsorption rate of the phosphate to magnetite in the rapid or slow adsorption step, respectively. nFe3O4 shows much stronger adsorption of phosphate compared to Fe3O4, which may be attributed to the larger surface area of the magnetite with a smaller particle size. In addition, the amount of functional groups and the surface electrical properties may also affect the adsorption of phosphate to magnetite by influencing the formation of the outer-sphere and/or inner-sphere complexes. The adsorption/desorption of phosphate to/from the magnetite decreases/increases with increasing pH, and the extent of change is more marked for nFe3O4. Increasing the ionic strength of the solution increases the adsorption of phosphate to the two differently sized magnetite particles, whereas the presence of humic acid only increases the adsorption of phosphate to Fe3O4. These trends may be caused by the different extents of change of the surface properties or the dispersion state of the two differently sized magnetite particles under different solution chemistry conditions. The results imply that when the synthesis of magnetite-based materials for phosphate sorption is performed, both the particle size and surface properties should be considered in order to realize the efficient and economical removal of phosphate from wastewater.
1. Introduction
The excessive use of phosphate, a life essential element, has caused serious water pollution problems such as the eutrophication of lakes, rivers and oceans, and the effective removal of phosphate from wastewater has always been an important issue.1 Several methods have been used to deal with this issue, including chemical precipitation,2,3 biological uptake,4 the sorption method5,6 and so on. Among these approaches, sorption stands out owing to its effectiveness, low-cost, ease of handling and the environmentally friendly method. Plenty of materials including natural minerals,7,8 waste products,9–11 metal oxide compounds,12–16 synthesized nanomaterials17–22 and so forth have been tested for phosphate adsorption. It has been proven that iron oxide-based materials are quite effective at adsorbing phosphate and that the magnetic iron oxide-based materials, such as magnetite are favored owing to their superiority in separation.5The adsorption of phosphate on iron oxide-based materials including ferrihydrite, goethite, hematite, magnetite, and other iron containing compound has been widely studied.6,8,16,23 Wang et al. revealed that the particle size of the ferrihydrite could control the adsorption kinetic behavior for phosphate, and further demonstrated the formation of a deprotonated bidentate complex between the phosphate and the surface of ferrihydrite could be the dominant adsorption mechanism.6 Yoon et al. also discussed the adsorption mechanism of phosphate to magnetite nanoparticles and indicated that phosphate could form the inner-sphere complexes with the surface of iron oxide through ligand exchange, which is affected by the amount of surface functional groups on the magnetite.5 The effects of solution chemistry, including the solution pH, coexistence of ions, humic acid (HA) and so forth, on the adsorption behavior of phosphate to metal oxide materials have also been studied, but the results varied among different studies.5,6,8,13,20,21 The adsorption of phosphate to iron oxide-based materials was found to decrease with the increasing pH, however, the extent of the decrease may depend on the point of the zero charge of the material.5,6,8 It was found that coexisting anions decrease the adsorption of phosphate to an iron–zirconium modified nanocomposite by competition adsorption,21 whereas an increase in the ionic strength of the solution can slightly enhance the adsorption of phosphate to zirconium oxide.13 In addition, the presence of HA was found to have a limited effect on the adsorption of phosphate to ferrihydrite and goethite,7 whereas Mahdavi et al. reported that 10 mg L−1 of HA decreased the adsorption of phosphate to copper oxide nanoparticles.15 Furthermore, several studies have tested the desorption percentage (i.e. the regeneration efficiencies) of phosphate from iron oxide-based materials, and rational recovery rates have been achieved.5,20 However, the combined effects of the particle size and the surface functional groups on the adsorption of phosphate towards magnetite particles have not been reported. Moreover, the effects of solution chemistry on the adsorption/desorption of phosphate to/from magnetites with different particle sizes and surface characteristics have not been systematically studied, and the dominating mechanisms have not been fully discussed. Understanding these issues is essential for designing a proper sorbent to remove phosphate from wastewater.
Therefore, nanometer-sized magnetite particles (nFe3O4) and micrometer-sized magnetite particles (Fe3O4) were selected as the sorbent, and the sacrificed sampling and batch experiment method was used to obtain the adsorption kinetics and adsorption isotherms of phosphate to magnetite, respectively. The objective of this study was to investigate the combined effect of the particle size and the amount of surface iron–oxygen groups on the adsorption behavior and to discuss the dominant mechanisms. In order to reveal the mechanisms, scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), and N2 sorption–desorption methods were used to characterize the morphology and surface properties of the magnetite. Meanwhile, the adsorption data were fitted to several models of adsorption kinetics and adsorption isotherms including the pseudo-first-order, Freundlich, Langmuir and Temkin models, and the fitting parameters were discussed. The effects of solution chemistry on the adsorption and desorption processes were also conducted, and the environmental implication of this study was then summarized.
2. Materials and methods
2.1 Chemicals and materials
Magnetite powders with nanometer-sized (nFe3O4) and micrometer-sized (Fe3O4) particle diameters were purchased from Shuitian Technology Co. (Shanghai, China). The two differently sized magnetite particles were synthesized using the sol–gel method. The particle size was tailored by varying the annealing temperature.24 HA (98%) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Other reagents of chemically pure grade, including monopotassium phosphate (KH2PO4), sodium chloride (NaCl), hydrochloric acid (HCl) and sodium hydroxide (NaOH) were all purchased from Guanghua Technology Co. (Guangzhou, China).2.2 Characterization of magnetite
The morphological structures and size dimensions of magnetites were explored using SEM (Sigma 500, Carl Zeiss Co., Germany), and the particle size was measured using Nano Measurer software (version 1.2, Fudan University, China). The surface element compositions of the magnetites were explored using XPS (Thermo K-Alpha, Thermo Fisher Scientific Inc., US), and the energy spectrum analysis of C1s and O1s was conducted using CasaXPS software (Version 2.3.0, Casa software Ltd., UK). Specific surface areas of the magnetites were obtained from the N2 adsorption–desorption isotherms using a physical adsorption instrument (ASAP 2020, Micrometrics Co., US) using the Brunauer–Emmett–Teller (BET) method, and the pore properties including the pore volume and size of the magnetite were calculated from the desorption isotherms using the Barrett–Joyner–Halenda (BJH) and Horvath–Kawazoe (HK) model, respectively. The point of zero charge values (pHPZC) of the magnetites were determined using the method reported by Kocharova et al.252.3 Adsorption experiments
The sacrificed sampling method was used to obtain the adsorption kinetics of phosphate to magnetite particles with different sizes. Based on the results of the preliminary experiment, a different mass was used for the two sized magnetite particles in order to guarantee the easy measurement of the aqueous concentration of a phosphate and the accuracy of the adsorption isotherm data. To start the experiment, 5 mg of nFe3O4 or 0.5 g of Fe3O4 were first added to a series of 40 mL amber glass vials containing 0.01 M NaCl solution. Then, a certain amount of KH2PO4 stock solution was added to make up the initial concentration of phosphate to 1 or 3 mg P L−1 for nFe3O4 and Fe3O4, respectively. Then, the vials were sealed and tumbled end-over-end at 5 rpm. At predetermined time intervals, certain vials were taken down and centrifuged at 3000 rpm for 5 min. The supernatant was withdrawn to analyze the concentration of freely dissolved phosphate, and then the vials were discarded. The adsorbed mass of phosphate to magnetite was calculated as the difference between the total mass and the mass in the dissolved phase.A batch experiment was conducted to obtain the adsorption isotherms of phosphate to magnetite with different particle sizes. First, a series of 40 mL amber glass vials each containing 10 mg nFe3O4 or 0.5 g Fe3O4 and 40 mL 0.01 M NaCl solution were prepared. Then, different amounts of the KH2PO4 stock solution were added to the vials to give initial concentrations of phosphate ranging from 0.5 to 20 mg P L−1. Then, the vials were sealed and tumbled end-over-end at 5 rpm for 3 d. After reaching equilibrium, the vials were centrifuged at 3000 rpm for 5 min, and the supernatant was withdrawn to analyze the concentrations of the freely dissolved phosphate. The adsorbed mass of phosphate was also calculated using a mass balance, using the same procedure as the kinetic experiment part.
Both the kinetic and adsorption isotherm data points were run in duplicate. The blank control samples showed negligible non-adsorptive losses, and the solution pH for the kinetic and adsorption isotherm experiment was set around neutral (7.00 ± 0.10).
2.4 Effects of solution chemistry
To consider the effects of the pH, ionic strength and HA on the adsorption and desorption of phosphate to magnetite particles with different sizes, a single point experiment in triplicate was conducted. A series of 40 mL amber glass vials each containing 10 mg nFe3O4 or 0.5 g Fe3O4 were prepared, and the background solution was varied for different purposes. For the experiment on the effect of the pH, the pH of the background solution was adjusted to a predetermined value ranging from 3 to 11 using HCl or NaOH solution. For the experiment on the effect of the ionic strength (IS), different NaCl background solutions with concentrations ranging from 0.01 to 1 M were used. For the experiment on the effect of HA, 5 and 10 mg L−1 HA background solutions were used. Then, a certain amount of KH2PO4 stock solution was added to the vials to give an initial concentration of phosphate of 1 mg P L−1. Then, the procedures for the tumbling, sampling and calculating the adsorbed mass of phosphate were performed using the same method as the experiment on the adsorption isotherms described above. Afterwards, the supernatant was removed as much as possible, and the corresponding background solution (e.g. 5 or 10 mg L−1 HA solution was used for the HA effect experiments) was added to the vials with magnetite residues. The vials were then tumbled at 5 rpm for 3 d to reach the desorption equilibrium. After centrifuging at 3000 rpm for 5 min, the supernatant was removed to analyze the desorbed concentration of phosphate, and the residue of the adsorbed mass of phosphate on the magnetite was calculated using the mass balance. The solution pH was around neutral (7.00 ± 0.10) for the effects of the IS and HA experiment.2.5 Determination of phosphate and data analysis
The concentrations of phosphate in the supernatant solution were measured using a spectrophotometric method modified from Murphy and Riley.26 This method depends on the formation of the complex during the reaction between the orthophosphate and molybdate, and ascorbic acid may reduce the complex and show absorption at the wavelength of 700 nm.The Freundlich, Langmuir and Temkin models were used to fit the adsorption isotherms of phosphate to the magnetite particles. The Freundlich model is given as eqn (1):
| Qe = KFCen | (1) |
The Langmuir model is given as eqn (2):
 | (2) |
The Temkin model is given as eqn (3):
 | (3) |
The adsorption kinetic data were fitted with a pseudo-first-order model. The model is given as eqn (4):
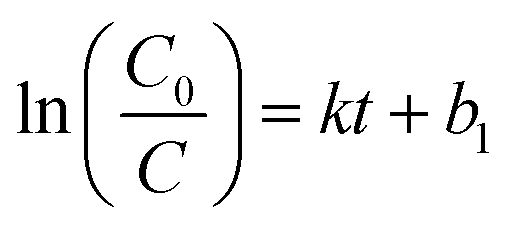 | (4) |
For the experiment on the effect of the solution chemistry, the solid–liquid distribution coefficient, Kd (L kg−1) was calculated to illustrate the effect of the solution chemistry conditions on the adsorption of phosphate on the magnetite particles. The desorption ratio (%) was calculated to compare the desorption trends of the phosphate from the magnetite particles in different solution chemistry conditions.
The solid–liquid distribution coefficient is given as eqn (5):
 | (5) |
The desorption ratio (DR) calculation equation is given as eqn (6):
 | (6) |
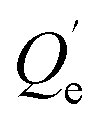 (mg kg−1) is the adsorption capacity when the desorption equilibrium is reached.
(mg kg−1) is the adsorption capacity when the desorption equilibrium is reached.
