
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon-based artificial SEI layers for aqueous lithium-ion battery anodes†
Usha Subramanya‡
a,
Charleston Chua‡a,
Victor Gin He Leonga,
Ryan Robinsona,
Gwenlyn Angel Cruz Cabiltesa,
Prakirti Singha,
Bonnie Yipa,
Anuja Bokareb,
Folarin Erogbogbob and
Dahyun Oh *a
*a
aChemical and Materials Engineering Department, Charles W. Davidson College of Engineering, San José State University, One Washington Square, San José, CA 95192-0080, USA. E-mail: dahyun.oh@sjsu.edu
bBiomedical Engineering Department, Charles W. Davidson College of Engineering, San José State University, One Washington Square, San José, CA 95192-0080, USA
First published on 2nd January 2020
Abstract
Replacing flammable organic electrolytes with aqueous electrolytes in lithium-ion batteries (LIB) can greatly enhance the safety of next-generation energy storage systems. With the extended electrochemical stability window of electrolytes, ‘water-in-salt’ (WIS) electrolytes containing LIB presented significant performance improvements. However, the solubility limits of lithium salts in water restrain the extent of kinetic protection offered by the high salt concentration. Here, we report design strategies of anode structure to improve the cycle life of LIB with WIS electrolytes. We introduced partially graphitic protective carbon layers on anode particles using a versatile coating method. This protective layer not only improved charge transfer kinetics but also minimized the exposure of anode surface for water electrolysis. The effectiveness of anode structure developed in this study was exemplified on TiO2 anodes, where cycle performance and coulombic efficiency improved by 11 times and 29% respectively over the base anode material.
1. Introduction
Developing safe and cost-effective batteries has become an important issue in scientific communities due to the high demand for storing electricity for mobile devices and electric vehicles. Currently, lithium-ion batteries (LIB) use highly flammable and toxic organic electrolytes that can lead to explosions. Aqueous LIB can address this safety issue by replacing flammable organic electrolytes.1–3 However, their poor cycle life and low energy density hinder their greater adoption in consumer products such as mobile electronics or electric vehicles. These issues stem from the narrow electrochemical stability window of water (1.23 V),2 limiting the selection of active materials that can stably operate within this potential range.4–7 This has not been an issue in organic electrolytes because of the formation of stable solid-electrolyte interphase (SEI) layers. These layers were known to be formed by the deposition of electrolyte decomposition products and prevent further degradation of electrolytes.8–12 However, aqueous electrolyte decomposition products like oxygen and hydrogen gas cannot form surface layers on electrode surfaces.1,13 Recent studies have shown that the use of a highly concentrated ‘water-in-salt’ (WIS) electrolyte results in preferential decomposition of lithium salts, forming SEI layers on the anode primarily consisting of LiF.14 This extends the electrochemical stability window of aqueous electrolytes from 1.23 V to 3.00 V, as the interphase layer suppresses further electrolyte decomposition.2,15,16While highly-concentrated aqueous electrolytes extend the stability window of aqueous batteries to 3 V, their cyclability and energy density are still inferior to organic electrolytes-based batteries.6,13,17 Thus, an alternative solution to achieve performance parity needs to be developed to overcome limitations arising from the solubility limit of salt in water and high cost of lithium salts in super-concentrated electrolytes. To that end, we propose to develop a design strategy for a protective layer on anode particles analogous to SEI layers to minimize battery performance degradation in aqueous LIB. Due to its high electronic conductivity and easy formation on diverse substrates, carbon-based protective layer has been widely used to improve the electrode performance in organic electrolyte.18,19 However, this has not been widely explored in ‘water-in-salt’ electrolyte batteries. Carbon–water interactions necessitate a balance between ‘graphitic’ and ‘amorphous’ bonding due to differing hydrophobicity and conductivity. Here we systematically determine and report the key parameters to develop an ideal carbon coating on anode as a form of artificial SEI layer (Fig. 1A) for aqueous LIB. We aim to build nanometer thick protective layers on anode particles that provide diffusion channels for lithium ions while minimizing the access of water molecules to electrode surfaces.
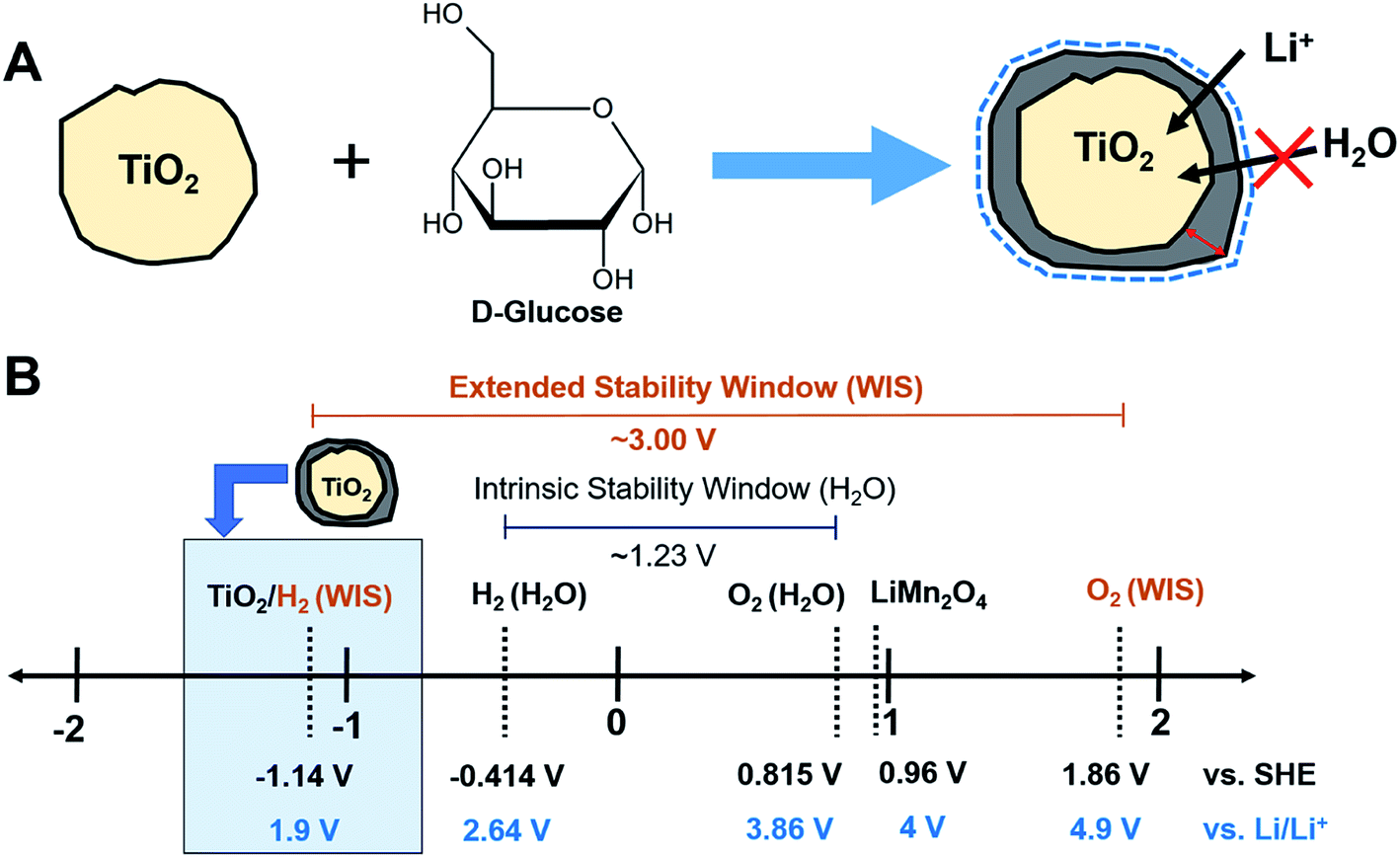 | ||
| Fig. 1 Schematics of (A) carbon coating process and (B) the stability window of water-in-salt (WIS) electrolyte with the electrochemical redox potentials of battery cell components in this study (anode: TiO2 and cathode: LiMn2O4). | ||
To exemplify the effectiveness of our artificially created SEI layer, we chose titanium dioxide (TiO2) as our model anode material (Fig. 1B). TiO2 presented low volumetric expansion during lithium intercalation in LIB with organic electrolytes, resulting in a good cycle life.20,21 Several polymorphs of TiO2 exist including anatase, bronze (TiO2-B), rutile, etc., each of which has shown different electrochemical reactions with lithium ions.22 For battery applications, the anatase phase is preferred as it is a thermodynamically stable phase and showed superior electrochemical performance to the majority of the other phases with good insertion kinetics (0.5 Li per formula unit).23–26 While TiO2 itself is stable in aqueous electrolyte batteries, due to its photocatalytic properties and close operating potential to hydrogen evolution, continual water decomposition can possibly occur on its surface without protective layers. Therefore, this work investigates the optimal thickness and composition of carbon needed to improve the performance of TiO2 anodes in aqueous LIB. By using glucose, an inexpensive common precursor, we developed a diverse range of artificial SEI layers on anodes to reduce the anode surface area available for water electrolysis and to improve the conductivity of anodes (Fig. 1A). Our findings presented synthesis guidelines for creating an artificial SEI layer designed to improve the aqueous LIB performance. This general anode design strategy to improve the aqueous battery performance can provide safer and higher performing next-generation aqueous LIB that can transform the current energy storage paradigm in a more sustainable manner.
2. Experimental section
Anatase TiO2 nanoparticles were synthesized using a sol–gel synthesis process. First, 8 mL of titanium isopropoxide (≥97%, Aldrich, called as TTIP in this work) was added into 33 mL of ethanol (>99.5%, Acros Organics) slowly while stirring. Four drops of nitric acid (>99%, Aldrich) were then added into the solution and stirred for 10 minutes. Deionized water was added dropwise for 2 min (3 mL) into this solution and a gel was formed. The resulting gel was aged for 24 hours at room temperature, then heated under vacuum for 48 hours at 95 °C. The dried products were ground into fine powder and annealed at 500 °C for 3 hours in a furnace.27 Coating of the sol–gel TiO2 (named as C-TiO2) was done using a carbothermal synthesis method. Different weight ratios of TiO2 and glucose (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 1:4, 1:6, 1:9 = TiO2:glucose) were mixed in 17.5 mL of deionized water for 45 minutes using a bath sonicator and then stirred for 3 hours on a magnetic stirring plate. The solution was then heated at 180 °C in a 23 mL Teflon-lined stainless-steel autoclave for 12 hours. The resulting brown mixture was washed with deionized water by centrifugation until the solution became clear.28,29 The solution was then dried at 80 °C in a vacuum oven for 6 hours followed by annealing in a tube furnace under Ar at varying temperatures (400–900 °C).
:2, 1:4, 1:6, 1:9 = TiO2:glucose) were mixed in 17.5 mL of deionized water for 45 minutes using a bath sonicator and then stirred for 3 hours on a magnetic stirring plate. The solution was then heated at 180 °C in a 23 mL Teflon-lined stainless-steel autoclave for 12 hours. The resulting brown mixture was washed with deionized water by centrifugation until the solution became clear.28,29 The solution was then dried at 80 °C in a vacuum oven for 6 hours followed by annealing in a tube furnace under Ar at varying temperatures (400–900 °C).
Anodes were fabricated with 8:1:1 mass ratio mixture of TiO2 powder, carbon black (Super P®, Alfa Aesar), and poly(tetrafluoroethylene) (Sigma Aldrich). The electrodes were punched using a 3/8′′ diameter hole punch. To indemnify the irreversible loss of cyclable lithium, the mass of the cathode was maintained at twice of the mass of the anode. The weight of anode was around 10 mg cm−2. The batteries were fabricated using a WIS electrolyte and LiMn2O4 cathode (MTI corporation, Al foil single side coated). The water-in-salt electrolyte was prepared by mixing 21 m (molality, mol kg−1) of lithium bis(trifluoromethanesulfonyl)imide (Gotion) in deionized water at 50 °C for 1 hour followed by continuous stirring at room temperature for 24 hours. The coin cells (CR2032) were assembled by placing the anode on the bottom cap of the cell followed by a glass microfiber separator (Whatman grade GF/A, 1.6 μm), 200 μL of WIS electrolyte, and cathode.
Galvanostatic measurements were done using the battery analyzer (MTI Corporation, BST8-WA) at a 0.2 or 0.5C rate for a voltage range of 0.8 V to 2.5 V at room temperature. Electrochemical impedance spectroscopy was done at OCV using an impedance analyzer (Gamry 1010E) using a frequency range of 0.01 Hz to 0.5 MHz with 10 mV of AC voltage. Before measurement, all battery samples were rested for a minimum of 5 hours. To determine the impedance contribution from individual electrode components, a symmetric cell approach was taken for impedance characterization, where a pair of coin cells were charged to a 100% state-of-charge (SOC) followed by storage at room temperature for a minimum of 4 days. After storage, each cell was opened and the anode of one cell was swapped with the cathode of the other to create two symmetric cells as shown in the ESI.†
X-ray diffraction (XRD) patterns were collected using a Rigaku Ultima III with Cu source (accelerating voltage and current of 40 kV and 44 mA) with a 1.5° min−1 of scan speed and a range taken as 2θ = 20–80°. Raman spectra were obtained using a Horiba LabRam Aramis Confocal Microscope with a 532 nm excitation laser and a 50× LWD objective lens. The spot size was 1–2 μm with power density of 0.64–2.6 mW μm−2. Transmission electron microscopy (TEM) images were collected using a JEOL 3010 electron microscope at an accelerating voltage of 300 kV. The thermogravimetric analyses (TGA, TA Instruments 5500) of the sol–gel TiO2 and C-TiO2 were done using 10 °C min−1 of heating rate under air.
3. Results/discussion
TiO2 was commonly used as a photoanode for photoelectrochemical water splitting30–33 and has low conductivity due to its wide bandgap (3.2 eV).34 To overcome these drawbacks for battery applications, an artificial SEI layer of carbon was introduced onto the TiO2 surface. The carbon coating prevents a direct electrochemical contact between the anode and water molecule thereby minimizing water electrolysis. We controlled the properties of this carbon layer by adjusting two key parameters, (1) the annealing temperature and (2) the mass ratio of active material to carbon precursor. The mass ratio of active material and glucose precursor determined the amount of carbon in C-TiO2 and the annealing temperature changed the amount of graphitic carbon as well as the conductivity. The annealing temperature must be carefully chosen because of the possibility of TiO2 phase transformation from anatase to rutile.We first determined the ideal range of annealing temperature of C-TiO2 by investigating the evolution of crystal phase using XRD (Fig. 2A). Only the annealing temperature was varied while keeping the ratio of TiO2 to glucose constant at 1:4 to find appropriate annealing temperature windows for C-TiO2 synthesis. Because anatase TiO2 showed a better lithium storage capacity than rutile TiO2,35 we aimed to preserve anatase phase during the carbon coating process. As some degree of phase transformation from anatase to rutile was observed in XRD after annealing C-TiO2 at 900 °C, annealing temperature below 900 °C was set as a limit of the C-TiO2 synthesis process in this study. Crystal sizes (Table 1) did not appreciably change within this annealing temperature range (400–900 °C), that could originate from the constrained growth due to the carbon coating.28 This provides a good similarity of TiO2 used for different coating conditions enabling us to evaluate the effect of carbon layer on aqueous LIB performance.
 | ||
| Fig. 2 The material properties of carbon coated TiO2 (C-TiO2). (A) XRD patterns of C-TiO2 measured with a scan speed of 1.5° min−1 for a range of 2θ = 20–80°. (B) The weight fractions of carbon in C-TiO2 synthesized with varied precursor ratios of TiO2 to glucose mixture using TGA with a heating rate of 10 °C min−1 under air. | ||
| Material | Annealing temperature | Phase | Crystalline size (Å) | Lattice parameters (Å) | ||
|---|---|---|---|---|---|---|
| a | b | c | ||||
| Sol–gel TiO2 | 400 | Anatase | 130 | 3.787 | 3.789 | 9.505 |
| C-TiO2 | 400 | Anatase | 161 | 3.789 | 3.789 | 9.505 |
| 500 | Anatase | 142 | 3.787 | 3.787 | 9.505 | |
| 600 | Anatase | 137 | 3.784 | 3.784 | 9.498 | |
| 700 | Anatase | 145 | 3.793 | 3.793 | 9.516 | |
| 800 | Anatase | 154 | 3.788 | 3.788 | 9.501 | |
| 900 | Rutile (69%) | >1000 | 4.593 | 4.593 | 2.958 | |
| Anatase (31%) | >814 | 3.785 | 3.785 | 9.509 | ||
The amount of the carbon shell present in C-TiO2 needs to be sufficient enough to enhance the conductivity of the battery electrode but also needs to be as small as possible to lower the diffusion barrier for lithium-ions to reach active materials. To develop an ideal composition of C-TiO2 for aqueous LIB, we varied mass ratios of TiO2 to glucose as 1:2, 1:4, 1:6, and 1:9 during the synthesis. The amounts of carbon in C-TiO2 synthesized with different ratios of glucose precursor to TiO2 were determined by TGA. TGA plots presented two plateaus for all different compositions of C-TiO2 (Fig. S1†), with the first plateau starting around 100 °C and the second plateau starting around 400 °C. At the first plateau, a total weight loss of around 2% was observed, corresponding to the evaporation of physically absorbed water. The second plateau (350–400 °C) is expected to originate from the carbon decomposition in C-TiO2. This second plateau is lower than the decomposition temperature of graphite36,37 possibly due to the incomplete graphitization of carbon in C-TiO2. In summary, the weight percentages of carbon in C-TiO2 composites were around 10, 18, 50, and 70% for C-TiO2 composites synthesized with 1:2, 1:4, 1:6, and 1:9 of precursor ratios (Fig. 2B).
The electrochemical performances of C-TiO2 anodes with different carbon amounts were investigated in aqueous LIB (Fig. 3). Fig. 3A shows the 1st and 40th cycling voltage profiles of aqueous LIB with sol–gel TiO2 or C-TiO2 anodes (fabricated with 1:2, 1:4, 1:6, and 1:9 of precursor ratios) tested against LiMn2O4 cathodes with 21 m LiTFSI aqueous electrolyte at 0.2C rates. During the first charging step, some irreversible loss of charges might be associated with the SEI layer formation on the surface of anode.12 As discussed earlier, this SEI layer acts as a passivation layer and blocks direct contact between electrode and water molecules, preventing further electrolyte decomposition. This irreversible capacity loss due to the SEI formation presented lower coulombic efficiencies for the initial 10 cycles for all compositions of anodes (Fig. 3C). Among C-TiO2 anodes fabricated with different weight fractions of carbon, C-TiO2 anodes made with 1:4 of precursor ratio showed better improvements in cycle life and coulomb efficiency than other C-TiO2 anodes. Having the carbon layer on anodes lowered the initial capacity, however, C-TiO2 anodes showed a better capacity retention in the following cycles than that of the battery tested with sol–gel TiO2 anodes (Fig. 3A and B). C-TiO2 anodes with higher carbon ratios (1:6 and 1:9) showed a lower coulombic efficiency than lower ratios (1:2 and 1:4) at 40th cycle. This may be attributed to the loss of lithium ions in thick interlayers made of large amounts of carbon precursors. Although the first discharge capacity of sol–gel TiO2 was higher than that of C-TiO2 with 1:4 ratio, C-TiO2 anodes with 1:4 ratio showed almost four times higher capacity at the end of 40th cycle than that of sol–gel TiO2. This suggests that 1:4 ratio is optimal to form stable SEI layers for aqueous LIB.
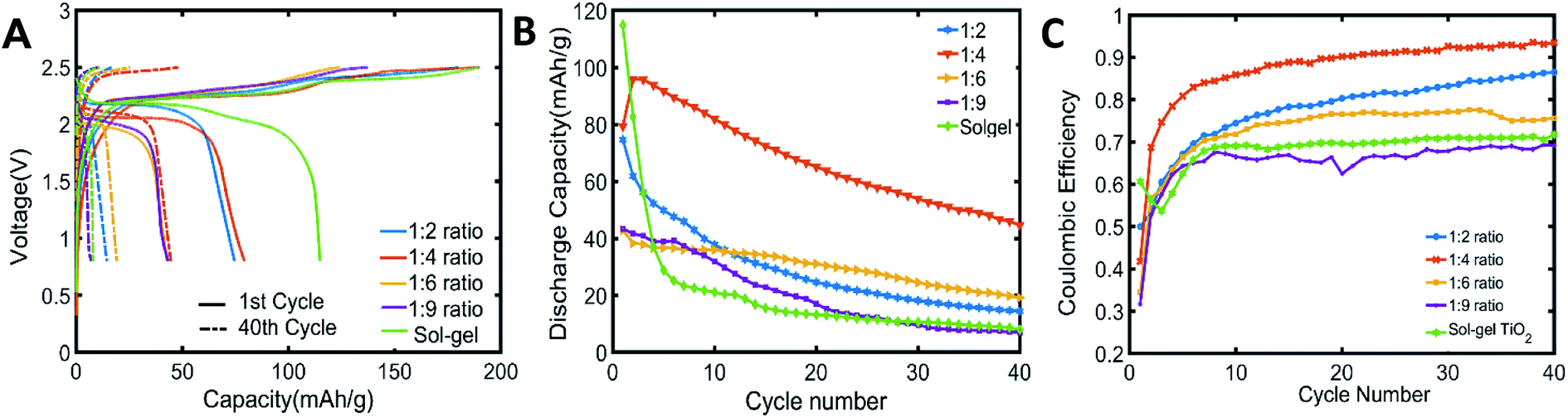 | ||
| Fig. 3 The aqueous LIB performance of C-TiO2 anodes with varied amounts of carbon shell in C-TiO2. The batteries were tested with WIS electrolyte (21 m LiTFSI in H2O) and LiMn2O4 cathode at C/5. The carbon layer on C-TiO2 was formed by annealing at 600 °C. (A) Voltage profiles of aqueous batteries at the 1st and 40th cycle with anodes made of sol–gel TiO2 or C-TiO2 synthesized with different weight fractions of precursors (1:2,1:4,1:6 and 1:9). (B) Cycling performances of aqueous LIB with sol–gel TiO2 anodes or C-TiO2 anodes fabricated with different mass ratios and corresponding (C) coulombic efficiencies. | ||
To investigate the influence of graphitic layer of C-TiO2 nanoparticles on aqueous LIB performance, the annealing temperatures were adjusted to control the fraction of graphitic carbon in C-TiO2 nanoparticles. The carbon amount in C-TiO2 was fixed the same as 18% or 70% (prepared with the precursor ratio of 1:4 and 1:9, respectively), and the annealing temperature was varied from 400 °C to 600 °C. The carbon layer distribution and its composition were investigated using TEM and Raman spectroscopy. While a clean edge of TiO2 crystal was observed for the TiO2 samples without carbon coatings (Fig. 4A), around 2–5 nm thick carbon layers were surrounding TiO2 crystals in C-TiO2 nanoparticles as can be seen in Fig. 4B. Using the Raman spectra of C-TiO2 nanoparticles, the effect of carbon layer annealing temperature on graphitization was studied. First, we observed the signature peak of TiO2 (Eg, A1g, and B1g peaks) in the Raman spectra of C-TiO2 confirming the presence of anatase TiO2 nanoparticles (Fig. S2†).38,39 The D and G peaks were found at ∼1360 cm−1 and ∼1560 cm−1 in the Raman spectra of C-TiO2 (Fig. 4C), with a noticeable absence of the second order D peak.40 This is characteristic of amorphous carbons with little to no long range order, consistent with literature using this carbon coating process.28 The effects of annealing temperature on the composition of the carbon coating were further analyzed using the D peak intensity divided by the G peak intensity. The G peak is associated with graphitic in-plane vibrations with E2g symmetry, while the D peak is associated with defective graphite.11 As such the ID/IG parameter is commonly used to characterize disorder in a carbon sample. Given that graphitic carbon has greater conductivity than amorphous carbon, it is desirable to minimize ID/IG to further improve the conductivity of the carbon layer. Using Fig. 4D, there is a clear tendency towards decreasing ID/IG as the heat treatment temperature of the coating process increases from 400 °C to 600 °C for both C-TiO2 samples prepared with 1:4 and 1:9 precursor ratios.
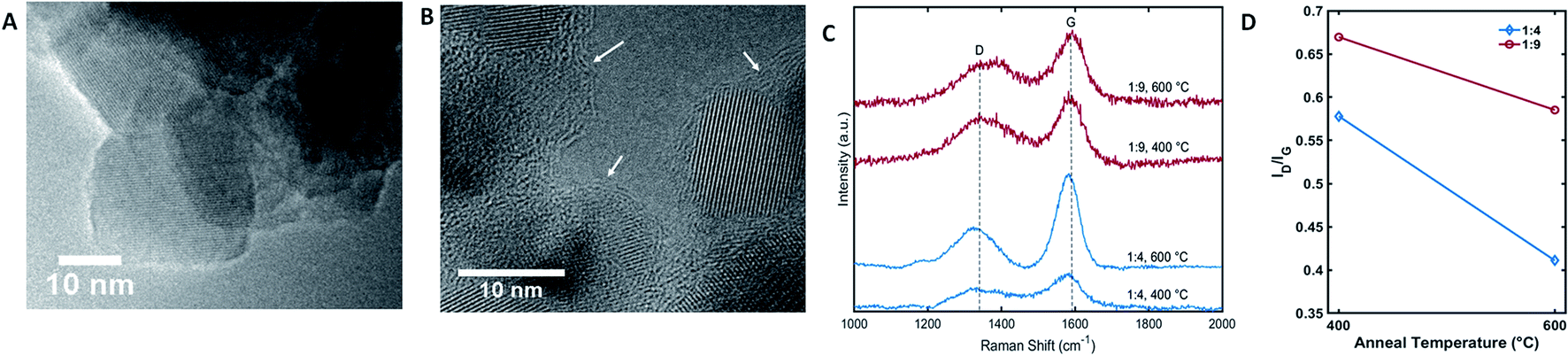 | ||
| Fig. 4 The properties of carbon layer on C-TiO2. TEM images (A) sol–gel TiO2 and (B) C-TiO2 with 1:4 of precursor ratio and annealed at 600 °C. Crystalline lattices corresponding to anatase TiO2 were observed in both (A) and (B). The arrows in (B) indicate the carbon layer surrounding TiO2 particles. Raman spectra (C) of C-TiO2 synthesized with different precursor ratios of TiO2:glucose and annealing temperatures. The intensities of ID and IG were compared by calculating ID/IG ratio (D) of C-TiO2 synthesized with different mass ratios and annealing temperatures. | ||
The variation in disorder of carbon layer had a significant influence on the aqueous LIB performance (Fig. 5). Annealing C-TiO2 at 600 °C was the most effective to improve the cycle performance compared to 400 °C, 800 °C or 900 °C (Fig. 5A, B and S3†). The first cycle coulombic efficiency of anodes with C-TiO2 annealed at 400 °C was similar to that of C-TiO2 annealed at 600 °C. However, the first discharge capacity of C-TiO2 annealed at 400 °C was only 60 mA h g−1 while C-TiO2 annealed at 600 °C showed about ∼35% higher first discharge capacity. The irreversible capacity loss during the initial cycles is associated with the formation of SEI, thus, C-TiO2 annealed at 400 °C did not form a stable SEI layer as can be seen from its lower coulombic efficiencies than that of 600 °C. Coulombic efficiencies of C-TiO2 anodes treated at 600 °C were maintained higher than that of 400 °C until 40th cycle (Fig. 5C), indicating a stable formation of SEI in aqueous LIB. We also expect that the carbon layer's increased conductivity might improve the battery performance compared to the C-TiO2 nanoparticles treated at 400 °C. However, further increasing the temperature to 900 °C resulted in the phase transformation of anatase TiO2 nanoparticles degrading the battery performance due to inferior electrochemical activity of rutile compared to anatase (Fig. S3†).23 Therefore, it is suggested to treat the C-TiO2 composite below 900 °C to maximize its aqueous LIB performance. In previous works, LiF, Li2O, LiOH, and Li2CO3 were observed on the anode surface in WIS based LIB after cycling.14,41 These are the main components comprising the SEI layer with WIS electrolytes. The defective carbon layer made by a hydrothermal carbonization process of carbohydrate could be a good source of precursor for Li2O, LiOH, and Li2CO3 formation. How the defective carbon layer and TFSI anions coordinate to produce LiF or the detail analysis of interface in WIS based LIB would require in situ analysis that remains as our future study.
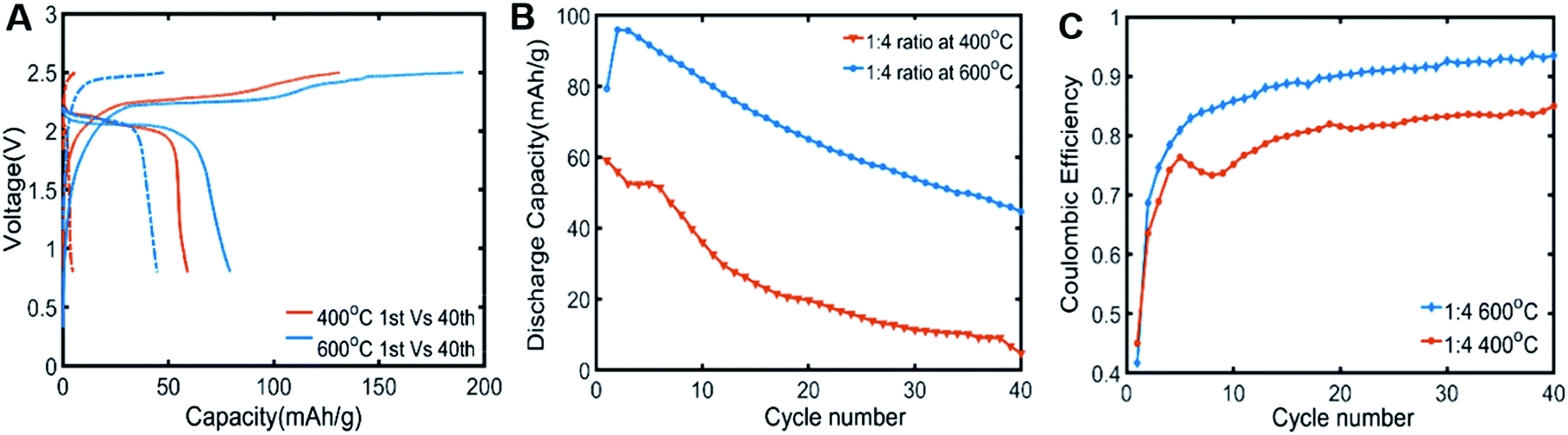 | ||
| Fig. 5 The full cell performance of C-TiO2 anodes with different annealing temperature of carbon layer. The precursor ratio for C-TiO2 synthesis was fixed as 1:4 (TiO2:glucose). (A) Voltage profiles of aqueous batteries at the 1st and 40th cycle tested with C-TiO2 anodes annealed at 400 °C and 600 °C. (B) Cycling performance and (C) corresponding coulombic efficiency of C-TiO2 anodes annealed at 400 °C and 600 °C. | ||
To better understand the effect of the artificial SEI on battery degradation, electrochemical impedance spectroscopy was used to characterize the surface layer and charge transfer impedance evolution over cycling as shown in Fig. 6. The equivalent circuit model used for fitting our system is shown in the inset of Fig. 6A, where RS and RCT are associated with resistances arising from surface layer formation and charge transfer of the anode material, respectively. We assigned these resistances to the anode based on the result from the symmetric cell approach, which shows that impedance from the cathode does not contribute greatly over cycling as shown in Fig. S4.† Typically, RS and RCT are associated with the high (>10 kHz) and medium (0.16–630 Hz) frequency semicircles42–44 and we applied a model that fits the high and medium frequency regions well.45 Overall, the carbon coating on anodes greatly reduced impedance evolution over cycling. The cycled electrodes were also investigated with SEM and XRD but no significant changes were observed (Fig. S5†). The surface layer impedance at 100 cycles for uncoated and coated TiO2 are 101 and 27.61 ohms, respectively, and the charge transfer resistances for uncoated and coated TiO2 are 982.2 and 167.6 ohms respectively, as summarized in Table 2. The decreased RS and RCT of C-TiO2 are attributed to the carbon coating that we applied on TiO2. For coated TiO2 anodes, RS did not greatly increase over cycling because carbon layers are expected to inhibit further growth of surface layer from electrolyte decomposition as it can be also supported by improved coulombic efficiencies. In addition, improved conductivity of anode particles with carbon layer might enhance the charge transfer during the battery reaction, thus enhancing overall battery performances.
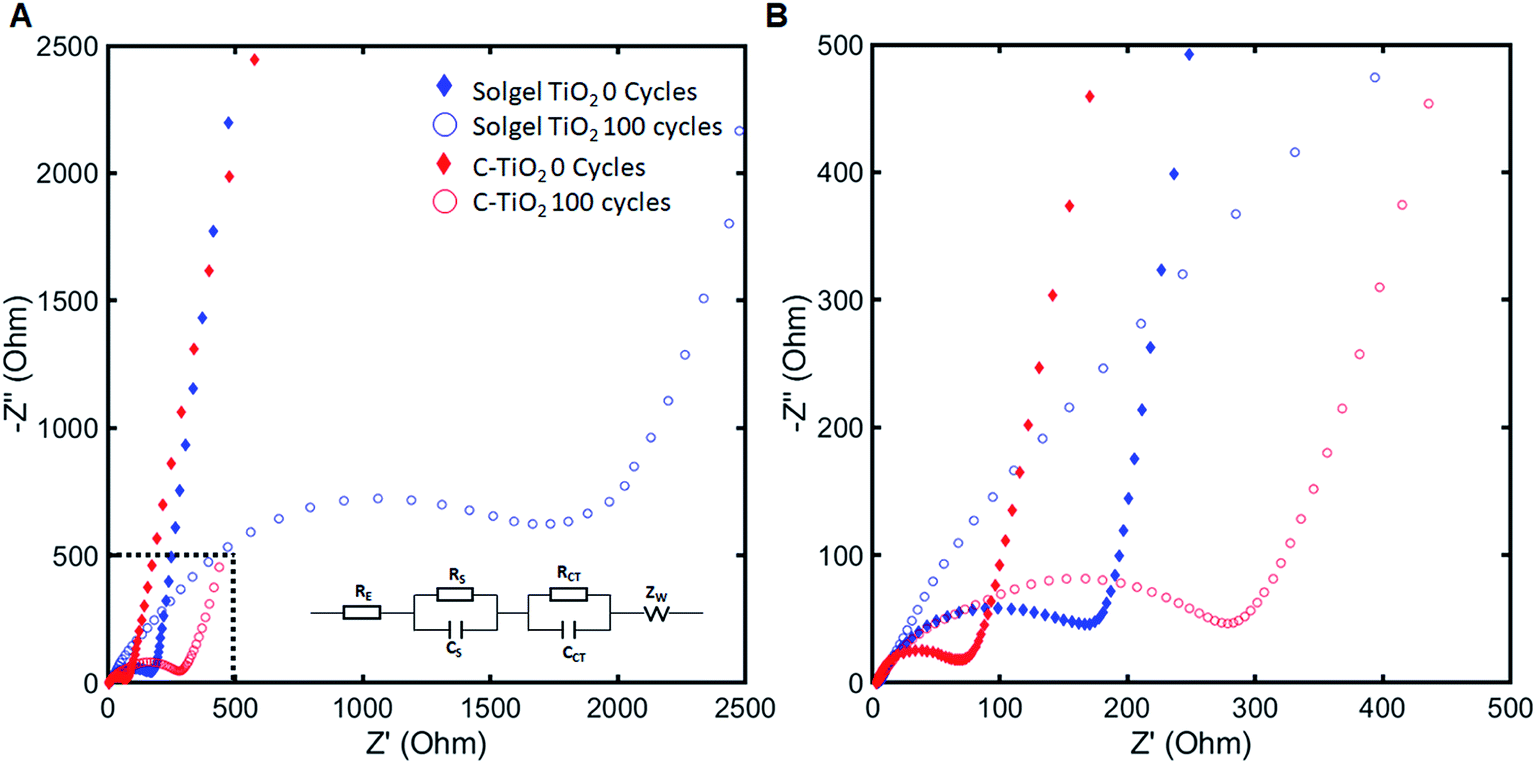 | ||
| Fig. 6 Impedance analysis of aqueous batteries with sol–gel TiO2 anodes and C-TiO2 anodes (1:4 of precursor ratio and annealed at 600 °C). (A) Nyquist plots of coated (C-TiO2) and uncoated (sol–gel TiO2) batteries for 0 and 100 cycles and proposed model (inset). (B) High and middle frequency regions of Nyquist plot in (A). | ||
| Material | Initial RS (ohm) | 100 cycle RS (ohm) | Initial RCT (ohm) | 100 cycle RCT (ohm) |
|---|---|---|---|---|
| Sol–gel TiO2 | 0 | 101.0 | 78.23 | 982.2 |
| C-TiO2, 600 °C | 38.08 | 27.61 | 1.048 | 167.6 |
4. Conclusion
In summary, the effect of carbon coating on anodes on aqueous LIB performance was investigated. Using a versatile carbon coating method, TiO2 nanoparticles were encapsulated, improving cycle performance and coulombic efficiency of the anode by 11 times and 29% respectively over sol–gel TiO2 anodes. We attribute these improvements to: (1) the conductive carbon layer, reducing charge transfer resistance and (2) the reduced contact between the water molecules and the anode, lowering the rate of electrolyte decomposition. Our findings show that 18 wt% of carbon fractions to encapsulate TiO2 is ideal to improve aqueous LIB performance. At this synthesis condition, the thickness of the carbon layer was sufficient to inhibit the electrolyte decomposition while allowing the charge transfer. We also found a calcination temperature of 600 °C to be ideal as the conductivity of the carbon layer was maximized while maintaining the anatase phase of the TiO2. To further enhance the energy storage capability of aqueous LIB, selecting anode materials with higher theoretical capacity and lower redox potential than TiO2 would be essential. For carbon-based artificial SEI, we found that the thickness and composition of carbon play a critical role in battery performance. Our findings suggest that controlling the property of the nanometer thick protective layer can greatly enhance the battery performance and an effective artificial SEI layer would enable us to develop safe and long-lasting next-generation aqueous LIB.Author contributions
U. S. and C. C. performed and designed the experiment. V. L, R. R. and A. B. synthesized sol–gel TiO2. G. A. C. C and P. S. performed carbon coating experiment. B. Y. and C. C. performed the impedance analysis. F. E. supervised the synthesis process of sol–gel TiO2. D. O. supervised the project. All authors have given approval to the final version of the manuscript.Funding sources
This work is supported by the professional development start-up fund from San José State University and LG Chem Global Innovation Contest.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the support from Prof. David Wagner and Prof. Craig England for performing TGA and XRD analysis and Dr Emory Chan and Dr Rohan Dhall for assisting TEM and Raman measurements. Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The authors thank LG Chem (GIC) and San José State University (Professional development grant) for funding supports.References
- W. Li, J. R. Dahn and D. S. Wainwright, Science, 1994, 264, 1115–1118 CrossRef CAS.
- J.-Y. Luo, W.-J. Cui, P. He and Y.-Y. Xia, Nat. Chem., 2010, 2, 760–765 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- C. Yang, J. Chen, T. Qing, X. Fan, W. Sun, A. von Cresce, M. S. Ding, O. Borodin, J. Vatamanu, M. A. Schroeder, N. Eidson, C. Wang and K. Xu, Joule, 2017, 1, 122–132 CrossRef CAS.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 RSC.
- Y. Wang, J. Yi and Y. Xia, Adv. Energy Mater., 2012, 2, 830–840 CrossRef CAS.
- W. Li, W. R. McKinnon and J. R. Dahn, J. Electrochem. Soc., 1994, 141, 2310–2316 CrossRef CAS.
- H. Buqa, A. Würsig, J. Vetter, M. E. Spahr, F. Krumeich and P. Novák, J. Power Sources, 2006, 153, 385–390 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- S. J. An, J. Li, C. Daniel, D. Mohanty, S. Nagpure and D. L. Wood, Carbon, 2016, 105, 52–76 CrossRef CAS.
- X. Yu and A. Manthiram, Energy Environ. Sci., 2018, 11, 527–543 RSC.
- H. Kim, J. Hong, K.-Y. Park, H. Kim, S.-W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed.
- L. Suo, D. Oh, Y. Lin, Z. Zhuo, O. Borodin, T. Gao, F. Wang, A. Kushima, Z. Wang, H.-C. Kim, Y. Qi, W. Yang, F. Pan, J. Li, K. Xu and C. Wang, J. Am. Chem. Soc., 2017, 139, 18670–18680 CrossRef CAS PubMed.
- L. Suo, O. Borodin, W. Sun, X. Fan, C. Yang, F. Wang, T. Gao, Z. Ma, M. Schroeder, A. von Cresce, S. M. Russell, M. Armand, A. Angell, K. Xu and C. Wang, Angew. Chem., Int. Ed., 2016, 55, 7136–7141 CrossRef CAS PubMed.
- C. Yang, L. Suo, O. Borodin, F. Wang, W. Sun, T. Gao, X. Fan, S. Hou, Z. Ma, K. Amine, K. Xu and C. Wang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6197–6202 CrossRef CAS PubMed.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS.
- H. Liu, W. Li, D. Shen, D. Zhao and G. Wang, J. Am. Chem. Soc., 2015, 137, 13161–13166 CrossRef CAS PubMed.
- G. Wang, H. Liu, J. Liu, S. Qiao, G. M. Lu, P. Munroe and H. Ahn, Adv. Mater., 2010, 22, 4944–4948 CrossRef CAS PubMed.
- M. Madian, A. Eychmüller and L. Giebeler, Batteries, 2018, 4, 7 CrossRef.
- H. Lahan, R. Boruah, A. Hazarika and S. K. Das, J. Phys. Chem. C, 2017, 121, 26241–26249 CrossRef.
- S. M. Gupta and M. Tripathi, Chin. Sci. Bull., 2011, 56, 1639 CrossRef CAS.
- S. S. El-Deen, A. M. Hashem, A. E. Abdel Ghany, S. Indris, H. Ehrenberg, A. Mauger and C. M. Julien, Ionics, 2018, 24, 2925–2934 CrossRef CAS.
- D. W. Murphy, R. J. Cava, S. M. Zahurak and A. Santoro, Solid State Ionics, 1983, 9–10, 413–417 CrossRef CAS.
- I. Issac, M. Scheuermann, S. M. Becker, E. G. Bardají, C. Adelhelm, D. Wang, C. Kübel and S. Indris, J. Power Sources, 2011, 196, 9689–9695 CrossRef CAS.
- D. Su, S. Dou and G. Wang, Chem. Mater., 2015, 27, 6022–6029 CrossRef CAS.
- A. Bokare, M. Pai and A. A. Athawale, Sol. Energy, 2013, 91, 111–119 CrossRef CAS.
- L.-W. Zhang, H.-B. Fu and Y.-F. Zhu, Adv. Funct. Mater., 2008, 18, 2180–2189 CrossRef CAS.
- M. T. García-Valverde, R. Lucena, F. Galán-Cano, S. Cárdenas and M. Valcárcel, J. Chromatogr. A, 2014, 1343, 26–32 CrossRef PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- J. H. Park, S. Kim and A. J. Bard, Nano Lett., 2006, 6, 24–28 CrossRef CAS.
- A. J. Cowan, J. Tang, W. Leng, J. R. Durrant and D. R. Klug, J. Phys. Chem. C, 2010, 114, 4208–4214 CrossRef CAS.
- Z.-W. Yin, S. B. Betzler, T. Sheng, Q. Zhang, X. Peng, J. Shangguan, K. C. Bustillo, J.-T. Li, S.-G. Sun and H. Zheng, Nano Energy, 2019, 62, 507–512 CrossRef CAS.
- C. Dette, M. A. Pérez-Osorio, C. S. Kley, P. Punke, C. E. Patrick, P. Jacobson, F. Giustino, S. J. Jung and K. Kern, Nano Lett., 2014, 14, 6533–6538 CrossRef CAS.
- V. Aravindan, Y.-S. Lee, R. Yazami and S. Madhavi, Mater. Today, 2015, 18, 345–351 CrossRef CAS.
- Y. Devrim and A. Albostan, J. Electron. Mater., 2016, 45, 3900–3907 CrossRef CAS.
- G. Zhang, M. Wen, S. Wang, J. Chen and J. Wang, RSC Adv., 2018, 8, 567–579 RSC.
- T. Ohsaka, F. Izumi and Y. Fujiki, J. Raman Spectrosc., 1978, 7, 321–324 CrossRef.
- W. F. Zhang, Y. L. He, M. S. Zhang, Z. Yin and Q. Chen, J. Phys. D: Appl. Phys., 2000, 33, 912–916 CrossRef CAS.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS.
- N. Dubouis, P. Lemaire, B. Mirvaux, E. Salager, M. Deschamps and A. Grimaud, Energy Environ. Sci., 2018, 11, 3491–3499 RSC.
- V. J. Ovejas and A. Cuadras, Batteries, 2018, 4, 43 CrossRef.
- A. Barai, K. Uddin, W. D. Widanage, A. McGordon and P. Jennings, Sci. Rep., 2018, 8, 1–13 CrossRef CAS PubMed.
- J. G. Zhu, Z. C. Sun, X. Z. Wei and H. F. Dai, RSC Adv., 2014, 4, 29988–29998 RSC.
- H. Dai, B. Jiang and X. Wei, Energies, 2018, 11, 220 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08268a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |