
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImmobilization of poly(lactide-co-glycolide) microspheres on bone implant materials for antibiotic release and the binding mechanisms†
Dongwei Wang‡
*a,
Dongqin Xiao‡ b,
Mengjie Luc,
Qing Liud,
Tao Xied,
Gang Fengb,
Jie Wengd and
Ke Duan*e
b,
Mengjie Luc,
Qing Liud,
Tao Xied,
Gang Fengb,
Jie Wengd and
Ke Duan*e
aCollaborative Innovation Center of Tissue Repair Materials of Sichuan Province, College of Life Sciences, China West Normal University, Nanchong, Sichuan, 637009 China. E-mail: wdw_swjtu@163.com; keduan@swmu.edu.cn
bResearch Institute of Tissue Engineering and Stem Cells, Nanchong Central Hospital, The Second Clinical College of North Sichuan Medical College, Sichuan, 637000 China
cDepartment of Pathology, Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, 646000 China
dKey Laboratory of Advanced Technologies of Materials, School of Materials Science and Engineering, Southwest Jiaotong University, Chengdu, Sichuan 610031, China
eSichuan Provincial Laboratory of Orthopaedic Engineering, Department of Bone and Joint Surgery, Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, 646000 China
First published on 18th February 2020
Abstract
Bone implants are susceptible to postoperative infections. Immobilization of antibiotic-loaded microparticles on implants is an effective approach to addressing this problem. Immobilization methods reported in earlier studies frequently used special or potentially harmful conditions. Therefore, the present study explored a new method to immobilize poly(lactide-co-glycolide) (PLGA) microspheres on bone implant materials. PLGA microspheres were prepared by an emulsion method using polyvinyl alcohol (PVA) as an emulsifier. The microspheres were immobilized on two commonly used orthopaedic biomaterials [hydroxyapatite-coated titanium (HA-Ti) and poly(methyl methacrylate) (PMMA)] by dispersing on the surface followed by vacuum drying. Microspheres were retained stably on both materials even after immersion in phosphate-buffered saline for 12 d. Pretreatment of microspheres with sodium borate (i.e., an eliminator of hydroxyl groups of PVA) substantially reduced their retention on HA-Ti, but only moderately reduced their retention on PMMA. This suggested that the binding of the residual PVA on the microspheres to the HA coating is the dominant contributor to their immobilization on HA-Ti, whereas other forces contributed substantially to their immobilization on PMMA. Microspheres containing ciprofloxacin (a water-soluble antibiotic) and triclosan (an oil-soluble antibiotic) were immobilized on HA-Ti and PMMA, respectively. They effectively killed adjacent bacteria. These results offer a simple and versatile method for immobilizing drug-release microspheres on some important bone implant surfaces.
Introduction
Skeletal disorders are an increasing global threat to human health. Implants manufactured from various materials are used for managing these problems. Titanium (Ti) alloys are widely used as artificial joints because of their ability to integrate with bones. Bioceramics, such as hydroxyapatite [HA, Ca10(OH)2(PO4)6], are chemically related to human bone mineral and are extensively applied as surface coatings on metallic artificial joints.1 Polymers are relatively easy to shape and frequently used when complex geometries are involved.1,2 For example, poly(methyl methacrylate) (PMMA) has been routinely used as bone cement in arthroplasty and more recently in craniofacial implants.3Bone implants face a common risk of postoperative infections.4 Implant-related infections (IRIs) are difficulty to treat by systemic therapies because bacteria colonizing the implant surface develop a biofilm to resist drug penetration, as well as because the surgery disrupts the blood circulation near the bone-implant interface.4,5 Consequently, local release of antimicrobial substances from the implant surface has been recognized as a promising approach to preventing IRIs.6
A number of methods have been reported for loading drugs on bone implant surfaces. Many works achieved this by soaking in a drug solution or codeposition of drugs in calcium phosphate coatings on implants.7,8 However, they either cannot control the drug release rate or require the drug molecules to have specific functional groups (e.g., carboxyl).7,8 Moreover, these aqueous-based methods are unsuitable for drugs insoluble or sparingly soluble in water. Polylactide (PLA) and related polymers are safe, biodegradable, and suited for the encapsulation of both water- and oil-soluble drugs.9,10 Studies thus have prepared gentamicin-loaded PLA coatings on Ti intramedullary nails for preventing IRIs,11 and these devices have been used clinically in Europe. However, PLA and related polymers are hydrophobic and do not integrate closely with bones,12 making them non-ideal for implants requiring strong skeletal fixation (e.g., artificial joints). Additionally, the coating process uses organic solvents, which can damage implants of certain polymers (e.g., PMMA). To address these disadvantages, several studies reported immobilization of polymer microparticles on implant surfaces. It is expected that, without fully covering the implant surface, the microparticles may not disrupt bone-implant integration. Mulia et al. attached poly(lactide-co-glycolide) (PLGA) microspheres to metals by a supercritical carbon dioxide (CO2) treatment.13 However, this method requires special equipment and may not be used for polymeric substrates, as supercritical CO2 is a solvent and plasticizer for many polymers.14 Son et al. treated PLGA microspheres with polyethyleneimine to create a positively charged surface, and immobilized them on HA-coated Ti via electrostatic attraction.15 However, polyethyleneimine is cytotoxic and it is unclear whether the method is applicable to less or non-ionic surfaces such as many polymers.16 Therefore, it is still necessary to develop more generally-suitable methods for immobilizing microspheres on biomaterials without the use of special or potentially harmful conditions.
Here, we report a new method of immobilizing PLGA microspheres on two biomaterials. By a simple vacuum-drying treatment, PLGA microspheres were immobilized on HA-coated Ti and PMMA to allow antibiotic release. By selecting chemical blocking of functional groups associated with microsphere surface, we also probed possible mechanisms underlying immobilization on these two surfaces.
Experimental
Preparation of HA-coated Ti discs
Triethyl phosphite (28.0 g) was mixed with 12.0 g of water and stirred (1200 rpm, 24 h) to form a sol.17 A 4 mol l−1 calcium nitrate [Ca(NO3)2] solution was added to the sol to a Ca/P molar ratio of 1.67. After standing for 24 h, the sol was diluted with water (sol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water = 1:3; v/v). Ti alloy discs (Ti6Al4V, Φ10 × 2 mm; Zhirui Metals, Baoji, China) were abraded to a 1200-grit finish and sonicated in acetone, ethanol, and water. They were soaked in the sol, withdrawn (1 cm min−1), dried (80 °C) overnight, and calcined (500 °C, 1 h) to form HA-coated Ti discs. These samples are abbreviated as HA-Ti.
:water = 1:3; v/v). Ti alloy discs (Ti6Al4V, Φ10 × 2 mm; Zhirui Metals, Baoji, China) were abraded to a 1200-grit finish and sonicated in acetone, ethanol, and water. They were soaked in the sol, withdrawn (1 cm min−1), dried (80 °C) overnight, and calcined (500 °C, 1 h) to form HA-coated Ti discs. These samples are abbreviated as HA-Ti.
Preparation of microspheres
Drug-free PLGA microspheres were prepared by an emulsion method. PLGA (400 mg; Mw: 60 K; lactide:glycolide = 75:25; Daigang Biotech, Jinan, China) was dissolved in 16 ml of dichloromethane (DCM), dropped in 100 ml of a 10 mg ml−1 polyvinyl alcohol solution (PVA, polymerization degree: 1700, 99% hydrolysis) over an ice bath, and stirred (800 rpm, 10 min) to form an emulsion. The ice-bath was removed, and the emulsion was stirred (400 rpm, 4 h) to remove DCM. Microspheres were collected by centrifugation (4000 rpm, 3 min), rinsed thrice with water, and lyophilized (Advantage, SPS Scientific, NY, USA).
PLGA microspheres containing ciprofloxacin (CIP), a water-soluble antibiotic used for treating bone infections,18 was prepared by adding 1 ml of a 25 mg ml−1 CIP solution (Bio Basic, Shanghai, China) to 4 ml of DCM containing 175 mg of predissolved PLGA. It was sonicated (power: 150 W; VCX 500; Sonics, Newtown, CT, USA) for 1 min to form an emulsion, and dropped in 60 ml of a 10 mg ml−1 PVA solution over an ice bath. After removal of ice bath, the emulsion was stirred (400 rpm) for 3 h to evaporate off DCM. Subsequent procedures were same as above. These microspheres were termed CIP-PLGA.
Immobilization of PLGA microspheres on HA-coated Ti discs
PLGA microspheres (15 mg) were suspended in 1 ml of ultrapure water, and 100 μl of the suspension was pipetted on each HA-Ti disc. The disc was transferred to a desiccator and vacuum-dried for 2 h, thereby immobilizing the microspheres on the surface.The stability of microspheres immobilized on HA-Ti discs was studied by immersion in phosphate buffered saline (PBS, pH 7.4). Discs were placed in centrifuge tubes containing 3 ml of PBS and kept in a shaking incubator (37 °C, 70 rpm) for up to 12 d. The discs were regularly transferred to new tubes containing 3 ml of fresh PBS. The liquid in the original tube was digested with 3 ml of 1 mol l−1 sodium hydroxide (NaOH; 37 °C, 48 h) and analyzed for total organic carbon content (TOC; Vario TOC, Elementar GmbH, Hanau, Germany). In the TOC analyzer, all organic matter was burned to CO2, and the CO2 level was measured with an infrared detector and converted to total carbon against a calibration curve established with standard potassium hydrogen phthalate solutions. At the end of the experiment, the microspheres remaining on the disc was shaken off by prolonged sonication in PBS, with repeated examinations under an optical microscope to ensure complete detachment of microspheres. This PBS was also analysed for TOC. Then, the proportion of microspheres retained on the disc at each timepoint was calculated based on the ratio of relevant TOCs.
To examine the contribution of vacuum drying to the immobilization, control samples were prepared as follows. The microsphere suspension was pipetted on HA-Ti, allowed to stand for 5 min, and (without vacuum drying) studied for stability. Throughout this study, each experiment included three parallel samples.
To study the role of residual PVA (see Results) on microsphere surface in the immobilization, microspheres were treated with a 30 mg ml−1 sodium borate solution (Na2B4O7, pH 9.2) for 2 h before immobilization on HA-Ti by vacuum drying. Na2B4O7 reacts efficiently with the –OH groups of PVA.19 As the high pH of the borate solution may be a complicating factor, some microspheres were treated in a 30 mg ml−1 phosphate solution (Na2HPO4, pH adjusted to 9.2) for 2 h, and then immobilized on HA-Ti by vacuum drying to serve as a control.
In vitro CIP release
CIP-PLGA (30 mg) were kept in 3 ml of PBS (37 °C) for 15 d; 1 ml was periodically sampled for UV-Vis analysis of CIP (277 nm, Shimadzu UV-250), and the PBS was replenished. After 15 d, the microspheres were collected, dissolved with DCM, and analyzed for residual CIP. The encapsulation efficiency (i.e., percentage of drug found in microspheres relative to drug added into the synthetic reactor) and cumulative release were calculated.In vitro antibacterial property
HA-Ti discs carrying immobilized CIP-PLGA microspheres were prepared and tested qualitatively and quantitatively for antibacterial property using two major pathogens responsible for IRIs: Staphylococcus aureus (S. aureus; ATCC 12228) and Escherichia coli (E. coli; ATCC 25922; both from Guangdong Provincial Microorganism Preservation Center, Guangzhou, China). In the qualitative test, sterile agar (Aobox Biotech, Beijing, China) was poured into Petri dishes. After agar solidification, 1 ml of the bacterial suspension (1 × 108 CFU ml−1) was inoculated on the agar, and discs were gently placed on the agar. All dishes were incubated at 37 °C for 24 h and examined. In the quantitative test, discs were placed in a 96-well plate; 1 ml of bacterial suspension (106 CFU ml−1) in Lura–Bertani medium (Hope Biotech, Qingdao, Shandong, China) was added to each well. After incubation (37 °C) for 6, 12, or 24 h, 100 μL of medium was aspirated into another 96-well plate; and 10 μL of Alamar blue reagent (Solarbio, Shanghai, China) was added to each well. After incubation for 3 h, absorbance at 570 nm was read.20 In all tests, discs carrying drug-free PLGA microspheres served as the control. The proportion of bacteria killed was represented by the difference in absorbances measured from experimental and control discs.Triclosan-loaded PLGA microspheres and immobilization on PMMA
PMMA discs were prepared by polymerization of methyl-methacrylate (MMA) as follows. MMA (40 ml) containing 2.5 g of predissolved benzoyl peroxide (2.5 g) was transferred in a sealed glass vial, and kept at 40 °C for 24 h to allow polymerization to a PMMA cylinder. The cylinder was cut into discs (Φ10 × 2 mm) and abraded to a 1200-grit finish. PLGA microspheres containing triclosan (triclosan-PLGA) were prepared similar to that of drug-free ones, except that 40 mg of triclosan (Xiya Reagent, Chengdu, China) was dissolved in DCM. For in vitro release, 30 mg of triclosan-PLGA microspheres were immersed in 3 ml of PBS containing 5 mg ml−1 sodium dodecylsulfate for 10 d,21 and 1 ml was regularly collected for UV-Vis analysis (280 nm). The microspheres were immobilized on PMMA discs by vacuum drying, and evaluated for immobilization stability and antibacterial property as described above.Material characterizations
Surface morphology and phase were studied by scanning electron microscopy (SEM; FEI QUANTA200 and Phenom G2 Pro backscattering electron microscope) and X-ray diffraction (XRD, Philips X′ Pert PRO XL-30), respectively. Residual PVA on drug-free PLGA microspheres were analyzed chemically.22,23 Briefly, 30 mg of microspheres were digested with 0.8 ml of 0.5 mol l−1 NaOH (80 °C, 20 min) and neutralized with 0.36 ml of 1 mol l−1 HCl; the liquid was adjusted to 2 ml with ultrapure water. Then 1.2 ml of 0.65 mol l−1 boric acid, 0.2 ml of an solution of iodine(0.05 mol l−1) and potassium iodide (0.15 mol l−1), and 0.6 ml of water was added to the liquid, forming a colored compound. The solution was allowed to rest for 15 min, and then the absorbance at 650 nm was measured and converted to the PVA solution based on a calibration pre established with standard PVA solutions. For morphological change of bacteria after anti-bacterial test, after incubation for 12 h, discs were removed, rinsed thrice with 2 ml of PBS, fixed with 4% (w/w) paraformaldehyde, dehydrated in ethanol series (30–100%; w/w), critical-point dried with CO2 (K850, Emitech, Ashford, UK), and studied by SEM.Results and discussions
Sol–gel HA coating on Ti
The sol–gel coating prepared on Ti (Fig. 1a) was smooth and dense with grinding marks (width: 0.4–2 μm) revealing the topography of the underlying substrate, indicating that the coating was thin. Higher-magnification observation (Fig. 1b) found that the coating consisted of nano-sized clusters. EDS mapping of Ca and P at low magnifications (not shown) confirmed that the coating fully covered the substrate without noticeable defects. XRD (Fig. 1c) peaks of calcined gel matched the standard diffraction pattern of HA. These confirmed that, the Ti disc was covered by a thin smooth HA coating.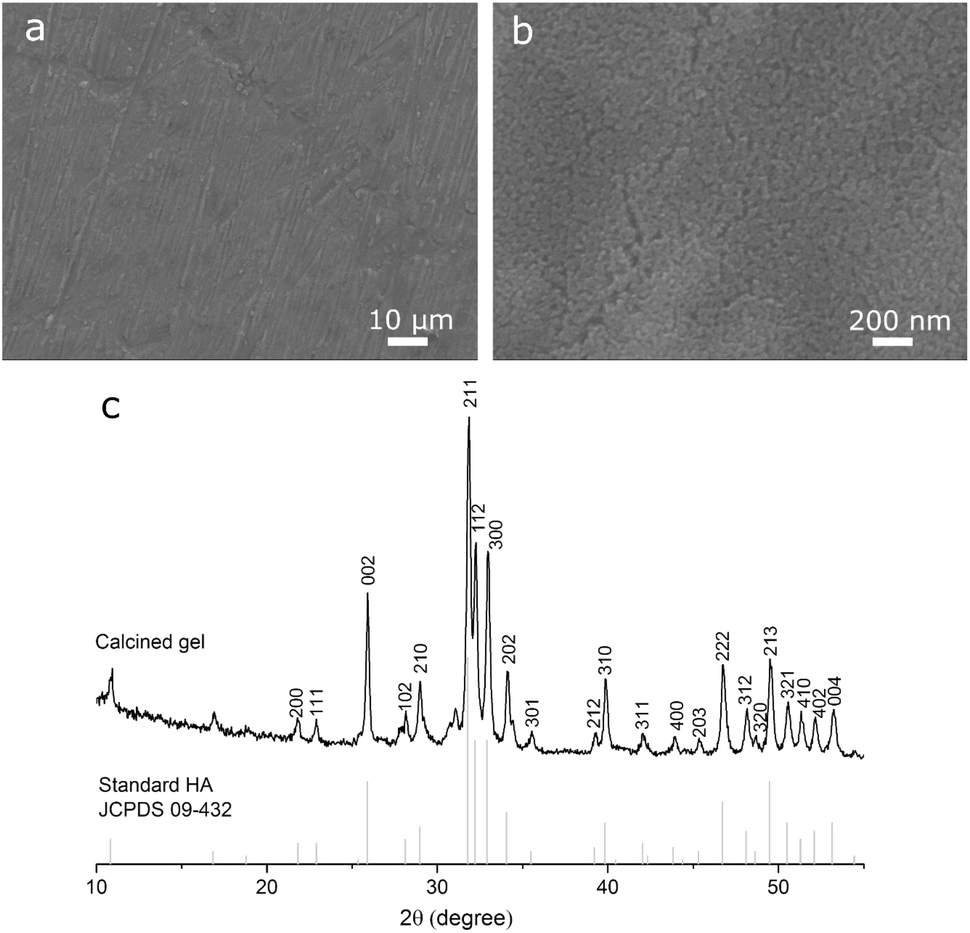 | ||
| Fig. 1 (a and b) Scanning electron micrographs of HA coating prepared on a Ti disc by the sol–gel method; (c) XRD pattern of calcined gel prepared by the method (bars: standard powder diffraction patterns of HA; JCPDS 09-432). | ||
PLGA microspheres
Drug-free PLGA microspheres (Fig. 2a) were spherical and smooth, with diameters ranging 5–40 μm (mean ± standard deviation: 20 ± 9 μm). CIP-PLGA microspheres (Fig. 2b) were less spherical and 51 ± 20 μm in size. Black spots on the microspheres (0.5–15 μm) disclosed the interior aqueous phase (i.e., CIP solution droplets) in the microspheres, which originated from the double-emulsion preparation process. Interestingly, embedded in the PLGA matrix, the interior aqueous phase is normally invisible by the routinely used secondary electron imaging mode (as seen in Fig. 7c and e below). However, in Fig. 2b, the backscattering electron (BSE) imaging mode used was able to disclose these inner liquid droplets thanks to the higher energy and, thus, escape depth of backscattered (vs. secondary) electrons. Evidently, because of the sensitivity of BSE imaging to atomic number, the inner aqueous phase (primarily water) appeared darker than the PLGA matrix (predominantly C and O). The encapsulation efficiency of CIP was 3.0%. Chemical analysis found microspheres to contain 0.14 ± 0.01% (w/w) of residual PVA. | ||
| Fig. 2 Backscattered scanning electron micrographs of (a) drug-free PLGA microspheres and (b) CIP-PLGA microspheres immobilized on a HA-Ti disc by vacuum drying. | ||
Microsphere immobilization on HA-coated Ti and stability
Drug-free PLGA microspheres were pipetted as a suspension on HA-Ti discs and vacuum-dried. After immersion in PBS for 10 min (Fig. 3a), 99% of the microspheres were retained on the surface; after 1 d, 97% did; and after 12 d, 80%. In comparison, when the suspension was pipetted on HA-Ti but subsequent vacuum drying was omitted, after immersion for 10 min, only 23% of the microspheres were retained on the surface. Given this poor immobilization stability, microspheres immobilized without vacuum-drying were not further studied for stability. Natural air-drying was also initially used for microsphere immobilization with some success. However, it was sensitive to ambient temperature and humidity, and the time required to complete dry a disc was long (i.e., hours) and unpredictable. Consequently, vacuum-drying was developed as a rapid and robust replacement for air-drying.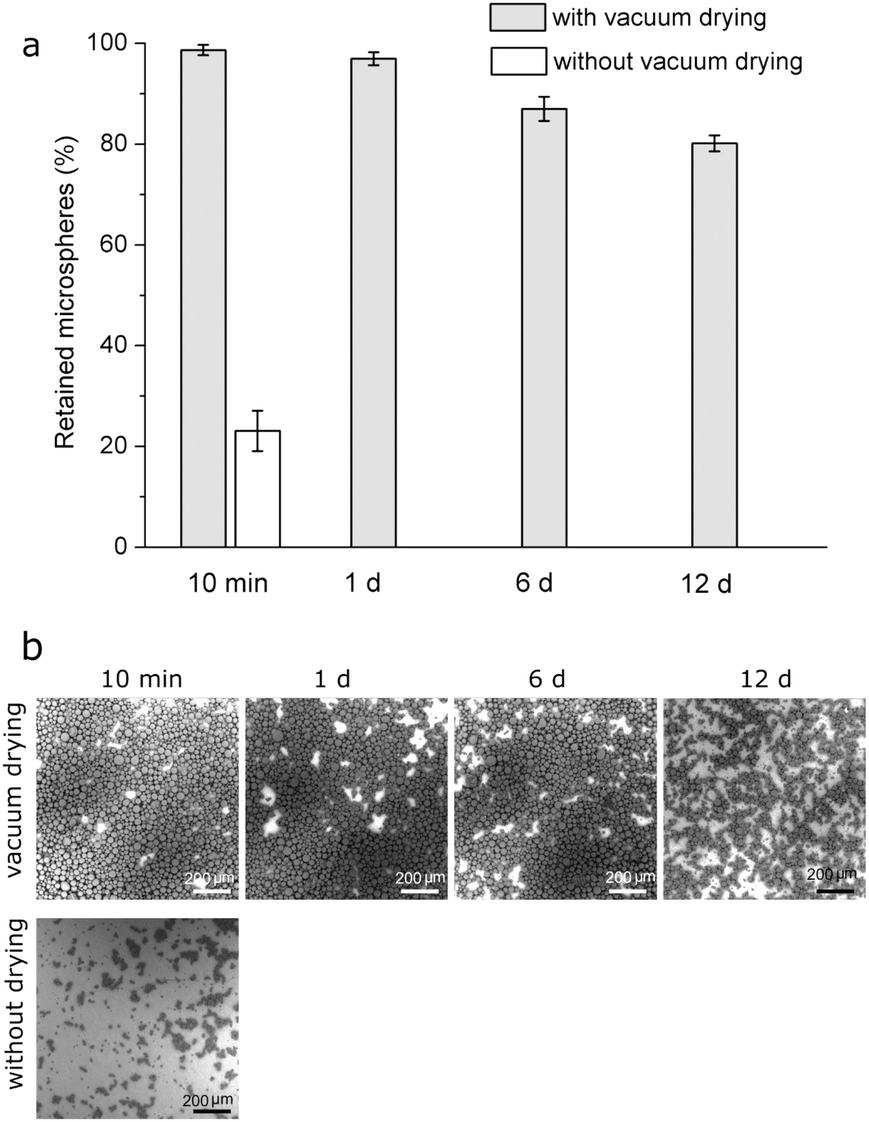 | ||
| Fig. 3 (a) Proportions of drug-free PLGA microspheres retained on HA-Ti discs after immersion for various periods in PBS (mean ± standard deviation, n = 3); (b) backscattered scanning electron micrographs showing microsphere retention during immersion. | ||
SEM observation found a similar trend (Fig. 3b). After immersion of vacuum-dried discs in PBS for 10 min, the surface was densely covered by microspheres. After 1 d, the surface changed negligibly. After 12 d, microspheres were partly lost, and the retained ones were smaller than those originally immobilized. By contrast, when microspheres were pipetted on discs without subsequent vacuum drying, after only 10 min in PBS, substantially fewer microspheres were still on the discs and the retained ones were smaller.
Earlier studies have shown that, PVA binds irreversibly to the surface of PLGA microspheres despite repeated rinsing.23,24 PVA was also reported to bind to Ca2+, metals, ceramics, and polymers via interactions (e.g., complexing, hydrogen bonding) between its –OH groups and surface groups of those materials.25–30 Several studies suggested binding of PVA to HA.31,32 Thus, we hypothesized that, the residual PVA on the PLGA microspheres may have contributed to their immobilization on HA-Ti by binding to the HA coating. To test the hypothesis, we treated PLGA microspheres with sodium borate before immobilization on HA-Ti by vacuum drying. Borate ions react efficiently with hydroxyl groups of PVA;19 in fact, their reaction is used to remove borate from wastewater.33 After immersion of borate-treated microspheres in PBS for 10 min (Fig. 4), 71% of the microspheres were retained on the surface; after 1 d, 46% were retained; after 6 d, 27%; and after 12 d, only 9.6%. In comparison, the microspheres treated with the phosphate solution of identical pH behaved similarly to untreated microspheres, with 72% retained after 12 d. Therefore, borate ions (rather than the high pH of the solution) substantially reduced the immobilization stability of the microspheres, indicating that residual PVA on the microsphere surface was a major contributor to their immobilization on HA-Ti.
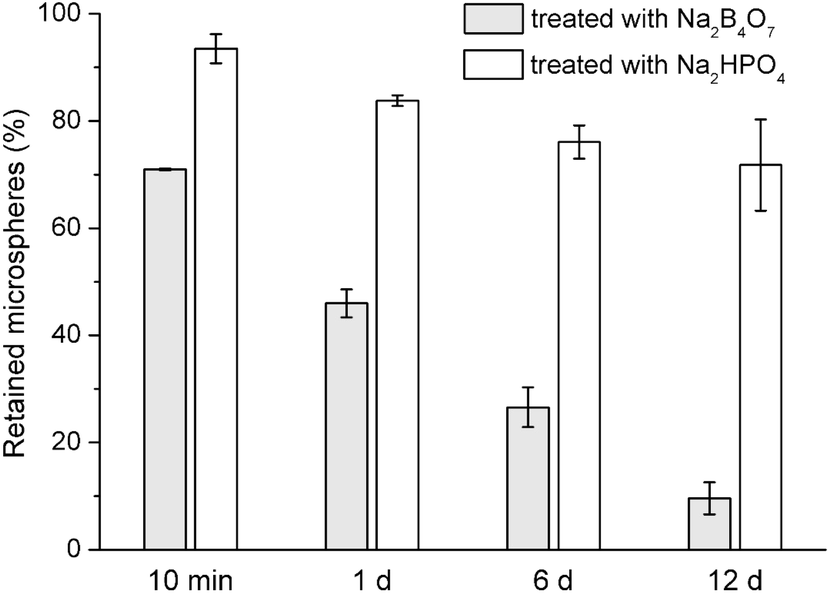 | ||
| Fig. 4 Proportions of borate- and phosphate-treated drug-free PLGA microspheres retained on HA-Ti discs after immersion for various periods in PBS (mean ± standard deviation, n = 3). | ||
Based on these, the immobilization mechanisms may be suggested as follows (Fig. 5). Vacuum-drying of the suspension on an HA-Ti disc forced the microspheres to form direct point-contacts with the HA coating. The residual PVA on the microsphere surface bound to the coating via interaction between its –OH groups and ions on the HA surface (possibly Ca2+), resulting in microsphere immobilization. In this PVA-mediated attachment mechanism, each polymer chain simultaneously interacts with the HA coating via numerous –OH groups. Thus, although each –OH group produces only a weak binding, the overall force resulting from numerous groups can be strong enough for the immobilization.34 In comparison, when the vacuum drying treatment is omitted, microspheres remained separated from the HA coating surface by water (i.e., wetting film) and could not attach stably. Borate ions also reduced the number of –OH groups available for surface binding, weakening their immobilization.
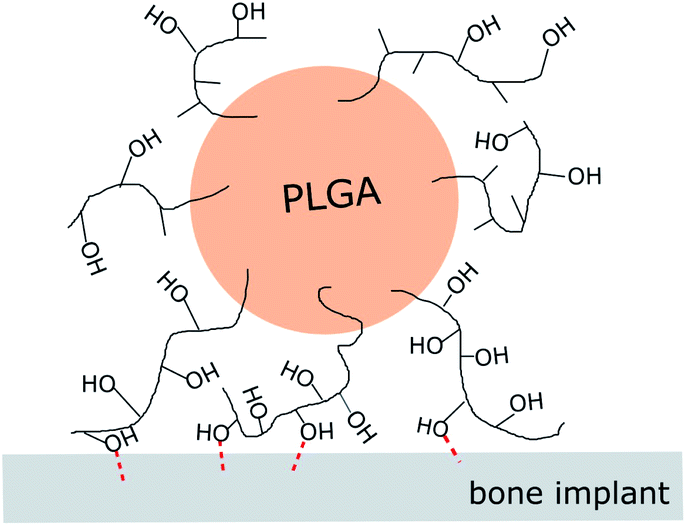 | ||
| Fig. 5 Schematic of binding between –OH groups of residual PVA associated with PLGA microspheres and HA (possible interaction sites: Ca2+) as a suggested mechanism leading to their immobilization on HA-coated Ti discs. | ||
In vitro drug release and antibacterial property
CIP-PLGA microspheres (Fig. 6a) rapidly released 62% of the CIP load in 7 h, followed by a slow sustained release for at least 15 d. This biphasic pattern is commonly observed for microsphere release systems. Presumably, CIP molecules located in/near the microsphere surface were released quickly, and those residing deeply were released more slowly. Other studies reported that, the first 6 h after implantation is critical for preventing IRIs, and an optimal antibiotic-release surface should release rapidly for ∼6 h followed by a slower sustained release.35 The release pattern of CIP-PLGA generally meets this requirement, but further improvement is needed. CIP-PLGA microspheres were immobilized on HA-Ti discs (Fig. 2b) and cocultured with S. aureus or E. coli for 24 h. In the qualitative test, the discs carrying CIP-PLGA produced clear inhibition zones against both bacteria (Fig. 6b and c), whereas discs carrying drug-free ones (control) formed none.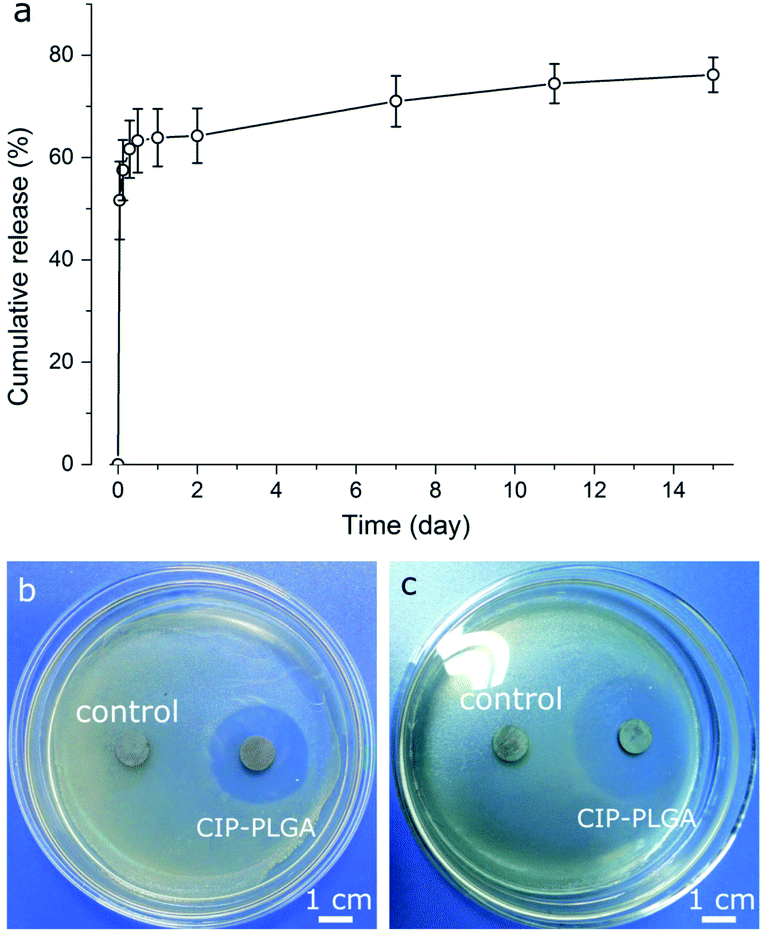 | ||
| Fig. 6 (a) Cumulative CIP release from CIP-PLGA microspheres (mean ± standard deviation, n = 3); (b and c) photographs of HA-Ti discs carrying immobilized drug-free (control) and CIP-PLGA microspheres after coculture with (b) S. aureus and (c) E. coli (c) for 24 h. | ||
In the quantitative test, between 6–24 h, the CIP-PLGA reduced the absorbance by 87.6–91.3% (for E. coli) and 52.3–81.7% (for S. aureus) relative to the controls (Fig. 7a). SEM found that, after anti-bacterial test for 12 h, both bacteria formed patches on some region in the control (i.e., discs carrying drug-microspheres) (Fig. 7b and d), whereas only isolated and substantially fewer bacteria were seen on the discs carrying CIP-PLGA microspheres (Fig. 7c and e). Therefore, both qualitative and quantitative tests confirm the bactericidal property of the immobilized HA-Ti discs. Obviously, this immobilization technique can be extended to other biomaterials and drugs to enable more biological functions. For example, we recently immobilized PLGA microspheres containing growth differentiation factor-5 (GDF-5) on porous calcium phosphate scaffolds, and found that GDF-5 release significantly increased the osteogenic differentiation of bone marrow stem cells and repair of segmental radial defects in rabbits.36