2.6 Statistical analysis
The solid–liquid distribution coefficients (Kd) and the desorption ratios (DR) were presented as the mean of three replicates with the standard error. Analysis of Duncan's multiple range tests were used to determine the significance of the difference for the Kd and DR values of the different groups (e.g. pH, ionic strength and HA). All of the statistical analysis was performed with SPSS software (version 19.0, IBM Co., US) and all of the figures were drawn using Origin (version 2017, OriginLab Co., US).3. Results and discussion
3.1 Characterization of the magnetite particles
The morphological characteristics of the magnetite particles with different sizes are shown in Fig. 1. Obvious differences in the particle size can be noted from the images. The nanometer-sized magnetite (nFe3O4) shows a diameter of 23.1 ± 3.4 nm (Fig. 1a), whereas the micrometer-sized magnetite (Fe3O4) shows a diameter of 0.35 ± 0.1 μm (Fig. 1b). Furthermore, the aggregation of the nFe3O4 particles is much tighter than that of Fe3O4, and the intervals of the nFe3O4 aggregates may form a pore structure, which Fe3O4 cannot. The structural properties of the two sized particles are compared in Table 1. It is not surprising that nFe3O4 has a much higher surface area and pore volume than that of Fe3O4. Furthermore, the average pore size of nFe3O4 is much smaller than that of Fe3O4. The structural properties are consistent with the findings in the SEM images. | ||
| Fig. 1 SEM images of magnetite particles with different sizes: (a) nanometer-sized magnetite particles; and (b) micrometer-sized magnetite particles. | ||
| Parameter | Material | ||
|---|---|---|---|
| Aspect | Details | nFe3O4 | Fe3O4 |
| a Determined using XPS.b Determined using BET and calculated using the BJH and HK models.c Determined using the method reported by Kocharova et al.25d k1 and k2 are the adsorption rate constants from 0–240 and 240–4320 min, respectively (see Fig. 3). | |||
| Surface composition (%)a | Fe | 23.34 | 16.12 |
| O | 39.09 | 35.74 | |
| C | 37.56 | 48.14 | |
| Structural propertiesb | Specific surface area (m2 g−1) | 106.50 | 5.93 |
| Pore volume (m3) | 0.295 | 0.030 | |
| Average pore size (nm) | 11.1 | 20.5 | |
| pHPZCc | 6.2 | 9.1 | |
| Kineticsd | k1 (min−1) | 1.2 × 10−3 ± 9.3 × 10−5 | 2.0 × 10−4 ± 8.9 × 10−5 |
| k2 (min−1) | 6.6 × 10−5 ± 6.3 × 10−6 | 5.3 × 10−5 ± 1.1 × 10−6 | |
| Freundlich model | KF (L kg−1) | 2431 ± 47 | 209 ± 9.5 |
| n | 0.202 ± 0.012 | 0.315 ± 0.021 | |
| R2 | 0.980 | 0.974 | |
| Langmuir model | A (mg L−1) | 0.208 ± 0.064 | 1.57 ± 0.35 |
| Q0 (mg kg−1) | 3454 ± 191 | 500 ± 27 | |
| R2 | 0.808 | 0.916 | |
| Temkin model | aT (L kg−1) | 162 ± 69 | 8.88 ± 2.47 |
| bT (kJ mol−1) | 0.0048 ± 0.0004 | 0.0254 ± 0.0019 | |
| R2 | 0.956 | 0.957 | |
The surface properties of the magnetite particles characterized using XPS are shown in Table 1. It was found that in addition to the existence of iron and oxygen, carbon also existed, and nFe3O4 shows a higher surface composition of iron and oxygen and a lower composition of carbon than that of Fe3O4. It should be noted that ethylene glycol was used in the preparation of the two sized magnetite particles, and therefore the relatively high content of C may be due to the residue of the organic reagent. Hasanpour et al. used the same method to synthesize the nanocomposite of Fe3O4–ZnO. From their characterization results using Fourier transform infrared spectroscopy (FTIR), the strong absorption peaks of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO (2366.99 cm−1) and CC (1430.24 cm−1) may indicate the residue of an organic reagent on the surface of the material.27 Based on the analysis results of the C1s and O1s binding energy and the corresponding carbon, oxygen and iron bonds on the magnetite surface (Fig. 2), the same surface composition was found for the two sized magnetite particles. However, the ratio of each chemical bond varies. For carbonaceous bonds, nFe3O4 has a greater amount of C–O and CO polar functional groups than that of Fe3O4. For oxygenous bonds, nFe3O4 also has a higher amount of iron–oxygen bonds than that of Fe3O4. The greater amount of oxygen involving bonds may result in a lower value for the point of zero charge (pHPZC). This is consistent with the measurement that shows that nFe3O4 exhibits a lower value of pHPZC (6.2) compared to that of Fe3O4 (9.1), which indicates that when the background solution is set at a neutral pH, the surface of the two sized magnetite particles carry the opposite charge.
CO (2366.99 cm−1) and CC (1430.24 cm−1) may indicate the residue of an organic reagent on the surface of the material.27 Based on the analysis results of the C1s and O1s binding energy and the corresponding carbon, oxygen and iron bonds on the magnetite surface (Fig. 2), the same surface composition was found for the two sized magnetite particles. However, the ratio of each chemical bond varies. For carbonaceous bonds, nFe3O4 has a greater amount of C–O and CO polar functional groups than that of Fe3O4. For oxygenous bonds, nFe3O4 also has a higher amount of iron–oxygen bonds than that of Fe3O4. The greater amount of oxygen involving bonds may result in a lower value for the point of zero charge (pHPZC). This is consistent with the measurement that shows that nFe3O4 exhibits a lower value of pHPZC (6.2) compared to that of Fe3O4 (9.1), which indicates that when the background solution is set at a neutral pH, the surface of the two sized magnetite particles carry the opposite charge.
 | ||
| Fig. 2 XPS analysis of the C1s and O1s binding energies and the corresponding carbon, oxygen and iron bonds on the surface of the magnetite particles with different sizes. (a) and (c) show the nanometer-sized magnetite particles, whereas (b) and (d) show the micrometer-sized magnetite particles. | ||
3.2 Adsorption of phosphate to the magnetite particles
The adsorption kinetics of phosphate to the two differently sized magnetite particles are shown in Fig. 3. It can be seen that it is much slower for nFe3O4 to reach the adsorption equilibrium than that for Fe3O4 (Fig. 3a). In addition, the Qe of nFe3O4 (5890 mg kg−1) is more than 30 times higher than that of Fe3O4 (184 mg kg−1). The adsorption kinetics data was fitted to a pseudo-first-order model. The results are shown in Fig. 3b, and the fitting parameters are summarized in Table 1. Wang et al.6 has reported that the adsorption progress of phosphate to the ferrihydrite surface could be divided into three stages: a relatively rapid adsorption step (the initial 20 min), followed by a slow adsorption step (from 20 to 240 min), and finally a very slow progression in the amount adsorbed (after 240 min). As there was no sampling point within the initial 20 min in our research, only two steps are discussed, a rapid adsorption step (0–240 min), followed by a relatively slow adsorption step (240–4320 min). The pseudo-first-order model can easily fit the two steps of the adsorption of phosphate for both nFe3O4 and Fe3O4. When the adsorption starts (0–240 min), the adsorption sites are quite adequate, with high rate constants (k1): 1.2 × 10−3 min−1 (nFe3O4) and 2.0 × 10−4 min−1 (Fe3O4). Clearly, nFe3O4 shows an adsorption rate that is almost 10 times greater than that of Fe3O4, which may be attributed to the much higher adsorption capacity required to reach for nFe3O4. As the adsorption proceeds (240–4320 min), the surface charge become more/less negative/positive, and the sites available for phosphate adsorption further decrease.6,28 Thus, the adsorption rate slows down, and the rate constants (k2) for this step also decreases with the increasing particle size from 6.6 × 10−5 min−1 (nFe3O4) to 5.3 × 10−5 min−1 (Fe3O4). It can be noted that compared with the k1, the k2 decreases more than 100 times for nFe3O4, whereas it only decreases 20 times for Fe3O4. Based on the literature studies, the slow adsorption of phosphate can be ascribed to the further accumulation of the negatively charged phosphate at the external surface of the iron oxide-based particles, which are also negatively charged, and to the diffusion into the internal pore structure formed between the aggregates of the iron oxide-based particles.6,29 This may account for the slower adsorption kinetics and higher decreasing times of the adsorption rate constant of nFe3O4 compared to that of Fe3O4, as nFe3O4 has more pore structures and a more negatively charged surface as indicated by the characterization results of the pore volume and the pHPZC of the two differently sized magnetite particles. | ||
| Fig. 3 The adsorption kinetics of phosphate on magnetite particles with different sizes (a), and the corresponding fitting results of the pseudo-first-order model using two adsorption steps (fast versus slow adsorption) (b). | ||
It has been reported that both the amount of adsorption sites and the surface charge may control the adsorption kinetics of phosphate to the iron oxide-based surface.6,30 Magnetite particles with a nanometer size obviously have a greater amount of adsorption sites for phosphate compared to Fe3O4, indicated by the much higher surface area. The final pH of the background solution was around neutral for the adsorption kinetic experiment. According to the pHPZC values of the two differently sized magnetite particles, nFe3O4 is negatively charged whereas Fe3O4 is positively charged. The phosphate is negatively charged, therefore, if the electrostatic interaction dominates the adsorption process it may be easier for it to adsorb onto the surface of Fe3O4 compared to that of nFe3O4. Thus, for the rapid adsorption step, the rate constant of nFe3O4 is much higher than that of Fe3O4. It can be speculated that in this step, the amount of adsorption sites is more important for control of the adsorption progress. When it proceeds to the slow adsorption step, the rate constant of nFe3O4 is just a little bit higher than that of Fe3O4, which indicates that the electrostatic interaction (attraction between the phosphate and the positively charged surface of Fe3O4) may play the dominant role and will counterbalance part of the weak adsorption because of the smaller amount of adsorption sites for Fe3O4.
The adsorption isotherms of phosphate for the two sized magnetite particles are shown in Fig. 4, and the fitting parameters of the chosen Freundlich, Langmuir and Temkin models are summarized in Table 1. It was found that the adsorption of phosphate on nFe3O4 is at least one order of magnitude higher than that on Fe3O4 (Fig. 4a). Compared to the Langmuir model, the adsorption of phosphate on magnetite can be better described by the Freundlich and Temkin models, which may be attributed to the heterogeneity of the magnetite surface.8 Parameters indicating the sorption capacity from the three models including KF, Q0 and aT all show the same trend, in that they decrease with the increasing size of the magnetite particles. This is consistent with the trend for the adsorption isotherms. The nonlinear index of the Freundlich model shows very low values (0.202 for nFe3O4 and 0.315 for Fe3O4) indicating a strong heterogeneous distribution of the adsorption sites. Nanometer-sized magnetite particles may have more hydroxyls attached to the iron or carbon atoms to interact with the phosphate compared to that of the micrometer-sized particles, which can be inferred from the results of the XPS characterization (Fig. 2). The values of the bT (Temkin model parameter) can indicate the adsorption mechanism, and if it is in the range of 8 to 16 kJ mol−1, the sorption is governed by ion exchange.30 The values of fitting bT are far less than 8 kJ mol−1 for the two differently sized particles, thus the adsorption process may involve both physical and chemical adsorption mechanisms.21
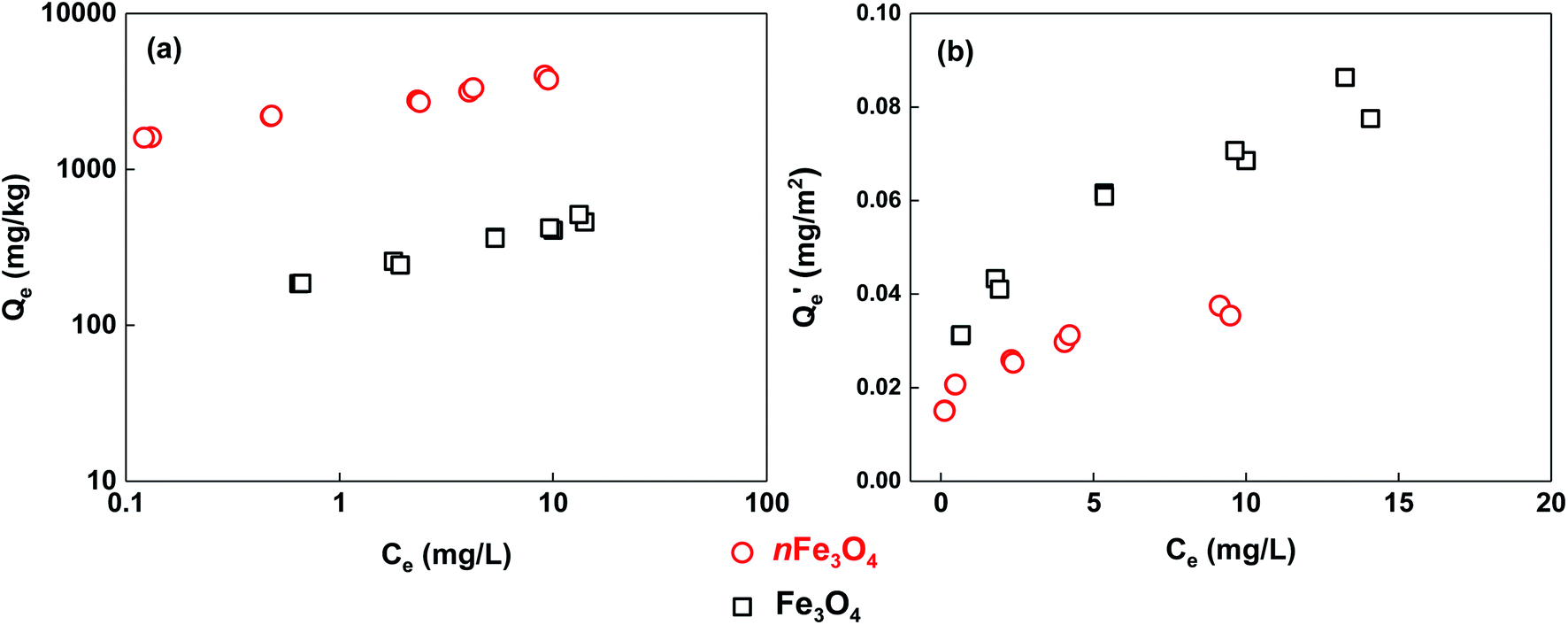 | ||
| Fig. 4 The adsorption isotherms of phosphate on magnetite particles with different sizes (a), and the data normalized using the BET specific surface area of the magnetite particles (b). | ||
To better understand the mechanisms dominating the adsorption of phosphate to these two differently sized magnetite particles, the BET-surface-area normalized adsorption isotherms are shown in Fig. 4b. It can be noted that the adsorption of phosphate to Fe3O4 becomes stronger than that to nFe3O4. Meaning that for the same unit area of adsorption surface of the two differently sized magnetite particles, phosphate has a higher affinity to adsorb on the surface of Fe3O4 compared to that of nFe3O4, whereas the adsorption capacity (Qe) is only 1.5 to 2 times higher when compared at the same aqueous phosphate concentration (Ce). In addition to the surface area (i.e. the amount of adsorption sites) of the magnetite particles, there are two further effects which may control the adsorption process. The first one is the electrostatic interaction which drives the formation of the outer-sphere complex between the phosphate and the surface of the magnetite particles.5,31 At certain pH conditions (around neutral) for the adsorption isotherm experiment, phosphate and nFe3O4 are both negatively charged, whereas Fe3O4 is positively charged, which is speculated from their values of pHPZC. The electrostatic attraction dominates the adsorption of phosphate to Fe3O4, whereas the electrostatic repulsion dominates that to nFe3O4, which makes phosphate form an outer-sphere complex on the surface of the micrometer-sized particles more easily and adsorb more strongly. The other effect is the formation of the inner-sphere complexes including the monodentate, bidentate, mononuclear or binuclear complexes through phosphate replacing hydroxyl on the surface of the magnetite particles.5,31–33 According to the results of XPS characterization, the magnetite particles with a nanometer-size have higher contents of oxygen and iron–oxygen bonds than the micrometer-sized particles, and may provide further amounts of iron containing groups to form inner-sphere complexes with phosphate. However, it can be inferred from the normalized adsorption isotherms that the stronger inner-sphere-complex effect between nFe3O4 and phosphate does not overcome the outer-sphere-complex effect between Fe3O4 and phosphate.
3.3 Effects of solution chemistry
The effects of the solution pH on the adsorption/desorption of phosphate to/from magnetite particles with different sizes are shown in Fig. 5a and b. A general trend can be found in which the adsorption of phosphate to nFe3O4 decreases significantly with the increasing pH (p < 0.05), but no such obvious trend can be found for the adsorption of phosphate to Fe3O4. The desorption ratios of the phosphate from these two differently sized magnetite particles show the same trend that increases with the increasing pH values, and the extent of the increase is more marked for the nanometer-sized magnetite particles. | ||
| Fig. 5 The effects of solution chemistry on the adsorption ((a), (c) and (e)) and desorption ((b), (d) and (f)) of phosphate on/from the magnetite particles with different sizes. Bars of the same style are marked with different letters to indicate significant differences (p < 0.05). | ||
It has been reported that the solution pH can alter the adsorption/desorption of phosphate to/from iron oxide-based materials by changing the amount of charge and the charge characteristics of the phosphate and the surface of the materials and further changing the dominant adsorption/desorption mechanisms.5,13,21,34,35 When the solution pH is around 3, the electrostatic attraction between the positively charged magnetite surface and the negatively charged phosphate will drive the formation of an outer-sphere complex,31 which provides the strong adsorption and weak desorption behavior of the phosphate. When the solution pH rises, the amount of positive charge on the surface of the magnetite particles with both sizes decreases, and the surface will finally become negatively charged when the solution pH is above the value of pHPZC (6.2 for nFe3O4 and 9.1 for Fe3O4). When the solution pH is above 6.2, the electrostatic attraction will change into electrostatic repulsion, which will significantly decrease/increase the adsorption/desorption of the phosphate to/from nFe3O4. The higher the pH, the lower/higher the adsorption/desorption of phosphate to/from the nanometer-sized magnetite particles. The effect of the solution pH is more powerful for the nanometer-sized magnetite particles than for the micrometer-sized magnetite particles. This is because the nFe3O4 has larger amounts of hydroxyl which can be deduced from the higher surface oxygen content and also the higher ratios of the C–O and Fe–O bond for nFe3O4 compared to that for Fe3O4 in the XPS analysis (Table 1 and Fig. 2). Furthermore, the protonation and deprotonation of the radical can alter the charge characteristics of the surface and further alter the related adsorption/desorption behavior.5,21 Moreover, phosphate still has a certain degree of adsorption on magnetite when the solution pH is above the pHPZC and the outer-sphere complex effect cannot be formed. This is because under a certain pH, the protonated iron surface groups with phosphate can form an inner-sphere complex such as the bidentate binuclear phosphate complex,32,33 which may result in a reduced desorption of phosphate from the magnetite particles.
The effects of the IS of the solution on the adsorption/desorption of phosphate to/from magnetite particles are shown in Fig. 5c and d. It can be found that the adsorption of phosphate on both sizes of magnetite increases with the increasing IS (p < 0.05), and changing the IS of the solution does not significantly alter the desorption behavior of the phosphate on both of the sizes of magnetite (p > 0.05). nFe3O4 shows a higher adsorption of phosphate for all of the three IS conditions compared to that of Fe3O4 respectively. It has been reported that if the adsorption of phosphate to the metal oxide material decreases with the increasing IS of the solution, the adsorbed phosphate may form outer-sphere complexes with the surface of the material, and if the adsorption increases with the increasing solution IS, inner-sphere surface complexes may be formed instead.13,14 Based on the higher ratios of the surface Fe–O and C–O bond and the lower values of the pHPZC of the nanometer-sized magnetite particles than that of the micrometer-sized magnetite particles from the XPS and pHPZC analysis, the neutral solution pH of the IS effect experiment will result in the surface of nFe3O4 being negatively charged, whereas it will result in the surface of Fe3O4 being positively charged. In these conditions, the inner-sphere complexes may be dominantly formed between the adsorbed phosphate and the nFe3O4 surface, whereas the outer-sphere complexes may be formed on the surface of Fe3O4.31–33 Thus, the increase in the solution IS will improve the formation of the protonated Fe–OH groups, which may favor the further formation of the inner-sphere complex between the adsorbed phosphate and the surface functional groups of nFe3O4, and then the adsorption of phosphate will be increased.13,36 However, the abovementioned effect is not significant for the formation of the outer-sphere complex, resulting in a reduced promoting effect in the adsorption of phosphate to Fe3O4. The increasing solution IS may not change the amount of adsorption sites and the dominant adsorption mechanism, thus the desorption ratios do not change significantly.
The effects of HA on the adsorption/desorption of phosphate to/from magnetite particles are shown in Fig. 5e and f. The presence of HA does not change the adsorption of phosphate on nFe3O4, but increases that on Fe3O4, especially when the HA concentration is 10 mg L−1. The desorption behavior is not significantly changed for both sizes of magnetite particles (p > 0.05). It has been reported that HA may decrease the adsorption of phosphate to metal oxides by covering the adsorption sites.15 At the same time, the presence of HA may help disperse the aggregates of the particles and expose more adsorption sites, which may result in increasing the adsorption.37,38 Also, based on the higher ratios of the surface Fe–O and C–O bonds and the lower values of pHPZC of the nanometer-sized magnetite particles compared to that of the micrometer-sized magnetite particles from XPS and pHPZC analysis, the neutral solution pH for the HA effect experiment makes the surface of nFe3O4 negatively charged, whereas the surface of Fe3O4 becomes positively charged. Thus, the negatively charged HA will interact more strongly with the positively charged Fe3O4, and the dispersion effect which can increase the available adsorption sites for phosphate may be stronger than the effect of covering the adsorption sites which can decrease the adsorption. Interestingly, the improvement resulting from HA on the adsorption of phosphate to Fe3O4 only shows a significant effect (p < 0.05) when the HA concentration is 10 mg L−1 rather than 5 mg L−1. This fact further demonstrates that only a certain amount of HA can expose a significant amount of adsorption sites for phosphate and increase the adsorption significantly. However, the interaction between both the negatively charged nFe3O4 and HA is much weaker, thus HA shows no obvious effects on the adsorption behavior of the phosphate to nFe3O4. Moreover, as HA does not alter the dominant interaction effect between the magnetite and phosphate, the desorption of phosphate from the two sized magnetite particles with the presence of HA does not change significantly.
3.4 Environmental implications
The removal of phosphate from wastewater using iron oxide-based materials as the sorbent has always been a good choice. If a sorbent that has fast sorption kinetics, a high sorption capacity, easy regeneration and an easy separation approach, that could also retain these advantages in a wide range of solution chemistry conditions (e.g. wastewater with extreme acidity or alkalinity, and very high concentrations of ionic strength and/or natural organic matter) could be synthesized, the problems caused by phosphate will be solved perfectly. However, based on our research, both the particle size and the surface chemistry properties of magnetite could control its capability to adsorb phosphate (summarized in Fig. 6). Thus, it is difficult to synthesize such an ideal material. Although magnetite with nanometer-sized particles has a very strong adsorption capacity for phosphate, it takes much more time to reach the adsorption equilibrium, and the solution chemistry conditions (e.g. the solution pH and IS) have a great effect on its adsorption and desorption behavior. Furthermore, it has been reported that the nanometer-sized particles of magnetite are very hard to separate from aqueous solution using the magnetic method as the magnetic force decreases with the decreasing size of the particles.39 In order to conquer these disadvantages, significantly more effort should be made and the benefits brought by “nanometer technologies” may be decreased, not to mention the price disadvantages of nanometer materials.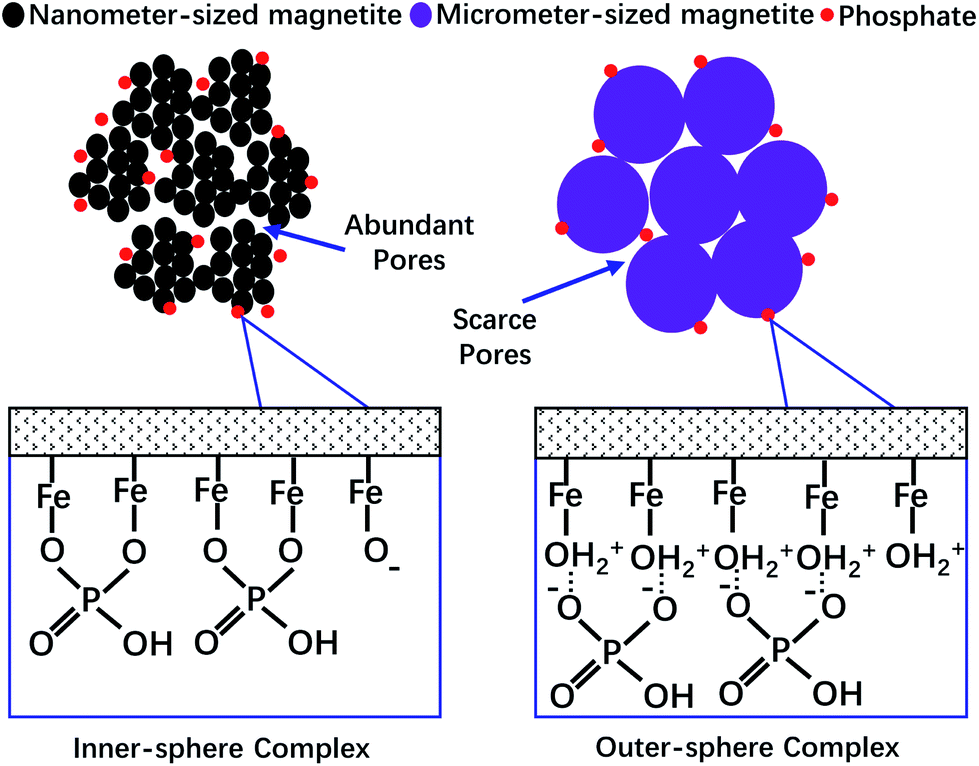 | ||
| Fig. 6 A schematic diagram of the mechanisms controlling the adsorption of phosphate to magnetite with different particle sizes and surface chemical characteristics. | ||
Therefore, in order to realize the effective and green removal of phosphates from wastewater, the proper surface area, the amount of surface functional groups and the charge characteristics of the sorbent should all be considered to improve the regeneration capacity, resistance to the interference of solution chemistry conditions, and excellent magnetic separation abilities in future research. Moreover, in order to discuss the sorption mechanisms dominated by sorbent properties more precisely and to avoid the uncertainties brought about by different synthesis processes or the conditions of the sorbents, choosing sorbents with different properties, but prepared using the same synthesis processes or conditions as possible is another major concern for the future research.
4. Conclusions
Nanometer and micrometer sized magnetite particles were chosen for studies of their adsorption kinetics and isotherms towards phosphate. It was found that the nanometer-sized magnetite has a higher surface area, larger pore volume, more abundant surface oxygen and iron containing functional groups and a lower pHPZC value compared to the micrometer-sized magnetite. Phosphate adsorption equilibrium is reached faster in the case of Fe3O4 than nFe3O4, and the amounts of sorption sites and the electrostatic interactions may become the main factors determining the adsorption rate of phosphate on magnetite in the fast-sorption and slow-sorption steps, respectively. The adsorption of phosphate on nFe3O4 is one order of magnitude higher than that on Fe3O4. In addition, the dominant effects of the sorbent surface area, the electrostatic interactions and iron–oxygen–phosphate complex formation may be valid mechanisms. The effects of the solution chemistry (e.g., solution pH, and concentrations of IS and HA) on the adsorption/desorption of phosphate on magnetite vary with particle size. This may be attributed to differences in the amount of surface functional groups, the ability to form an outer-sphere and/or inner-sphere complex, and the interaction affinity with HA between these two differently sized magnetite particles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study is supported by the National Natural Science Foundation of China [21607120, 21707101], the Application and Fundamental Research Project of Yunnan Province, China [2016FD042, 2016FB072], and the Construction Funds of First-class Subject (Ecology) in Yunnan Province, China.References
- D. J. Conley, H. W. Paerl, R. W. Howarth, D. F. Boesch, S. P. Seitzinger, K. E. Havens, C. Lancelot and G. E. Likens, Science, 2009, 323, 1014–1015 CrossRef CAS PubMed.
- P. Wilfert, P. S. Kumar, L. Korving, G. J. Witkamp and M. C. van Loosdrecht, Environ. Sci. Technol., 2015, 49, 9400–9414 CrossRef CAS PubMed.
- D. Guaya, C. Valderrama, A. Farran, C. Armijos and J. L. Cortina, Chem. Eng. J., 2015, 271, 204–213 CrossRef CAS.
- H. Monclus, J. Sipma, G. Ferrero, I. Rodriguez-Roda and J. Comas, Bioresour. Technol., 2010, 101, 3984–3991 CrossRef CAS PubMed.
- S.-Y. Yoon, C.-G. Lee, J.-A. Park, J.-H. Kim, S.-B. Kim, S.-H. Lee and J.-W. Choi, Chem. Eng. J., 2014, 236, 341–347 CrossRef CAS.
- X. Wang, W. Li, R. Harrington, F. Liu, J. B. Parise, X. Feng and D. L. Sparks, Environ. Sci. Technol., 2013, 47, 10322–10331 CrossRef CAS PubMed.
- O. K. Borggaard, B. Raben-Lange, A. L. Gimsing and B. W. Strobel, Geoderma, 2005, 127, 270–279 CrossRef CAS.
- X. Wang, F. Liu, W. Tan, W. Li, X. Feng and D. L. Sparks, Soil Sci., 2013, 178, 1–11 CrossRef CAS.
- K. Sakadevan and H. J. Bavor, Water Res., 1998, 32, 393–399 CrossRef CAS.
- K. C. Cheung and T. H. Venkitachalam, Chemosphere, 2000, 41, 243–249 CrossRef CAS PubMed.
- J. Chen, H. Kong, D. Wu, Z. Hu, Z. Wang and Y. Wang, J. Colloid Interface Sci., 2006, 300, 491–497 CrossRef CAS PubMed.
- X.-h. Guan, G.-h. Chen and C. Shang, J. Environ. Sci., 2007, 19, 312–318 CrossRef CAS.
- H. Liu, X. Sun, C. Yin and C. Hu, J. Hazard. Mater., 2008, 151, 616–622 CrossRef CAS PubMed.
- S. A. Kang, W. Li, H. E. Lee, B. L. Phillips and Y. J. Lee, J. Colloid Interface Sci., 2011, 364, 455–461 CrossRef CAS.
- S. Mahdavi and D. Akhzari, Clean Technol. Environ. Policy, 2015, 18, 817–827 CrossRef.
- S. Moharami and M. Jalali, Environ. Prog. Sustainable Energy, 2014, 33, 1209–1219 CAS.
- Y. Su, H. Cui, Q. Li, S. Gao and J. K. Shang, Water Res., 2013, 47, 5018–5026 CrossRef CAS PubMed.
- Y.-F. Lin, H.-W. Chen, Y.-C. Chen and C.-S. Chiou, J. Taiwan Inst. Chem. Eng., 2013, 44, 45–51 CrossRef CAS.
- G. Pan, L. Li, D. Zhao and H. Chen, Environ. Pollut., 2010, 158, 35–40 CrossRef CAS PubMed.
- A. Abo Markeb, A. Alonso, A. D. Dorado, A. Sanchez and X. Font, Environ. Technol., 2016, 37, 2099–2112 CrossRef CAS PubMed.
- W. Xiong, J. Tong, Z. Yang, G. Zeng, Y. Zhou, D. Wang, P. Song, R. Xu, C. Zhang and M. Cheng, J. Colloid Interface Sci., 2017, 493, 17–23 CrossRef CAS PubMed.
- M. Kondalkar, U. Fegade, S. Attarde and S. Ingle, J. Dispersion Sci. Technol., 2018, 39, 1635–1643 CrossRef CAS.
- D. Martinez-Fernandez, D. Bingol and M. Komarek, J. Hazard. Mater., 2014, 276, 271–277 CrossRef CAS.
- J. Xu, H. Yang, W. Fu, K. Du, Y. Sui, J. Chen, Y. Zeng, M. Li and G. Zou, J. Magn. Magn. Mater., 2007, 309, 307–311 CrossRef CAS.
- N. Kocharova, T. Aaritalo, J. Leiro, J. Kankare and J. Lukkari, Langmuir, 2007, 23, 3363–3371 CrossRef CAS PubMed.
- J. Murphy and J. P. Riley, Anal. Chim. Acta, 1962, 27, 31–36 CrossRef CAS.
- A. Hasanpour, M. Niyaifar, M. Asan and J. Amighian, J. Magn. Magn. Mater., 2013, 334, 41–44 CrossRef CAS.
- Y. Arai and D. L. Sparks, J. Colloid Interface Sci., 2001, 241, 317–326 CrossRef CAS.
- I. R. Willett, C. J. Chartres and T. T. Nguyen, Eur. J. Soil Sci., 1988, 39, 275–282 CrossRef CAS.
- A. M. El-Kamash, A. A. Zaki and M. A. El Geleel, J. Hazard. Mater., 2005, 127, 211–220 CrossRef CAS PubMed.
- T. J. Daou, S. Begin-Colin, J. M. Grenèche, F. Thomas, A. Derory, P. Bernhardt, P. Legaré and G. Pourroy, Chem. Mater., 2007, 19, 4494–4505 CrossRef CAS.
- H. Zeng, B. Fisher and D. E. Giammar, Environ. Sci. Technol., 2008, 42, 147–152 CrossRef CAS PubMed.
- B. Pan, J. Wu, B. Pan, L. Lv, W. Zhang, L. Xiao, X. Wang, X. Tao and S. Zheng, Water Res., 2009, 43, 4421–4429 CrossRef CAS PubMed.
- I. de Vicente, A. Merino-Martos, L. Cruz-Pizarro and J. de Vicente, J. Hazard. Mater., 2010, 181, 375–381 CrossRef CAS PubMed.
- E. Oguz, J. Hazard. Mater., 2004, 114, 131–137 CrossRef CAS PubMed.
- R. Giesler, T. Andersson, L. Lovgren and P. Persson, Soil Sci. Soc. Am. J., 2005, 69, 77–86 CAS.
- F. Mohd Omar, H. Abdul Aziz and S. Stoll, Sci. Total Environ., 2014, 468–469, 195–201 CrossRef CAS PubMed.
- D. Dickson, G. Liu, C. Li, G. Tachiev and Y. Cai, Sci. Total Environ., 2012, 419, 170–177 CrossRef CAS PubMed.
- S. C. Tang and I. M. Lo, Water Res., 2013, 47, 2613–2632 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |