
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of an isatin derivative on tumor cell migration and angiogenesis
Yunsong Chang†
ab,
Yuan Yuan† a,
Qian Zhanga,
Yao Ronga,
Yang Yanga,
Ming Chic,
Zhen Liua,
Yongmin Zhang*ad,
Peng Yu*a and
Yuou Teng*a
a,
Qian Zhanga,
Yao Ronga,
Yang Yanga,
Ming Chic,
Zhen Liua,
Yongmin Zhang*ad,
Peng Yu*a and
Yuou Teng*a
aChina International Science and Technology Cooperation Base of Food Nutrition/Safety and Medicinal Chemistry, College of Biotechnology, Tianjin University of Science and Technology, Tianjin 300457, China. E-mail: ryongmin.zhang@upmc.fr; yupeng@tust.edu.cn; tyo201485@tust.edu.cn
bJecho Biopharmaceuticals Co., Ltd., No. 2633 ZhongBinDaDao, Tianjin Eco-City, Tianjin, China
cQinghai Light Industry Institute Co., Ltd., Xining, 810000, Qinghai, China
dSorbonne Université, Institut Parisien de Chimie Moléculaire, CNRS UMR 8232, 4 Place Jussieu, 75005 Paris, France
First published on 8th January 2020
Abstract
Compound 5-61, a 5-(2-carboxyethenyl)isatin derivative was previously shown to have potent anticancer activity. Its effect on angiogenesis was further explored in this study. Notably, 5-61 showed selective cytotoxicity against liver hepatocellular carcinoma HepG-2 cells (IC50 = 7.13 nM). 5-61 powerfully induced apoptosis and G2/M phase arrest as well as inhibited the migration of HepG2 cells. Additionally, 5-61 clearly diminished tube formation and the actin arrangement in HUVECs. The physiological anti-angiogenic effects of 5-61 were further assessed by chick chorioallantoic membrane assays in vivo. The effects exerted by 5-61 were found to be mediated by VEGF along with its downstream signaling pathways including the PI3K/Akt/mTOR pathway and mitogen-activate protein kinase pathways (ERK). These results suggested that 5-61 is a potential tumor angiogenesis inhibitor that functions by interrupting the auto-phosphorylation of AKT, mTOR, and ERK 1/2.
Introduction
Isatin(indole-2,3-dione) is considered one of the most structurally versatile heterocycles and its core structure is present in many bioactive agents and pharmaceutical agents.1,2 Isatin has been incorporated into many synthetic drugs such as indirubin and sunitinib, which have been approved for treating various diseases.3–5 Isatin derivatives exhibit a wide spectrum of pharmacological activities including anti-fungal,6 anti-convulsant,7 anti-inflammatory,8 anti-depressant,9 anti-cancer,10 anti-microbial,11 anti-bacterial,1 and anti-human immunodeficiency virus12 activities.As destruction of ecological equilibrium and environmental deterioration has occurred worldwide, malignant tumors and other diseases have become more common, threatening human health. Therefore, new antitumor agents with high efficiency and low toxicity are urgently needed. Among the various anticancer drugs developed, targeting angiogenesis-related mechanisms is an attractive option for slowing or halting cancer progression.13 Angiogenesis is considered a hallmark of tumor progression as it supplies nutrients and oxygen for tumor cell growth and provides a route for tumor cell metastasis.14 Angiogenesis is regulated by multiple angiogenic stimulators including vascular endothelial growth factor (VEGF), hypoxia-inducible factor, fibroblast growth factor. During tumor neovascularization, the most prominent positive regulator, VEGF, shows high expression and functions by binding to VEGFR-2, which results in the development and maintenance of a vascular network and promotion of cancer cell survival, proliferation, adhesion, and migration.13,15,16 Apatinib, a novel VEGFR2 inhibitor approved for terminal gastric cancer treatment, significantly blocks the effects of the intracellular VEGF signaling pathway.17,18 Another small molecule anti-angiogenic agent, sunitinib, inhibits VEGF-dependent mitogenesis of human umbilical vein endothelial cells (HUVECs) which apparently inhibits tumor growth in different tumor types.19 However, previous studies have shown that these anti-angiogenic drugs are associated with adverse reactions such as teratogenicity and proteinuria.20 Therefore, exploring drugs that specifically target the VEGF pathway with high efficacy and few side effects may be a prospective strategy for cancer chemoprevention and therapy.
Previously, we designed and synthesized a series of 5-(2-carboxyethenyl)isatin derivatives and evaluated their cytotoxicity. We found that combining a 5-[trans-2-(methoxycarbonyl) ethen-1-yl] group and 1-(4-methoxybenzyl) group in isatin (referred to as compound 5-61, Fig. 1A) significantly enhanced its antitumor activity in vitro.21 However, the underlying antitumor mechanism and angiogenic effects exerted by this molecule remained unclear. In this study, we examined effects of compound 5-61 on 2 different cell lines: (1) HepG2 cells for the inhibition of growth, proliferation, apoptosis, and migration in vitro and (2) HUVECs for the inhibition of angiogenesis in vitro. We also studied its anti-angiogenic effects using a chick chorioallantoic membrane (CAM) model in vivo.
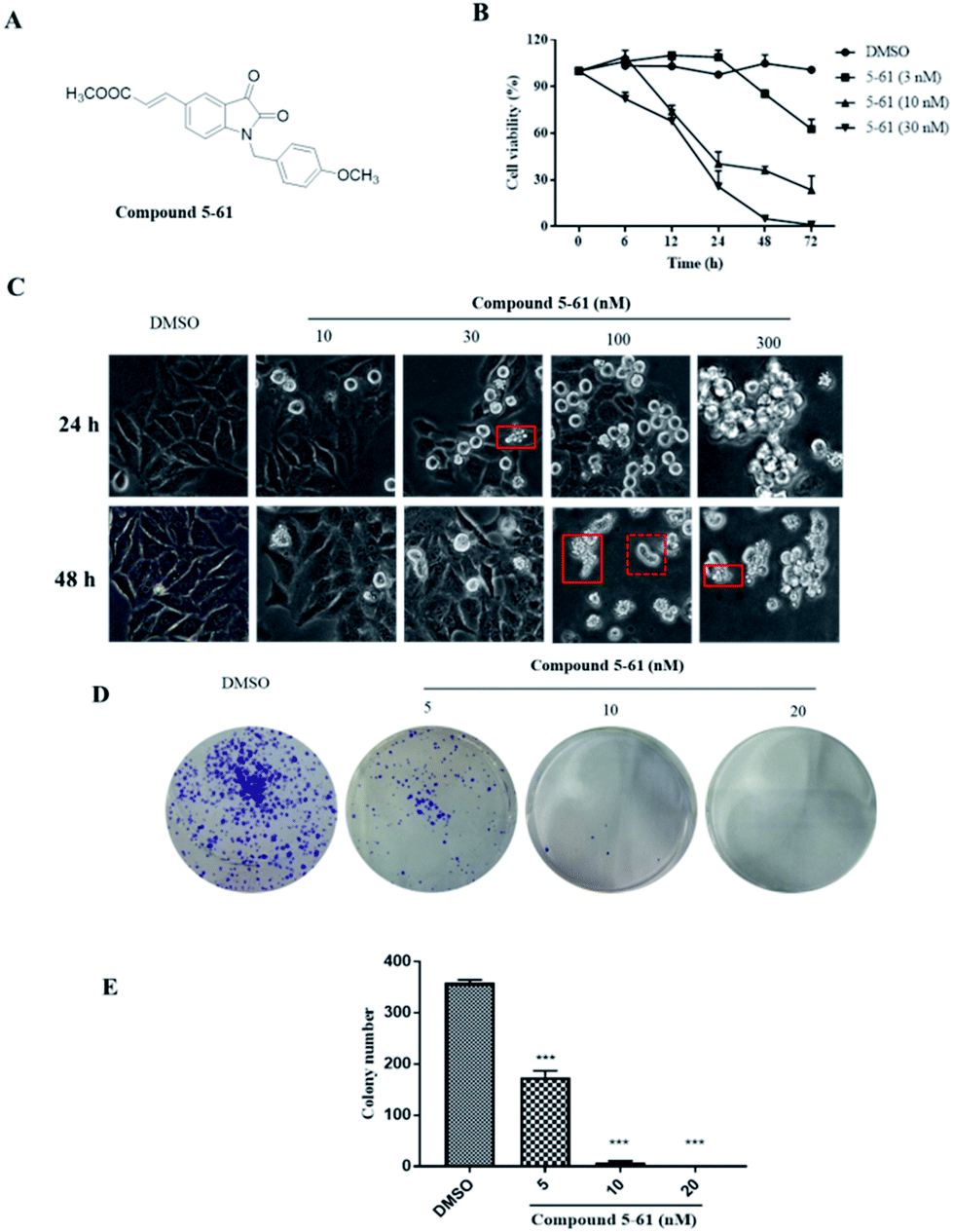 | ||
| Fig. 1 Inhibitory effect of compound 5-61 on tumor cell growth. (A) Structure of compound 5-61. (B) The proliferation effect of compound 5-61 on HepG2 cell growth. (C) Morphological changes in compound 5-61-treated HepG2 cells. (D and E) Representative dishes and quantitative data on the inhibitory effects of compound 5-61 on HepG2 cells proliferation evaluated by clonogenic assay. Data are shown as the mean ± S.D. of three independent experiments. | ||
Materials and methods
Cell lines and culture conditions
HepG-2 cells were cultured in DMEM supplied with 10% FCS and 1% penicillin/streptomycin. The primary HUVECs were purchased from Shanghai Haling Biotechnology and were grown in F-12 medium supplied with 15% FCS and 1% penicillin/streptomycin. HUVECs under passage number three to seven were used in the evaluations. All cells were cultured in a 5% CO2 humidified cell culture incubator at 37 °C.MTT assay
Cell proliferation inhibitory effect of 5-61 was assessed using MTT assay. Briefly, HepG2 cells (5 × 104 cells per mL) were seeded to 96-well microplate and cultivated at 37 °C for 0, 6, 12, 24, 48 and 72 h. Then various concentrations of compound 5-61 (3, 10, 30 nM) were added to 96-well microplates. MTT (10 μL, 5 mg mL−1) were added to each well after 48 h, then the plate was incubated without light at 37 °C for another 4 h. After that, 100 μL per well DMSO was added to dissolve formazan crystals. OD492 nm and 630 nm were recorded at dual wavelength of using multi-function microplate reader (Bio-Tek, USA) and the IC50 values were obtained using Graph Pad Prism 7.0 software.Clone survival assay
Clone formation assay was performed to further evaluate the activity of compound 5-61 on HepG2 cell proliferation. In brief, HepG2 cells (5 × 103 cells per mL) were planted in 6-well microplate and treated with compound 5-61 (5, 10, and 20 nM) and incubated for 5 days until colonies were clearly visible and countable. Then cell colonies were fixed, stained and observed using the Nikon Eclipse Ti microscope (Nikon, Tokyo, Japan).Apoptosis analysis using flow cytometric
The proapoptotic effect of compound 5-61 was determined by an annexin V-FITC/PI apoptosis detection kit. In brief, HepG2 (5 × 104 cells per mL) were planted in 6-well microplate and treated with DMSO or 5-61 (10, 30, 100, and 300 nM) for 24 and 48 h. Next, cells were collected and resuspended using 1× binding buffer. After staining cells for 10 min with 5 μL Annexin V-FITC and 5 μL PI (50 mg mL−1), the numbers of apoptotic cells were obtained using flow cytometry.Cell cycle analysis
After seeded and cultured for 24 h, HepG-2 (5 × 104 cells per mL) were treated with DMSO or 5-61 (10, 30, 100, and 300 nM) for another 24 h. Then, the cells were fixed with ice-cold ethanol 70% (v/v) ethanol and stored at −20 °C overnight. The fixed cells were incubated with 50 μg mL−1 PI, 100 μg mL−1 RNase A and 0.2% Triton-100 at 37 °C for 30 min in dark. The rates of different cell cycle phases were measured by FACS Calibur system (BD, USA, version 2.0).Cell migration analysis
The cell migration analysis was studied by wound healing assay and 8 μm pore size transwell chambers (EMD Millipore corporation, Germany). HepG2 cells (2 × 105 cells per well) were added in 6-well microplates and cultivated to grow approximately 80% confluence for 24 h. Then, a straight line was made in the middle of the dish by using a 10 μL sterile pipette tip to scratch HepG2 cells. De-attached cells were washed by PBS(−) for three times and treated with compound 5-61 (10 and 30 nM) for 0, 12, 24, and 48 h. The cells migrating into the wounded area were visualized right after the wound was scratched and 12, 24 and 48 h. The migration distance was calculated following the formula: migration distance (μm) = W0 − Wt, W0 was the cell width at 0 h; Wt was the cell width at different time (12–48 h).Transwell analysis
2 ×105 cells per well HepG2 cells (200 μL) were added into upper side membrane of the chambers without FCS while the lower chamber was filled with DMEM (600 μL) containing 10% FCS. Cells were cultured with DMSO or 5-61 (10, 30, and 100 nM) after 30 min. Cells were allowed migrating towards a chemotactic stimulus of 10% FCS for 24 h. The remaining non-migrated cells were removed from the upper chamber. After washed for three times by PBS(−), the migrated cells were fixed with absolute methanol, stained with 1 μg mL−1 DAPI and photographed. The pictures of five horizons were taken randomly.Tube formation analysis
In vitro capillary-like network formation of HUVEC cells was carried out in 96-well microplate precoated with Matrigel (BD Bioscience, USA).22 Matrigel (13.9 mg mL−1, 50 μL per well) was added to 96-well microplate and polymerized for 30 min to form a gel layer at 37 °C. Following the polymerization, the HUVECs (1 × 105 cells per well) were seeded to the Matrigel-coated wells and incubated at 37 °C for 40 min and then compound 5-61 (1, 3, and 10 nM) were added to incubate for 12 h. The images of capillary-like tube formation were taken by a Nikon Eclipse Ti microscope. The number of nodes, total branching lengths and number of meshes were measured using Image-J software.Phalloidin staining assay
Phalloidin staining assay was used to evaluate the activity of compound 5-61 on HUVEC cell cytoskeleton. In brief, 2 × 104 cells per mL HUVEC cells were planted in 24-well microplate and then treated with 5-61 (10 and 30 nM) for 24 h. Afterwards, cells were fixed for 20 min with 4% paraformaldehyde and then blocked with 1% BSA for 20 min at room temperature. Next, cells were stained for 30–60 min with FITC-phalloidin (5 μg mL−1, Sigma-Aldrich, Germany) at room temperature in dark environment and then stained with 5 μg mL−1 DAPI for 5 min. The samples washed by PBS(−) for three times in the middle of each step above. Finally, the images were captured by fluorescence microscope.Chicken embryo chorioallantoic membrane (CAM) analysis
Chorioallantoic membranes (CAM) from developing chicken eggs are usually used in biological and biomedical research to investigate angiogenesis in tissue level. The fertilized chicken eggs (55–60 g) were incubated in a humidified incubator at 37.8 ± 0.5 °C and rotated once in every 2 h. Until the seventh day, an air chamber (10 mm × 10 mm) was carefully produced on the broad side of the egg and continually incubate for another 1 day. Then the carrier of fertilized gelatin sponge (3 mm × 3 mm × 3 mm) with compound 5-61 (2, 6, and 20 μM) or sunitinib (as positive control) was put on well-vascularized sites of the CAM. The eggs were incubated in egg incubator for 72 h. Then CAM was separated from chick embryos, fixed with 1 mL methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetone = 1:1 fixing solution for 15 min and photographed using stereo microscope. The total numbers of vessels in the CAM were quantified by Image-J software.
:acetone = 1:1 fixing solution for 15 min and photographed using stereo microscope. The total numbers of vessels in the CAM were quantified by Image-J software.
Western blotting assay
Total protein from HUVECs treated with 5-61 (30, 100, 300, and 1000 nM) for 24 h was extracted using lysis buffer. Sample were evaluated by SDS-PAGE and was detected by Odyssey Infrared Fluorescence Imaging System (Li-cor company, USA). All antibodies (1:2000 dilution) were purchased from CST.
ELISA assay
Quantikine ELISA kit (Nanjing jiancheng Bio Institute, China) was used to measure the expression level of VEGF-A protein in the supernatant of HepG2 cells treated with various concentrations of 5-61. In brief, 1 × 105 cells per mL HepG2 cells were plated in 6-well plate and treated with 5-61 (30, 100, 300, and 1000 nM) for 24 h. Then cells supernatants were harvested and examined. A standard curve drawn by ELISA was used to calculate the OD and the expression level of VEGF-A protein.Statistical analysis
GraphPad Prism 7 (San Diego, CA) was used for statistical analysis. The minimum statistically significance level was set at a p value of 0.05. The differences between the mean values of tested groups were presented as mean ± SD and determined by one-way analysis of variance (ANOVA). All experiments were independently repeated more than three times.Results
Compound 5-61 inhibited the proliferation of HepG-2 cells in vitro
To evaluate the in vitro effectiveness of compound 5-61 on tumor growth, we conducted an MTT assay to analyze cell proliferation at different incubation times and at various concentrations of compound 5-61. As shown in Fig. 1B, compound 5-61 significantly inhibited the growth of HepG-2 cells in a time- and concentration-dependent manner with a 50% inhibitory concentration (IC50) of 7.13 nM. After treatment with 5-61, the cells presented the typical morphological characteristics of apoptosis, including nuclear pyknosis, sublobes, fragmentation, and apoptotic body generation (Fig. 1C). Additionally, clone formation was markedly decreased when HepG2 cells were treated with 5 and 10 nM of compound 5-61 (Fig. 1D), suggesting that this compound strongly prevented tumor cell proliferation in vitro.Compound 5-61 induced apoptosis and inhibited cell cycle progression in HepG-2 cells
To further evaluate whether the growth-inhibitory effects of compound 5-61 were related to apoptosis induction, HepG2 cells were treated with 5-61 (0, 10, 30, 100, and 300 nM) for 24 and 48 h (Fig. 2A). Based on Annexin V-FITC/PI double staining, the percentage of cell apoptosis induced by 5-61 was examined by flow cytometry. The data suggest that apoptotic HepG2 cells were significantly induced after treatment with 5-61 for 24 and 48 h; additionally, treatment with 5-61 at concentrations of 0, 10, 30, 100, and 300 nM resulted in dose-dependent increases in the numbers of late apoptotic cells from 3.6% to 43.7% and from 4.0% to 66.4%, respectively. These results demonstrate that apoptosis induction partially accounts for the growth inhibition of HepG2 cells.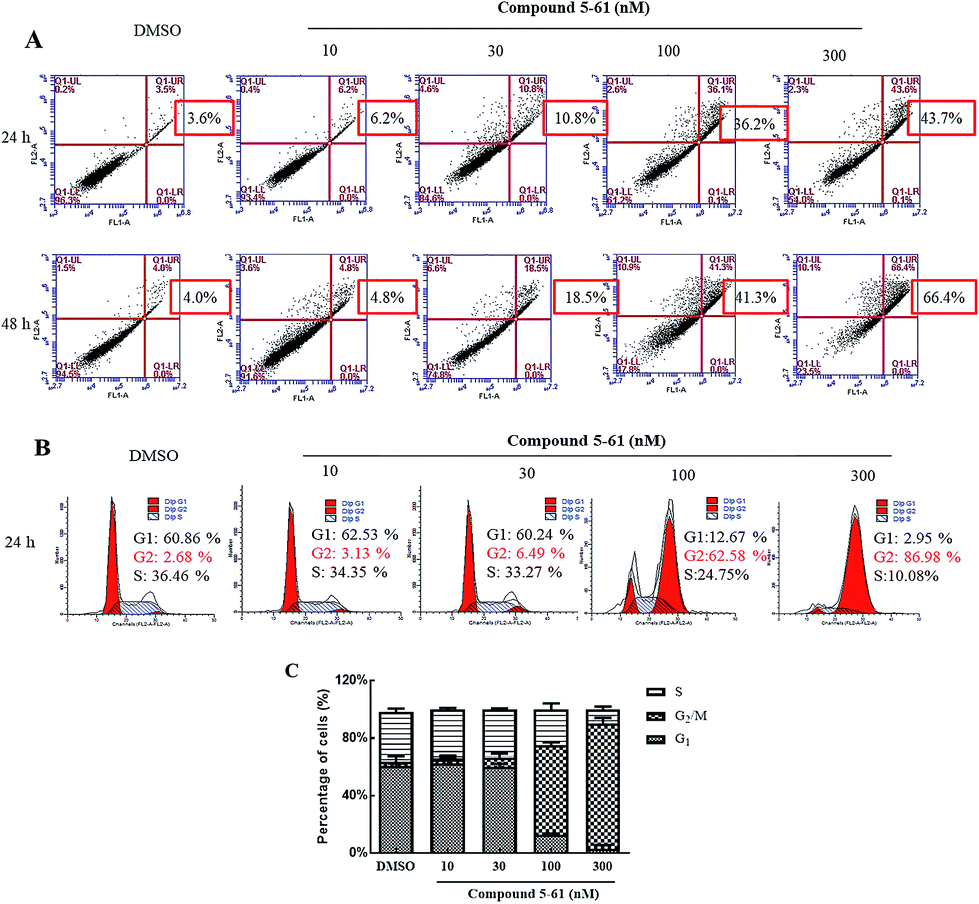 | ||
| Fig. 2 Effect of compound 5-61 induced apoptosis and cell cycle arrest in HepG2 cells. (A) Effect of compound 5-61 on Annexin V/PI dye HepG2 cells. (B) Cell cycle distribution analyzed by Modfit LT software. (C) Quantitative analysis of cell cycle. Annexin V−/PI− (lower left) cells represent survival, Annexin V+/PI− (lower right) cells were defined as early apoptotic cells, Annexin V+/PI+ (upper right) cells are late apoptotic cells, Annexin V+/PI− (upper left) cells are necrotic apoptotic cells. All experiments were performed in triplicate. | ||
To clarify the mechanism by which compound 5-61 inhibited HepG2 cell proliferation, propidium iodide (PI) staining was conducted to evaluate the DNA distribution.23,24 Cell cycle analysis indicated that HepG2 cells were notably arrested at G2/M phase after treatment with different concentrations of 5-61 for 24 h (Fig. 2B). These results confirm that compound 5-61 induces G2/M phase cell arrest.
Compound 5-61 suppressed the migration of HepG2 cells
Tumor cell metastasis is a complex process involving migration, invasion, or infiltration into blood vessels. We thus investigated whether 5-61 inhibits the migratory capacity of HepG2 cells by conducting an in vitro wound healing assay and transwell migration assay. In the wound healing assay, the migration distances were measured at 12, 24, and 48 h after the wounds were made. Fig. 3A and B show that 5-61 obviously decreased the ability of cells to cover the scratched area when HepG2 cells were incubated with 10 and 30 nM of 5-61 for different times compared to the control group.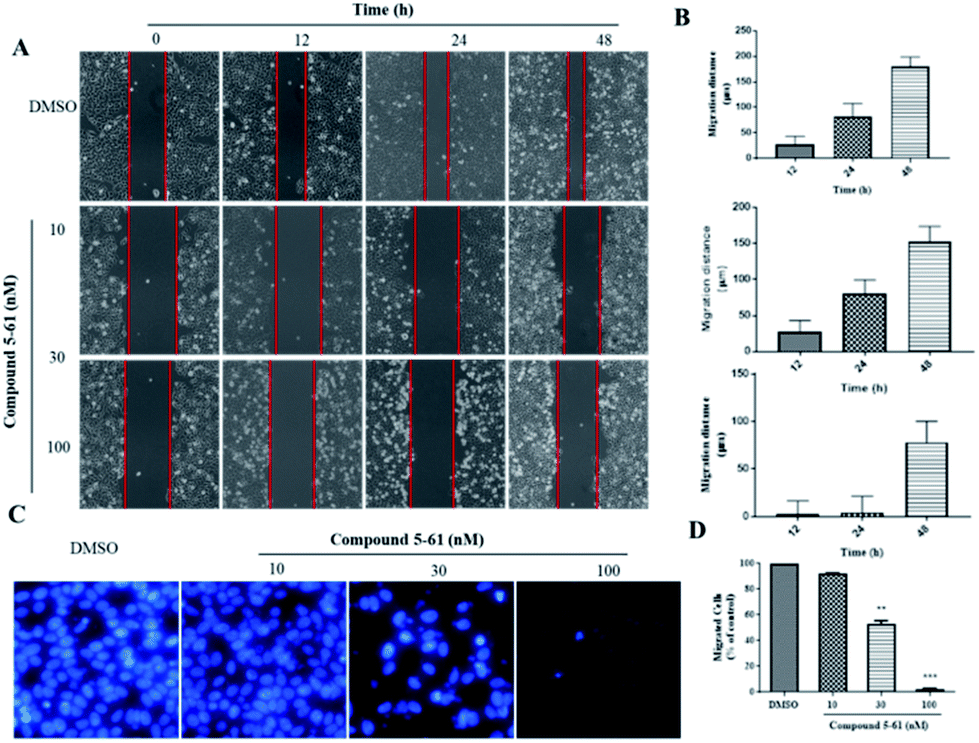 | ||
| Fig. 3 Effect of compound 5-61 on HepG2 cell migration. (A) Effect of wound healing assay. (B) Analysis of cell migration distance. (C) Effect of transwell migration assay. (D) Analysis of cell migration levels. | ||
To further verify these results and exclude the possibility that 5-61-reduced migration was correlative of decreasing cell proliferation, a transwell cell migration assay was also conducted to assess the capacity and rate of cell migration. The data revealed that 5-61 remarkably reduced the migration of HepG2 cells, specifically at doses of 30 and 100 nM, compared to the control group (Fig. 3C and D).
Compound 5-61 inhibited capillary tube formation by endothelial cells
HUVECs are widely used as an in vitro model system for studying the progression of neovascularization. During neovascularization, endothelial cells have been demonstrated to play a pivotal role in in vitro models.23,24 Therefore, a capillary-like tube formation assay was performed using endothelial cells on Matrigel to evaluate the antiangiogenic effects of 5-61.Firstly, an MTT assay combined with morphological imaging was performed to analyze cell proliferation and to evaluate the cytotoxic action of 5-61 on HUVECs. Fig. 4A and B showed no apparent reduction in cell viability or morphological changes after compound 5-61 treatment for 48 h compared to the control group.
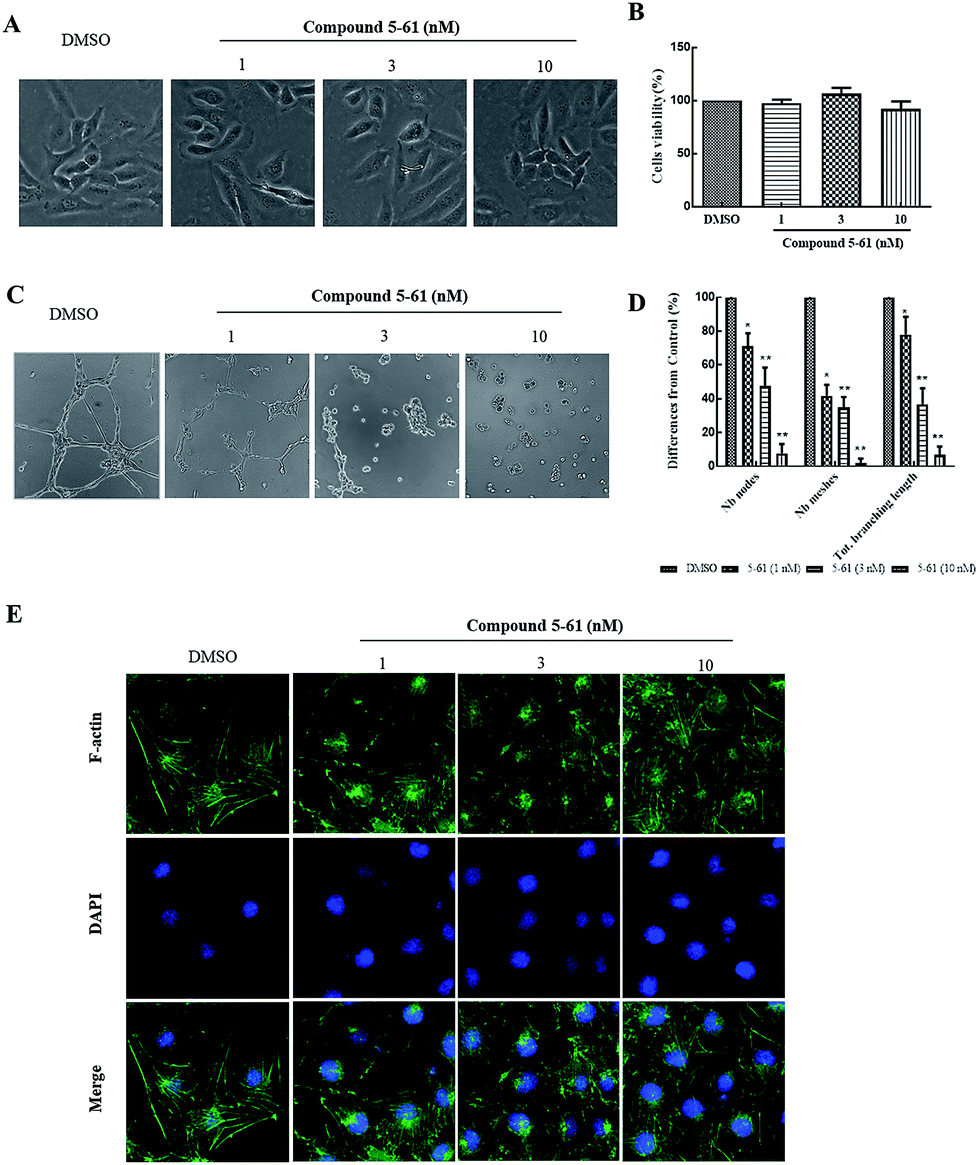 | ||
| Fig. 4 Suppressive effects of 5-61 on tube formation of HUVECs. (A) Morphological changes in 5-61-treated HUVECs. (B) Cell proliferation were measured by MTT assay after compound 5-61 treatment for 48 h. (C) Tube formation assay of HUVECs after compound 5-61 treatment for 12 h. (D) Number of nodes, meshes, and total branching length were quantified. (E) Phalloidin staining shows F-actin distribution in normal or infected HUVECs treatment with compound 5-61. | ||
After 12 h of incubation, the untreated group showed the formation of capillary-like enclosed tubular networks, whereas formation of the basal tube network in groups treated with 1, 3, and 10 nM of 5-61 was dramatically reduced (Fig. 4C). Quantitative image analysis confirmed that 5-61 significantly diminished both the dimensional (branching length per field) and topological parameters (number of nodes and meshes per field) of the capillary-like network (Fig. 4D).
Inhibitory effect of compound 5-61 in the CAM assay
The CAM assay is a widely-used model of neovascularization that can be extensively applied to investigate new vessel formation and inhibition in vivo.25,26 The CAM assay was thus used to further assess the potential anti-angiogenesis effects of 5-61. Blood vessels formed densely a branching vascular network in the negative control group (Fig. 5A and B). When chicken embryos were incubated with 300 μM sunitinib (positive control group) for 72 h, the number of capillaries in the CAM sections was modestly decreased. However, treatment with 6 and 20 μM 5-61 significantly attenuated neovascularization, showing comparable to sunitinib.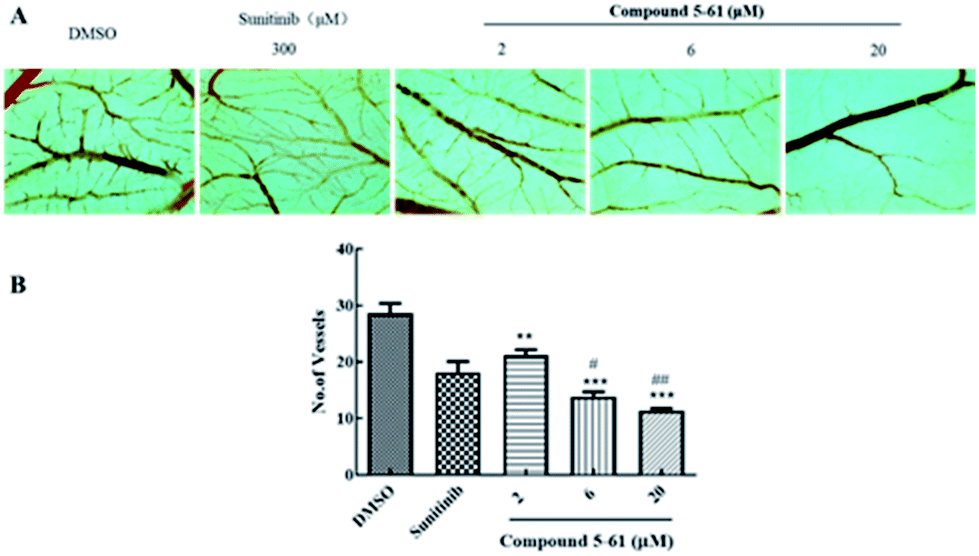 | ||
| Fig. 5 Effects of compound 5-61 on neovascular of CAM model. (A) CAM membranes were extracted from chick embryos after treatment with different concentrations of compound 5-61 for 72 h. (B) Quantitative analysis of average number of blood vessels. ***p < 0.001 and **p < 0.01 vs. DMSO group; #p < 0.05 and ##p < 0.01 vs. sunitinib group. | ||
Compound 5-61 suppressed the activation of signaling pathways downstream of VEGF
VEGF is one of the most pivotal mediators among angiogenesis-associated growth factors.27 Accordingly, we examined whether 5-61 directly inhibits VEGF-A secretion in vitro by ELISA. The results showed that 5-61 decreased VEGF-A levels in HepG2 cell supernatants, suggesting that 5-61 directly regulates the VEGF/VEGFR2 signaling pathway (Fig. 6A and B).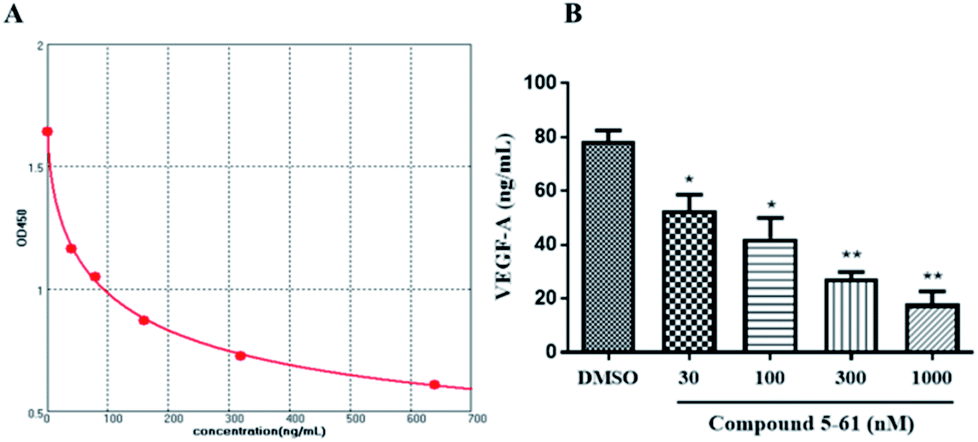 | ||
| Fig. 6 VEGF-A protein was determined by ELISA in HepG2 cells. (A) Standard curve. (B) Changes in VEGF-A protein in supernatant of HepG2 cells treated with 5-61 for 24 h. | ||
Akt, mTOR, and ERK mediate endothelial cell proliferation and migration downstream of VEGF.28 To identify the potential molecular mechanisms of 5-61 in tumor angiogenesis, we determined the expression levels of these proteins by western blot analysis. We found that treatment with 5-61 for 24 h could remarkably decrease the phosphorylation of Akt, mTOR, and ERK but had slight effects on their total protein expression levels (Fig. 7A–F).
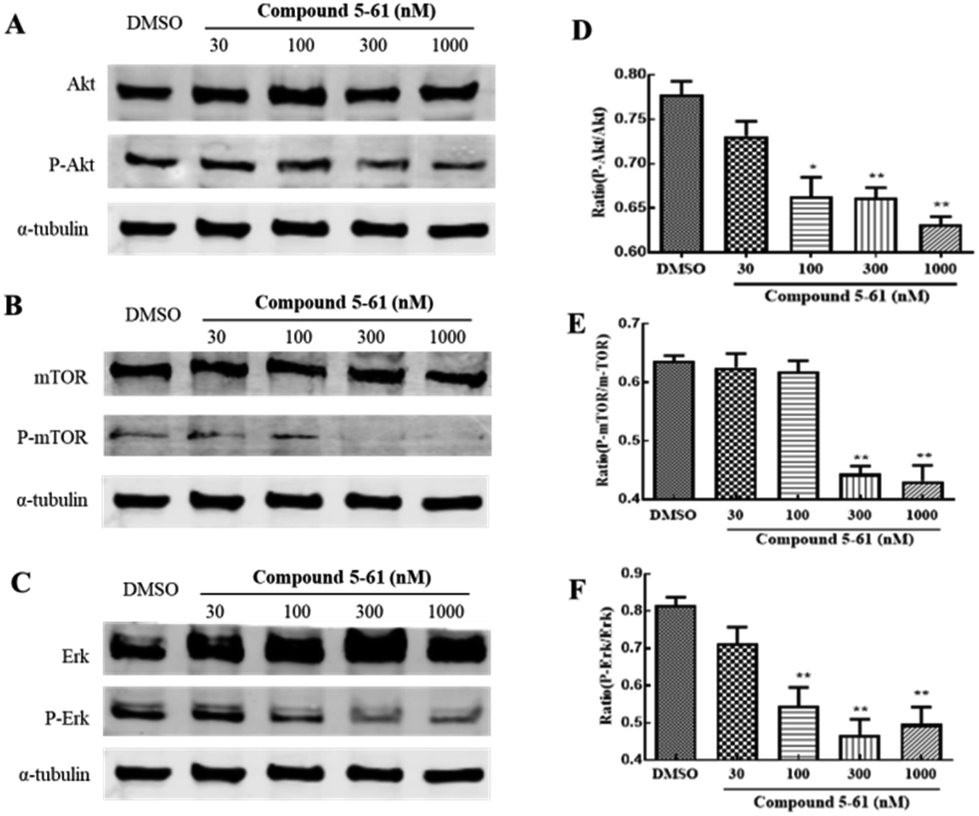 | ||
| Fig. 7 Changes in angiogenesis proteins treated with compound 5-61 in HUVECs detected by western blotting. (A–C) Changes in PI3K/Akt and MAPK pathway protein (D–F). Ratio of P-Akt/Akt, P-mTOR/mTOR, and P-Erk/Erk. | ||
Discussion
Indole has been introduced as an important structure in the discovery and development of new anticancer agents.29 In a previous study, a series of novel indole derivatives were designed and synthesized in 5–6 steps in our laboratory. Through structure–activity relationship analysis, compound 5-61 was found to strongly inhibit tumor cell proliferation.30 Based on the results displayed above, we confirmed that compound 5-61 inhibited the growth of tumor cells mainly via inducing cell cycle arrest in the G2/M phase and inducing apoptosis. We also showed that compound 5-61 exerts potent anti-angiogenic activities by preventing HepG2 cell migration and endothelial cell tube formation both in vitro and in vivo. Furthermore, compound 5-61 strongly inhibited small vessel development with similar effects as sunitinib according to an in vivo CAM assay. The possible mechanisms involved in the anti-angiogenic activities of 5-61 in endothelial cells include: (i) directly suppressed formation of HUVEC actin filaments, (ii) downregulation of VEGF-A expression in tumor cells, and (iii) direct inhibition of Akt, mTOR, and ERK tyrosine kinase activity. Our data clarify the molecular mechanisms through which 5-61 exerts its promising anti-angiogenic and anti-cancer effects.Our results confirm that compound 5-61 has excellent cytotoxic activity against HepG2 cells (IC50 = 7.13 nM) (Fig. 1B). As shown in Fig. 1C, apoptotic morphology (characterized by cell shrinkage and/or blebbing) and G2/M phase arrest (characterized by cell elongation) were clearly observed after HepG2 cells were treated with 30 nM 5-61 for 48 h. According to flow cytometry analysis, 5-61 strongly induced apoptosis in HepG2 cells in vitro and decreased the transition to the G1 phase (Fig. 2A and B), which is consistent with the results of morphological studies showing that 5-61 has pro-apoptotic effects and induces G2/M phase arrest of cancer cells.
Metastasis of tumor cells is a complex process involving numerous multi-step pathways including cell detachment from primary tumors and migration of invading tumor cells. In the present study, wound healing and transwell migration assays were performed to investigate the inhibitory effects of 5-61 on HepG2 cell migration. In wound healing assays, 10 nM 5-61 showed moderate inhibitory effects (Fig. 3A). The strongest effects were observed at a concentration of 30 nM 5-61. Additionally, the transwell migration assay showed that 5-61 inhibited cell migration in a dose-dependent manner.
Angiogenesis is a precisely regulated procedure involving an intricate cascade of events including high endothelial cell proliferation, migration, and tube formation in the final stage.31 Therefore, targeting of angiogenesis processes may be useful for cancer therapy. We found that 5-61 dose-dependently reduced the quality and quantity of capillary-like structures formed in the tube formation assay; however, the growth and number of HUVECs were not affected by treatment with the same concentrations of 5-61 (Fig. 4A). The anti-angiogenic effect of 5-61 was further verified in a CAM assay in vivo (Fig. 5A). These results suggest that 5-61 inhibits angiogenesis in endothelial cells.
VEGF-A, which is widely expressed in most malignant tumors, is thought to be the most crucial angiogenesis factor.32 Furthermore, among the three different VEGF receptors, VEGFR-2 is the main responder to VEGF-A in in vitro tumor angiogenesis.33 Here, we demonstrated that 5-61 suppresses the activity of VEGFA (Fig. 6A), as well as inhibits VEGF/VEGFR-2-mediated intracellular signaling pathways, including PI3K/Akt, mTOR, and ERK in HUVECs (Fig. 7A–C), which are essential for the mitogenic activities of VEGF in endothelial cells.
In conclusion, we demonstrated that compound 5-61 suppressed HepG2 cell proliferation, survival, and migration. Further, it has potent anti-angiogenic properties both in vivo and in vitro, by inhibiting capillary-like structure formation of HUVECs and reducing the neovascularization in the CAM. Moreover, 5-61 likely exerted these effects by targeting VEGFA and its downstream signal transduction pathways including PI3K, Akt, mTOR, and MAPKs (ERK). These findings support the anti-cancer effects and mechanisms of compound 5-61 (Fig. 8). Studies are underway to further clarify the anti-angiogenesis effect of 5-61 on other tumor cell lines, and various types of anti-angiogenic animal models are required for further studies on 5-61.
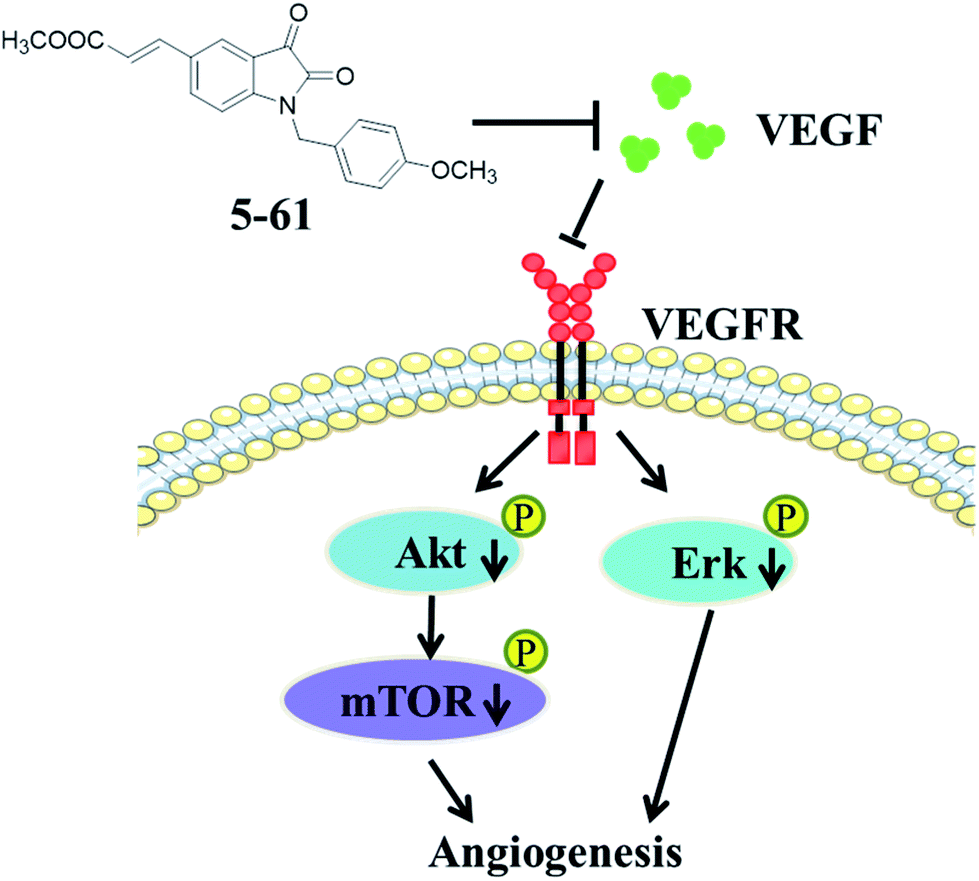 | ||
| Fig. 8 Schematic illustration of potential action mechanism of compound 5-61 in angiogenesis. | ||
These findings thus provide insight into the angiogenesis-targeting mechanism underlying 5-61 activity.
Conflicts of interests
The authors declare that there are no conflicts interest.Acknowledgements
This work was supported jointly by National Natural Science Foundation of China (31601203 and 81703014), International Science & Technology Cooperation Program of China (2013DFA31160).References
- H. Guo, Eur. J. Med. Chem., 2019, 164, 678–688 CrossRef CAS PubMed.
- Q. Zhang, Y. Teng, Y. Yuan, T. Ruan, Q. Wang, X. Gao, Y. Zhou, K. Han, P. Yu and K. Lu, Eur. J. Med. Chem., 2018, 156, 800–814 CrossRef CAS PubMed.
- Z. Xu, X. F. Song, Y. Q. Hu, M. Qiang and Z. S. Lv, Eur. J. Med. Chem., 2017, 138, 66–71 CrossRef CAS PubMed.
- Z. Xu, S. Zhang, X. Song, M. Qiang and Z. Lv, Bioorg. Med. Chem. Lett., 2017, 27, 3643–3646 CrossRef CAS PubMed.
- Y. A. Ammar, E. A. Sh, A. Belal, S. Y. Abbas, Y. A. Mohamed, A. Mehany and A. Ragab, Eur. J. Med. Chem., 2018, 156, 918–932 CrossRef CAS PubMed.
- S. N. Pandeya, P. Yogeeshwari, D. Sriram and G. Nath, Indian J. Pharm. Sci., 2006, 64, 209–212 Search PubMed.
- M. Verma, S. N. Pandeya, K. N. Singh and J. P. Stables, Acta Pharm., 2004, 54, 49–56 CAS.
- S. K. Sridhar and A. Ramesh, Biol. Pharm. Bull., 2001, 24, 1149–1152 CrossRef CAS PubMed.
- R. Rohini, P. M. Reddy, K. Shanker, K. Kanthaiah, V. Ravinder and A. Hu, Arch Pharm. Res., 2011, 34, 1077–1084 CrossRef CAS PubMed.
- H. S. Ibrahim, S. M. Abou-Seri, M. Tanc, M. M. Elaasser, H. A. Abdel-Aziz and C. T. Supuran, Eur. J. Med. Chem., 2015, 103, 583–593 CrossRef CAS PubMed.
- Y. Kia, H. Osman, R. S. Kumar, V. Murugaiyah, A. Basiri, S. Perumal, H. A. Wahab and C. S. Bing, Bioorg. Med. Chem., 2013, 21, 1696–1707 CrossRef CAS PubMed.
- P. Selvam, N. Murugesh, M. Chandramohan, Z. Debyser and M. Witvrouw, Indian J. Pharm. Sci., 2008, 70, 779–782 CrossRef CAS PubMed.
- N. Ai, C. M. Chong, W. Chen, Z. Hu, H. Su, G. Chen, W. Q. Lei and W. Ge, Oncotarget, 2018, 9, 31958–31970 Search PubMed.
- H. M. Verheul, E. E. Voest and R. O. Schlingemann, J. Pathol., 2004, 202, 5–13 CrossRef CAS PubMed.
- C. Fontanella, E. Ongaro, S. Bolzonello, M. Guardascione, G. Fasola and G. Aprile, Ann. Transl. Med., 2014, 2, 123 Search PubMed.
- J. Welti, S. Loges, S. Dimmeler and P. Carmeliet, J. Clin. Invest., 2013, 123, 3190–3200 CrossRef CAS PubMed.
- M. Huang, B. Huang, G. Li and S. Zeng, BMC Gastroenterol., 2018, 18, 169 CrossRef PubMed.
- H. Peng, Q. Zhang, J. Li, N. Zhang, Y. Hua, L. Xu, Y. Deng, J. Lai, Z. Peng, B. Peng, M. Chen, S. Peng and M. Kuang, Oncotarget, 2016, 7, 17220–17229 Search PubMed.
- K. L. Osusky, D. E. Hallahan, A. Fu, F. Ye, Y. Shyr and L. Geng, Angiogenesis, 2004, 7, 225–233 CrossRef CAS PubMed.
- S. L. Beedie, C. Mahony, H. M. Walker, C. H. Chau, W. D. Figg and N. Vargesson, Sci. Rep., 2016, 6, 30038 CrossRef CAS PubMed.
- Y. O. Teng, H. Y. Zhao, J. Wang, H. Liu, M. L. Gao, Y. Zhou, K. L. Han, Z. C. Fan, Y. M. Zhang, H. Sun and P. Yu, Eur. J. Med. Chem., 2016, 112, 145–156 CrossRef CAS PubMed.
- T. W. Chung, S. J. Kim, H. J. Choi, C. H. Kwak, K. H. Song, S. J. Suh, K. J. Kim, K. T. Ha, Y. G. Park, Y. C. Chang, H. W. Chang, Y. C. Lee and C. H. Kim, J. Mol. Med. (Berl.), 2013, 91, 271–282 CrossRef CAS PubMed.
- Y. Pang, G. Qin, L. Wu, X. Wang and T. Chen, Exp. Cell Res., 2016, 347, 251–260 CrossRef CAS PubMed.
- Y. Jiang, X. Zhou, X. Chen, G. Yang, Q. Wang, K. Rao, W. Xiong and J. Yuan, Mutat. Res., 2011, 726, 75–83 CAS.
- H. J. Park, Y. Zhang, S. P. Georgescu, K. L. Johnson, D. Kong and J. B. Galper, Stem Cell Rev., 2006, 2, 93–102 CrossRef CAS PubMed.
- D. Ribatti, A. Vacca, L. Roncali and F. Dammacco, Int. J. Dev. Biol., 1996, 40, 1189–1197 CAS.
- Q. Li, Y. Wang, L. Zhang, L. Chen, Y. Du, T. Ye and X. Shi, Fitoterapia, 2016, 111, 78–86 CrossRef CAS PubMed.
- S. Rousseau, F. Houle, J. Landry and J. Huot, Oncogene, 1997, 15, 2169–2177 CrossRef CAS PubMed.
- S. Dadashpour and S. Emami, Eur. J. Med. Chem., 2018, 150, 9–29 CrossRef CAS PubMed.
- K. Han, Y. Zhou, F. Liu, Q. Guo, P. Wang, Y. Yang, B. Song, W. Liu, Q. Yao, Y. Teng and P. Yu, Bioorg. Med. Chem. Lett., 2014, 24, 591–594 CrossRef CAS PubMed.
- J. Folkman, Semin. Oncol., 2002, 29, 15–18 CrossRef CAS PubMed.
- Q. Li, Y. Wang, L. Zhang, L. Chen, Y. Du, T. Ye and X. Shi, Fitoterapia, 2016, 111, 78–86 CrossRef CAS PubMed.
- L. Claesson-Welsh and M. Welsh, J. Intern. Med., 2013, 273, 114–127 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |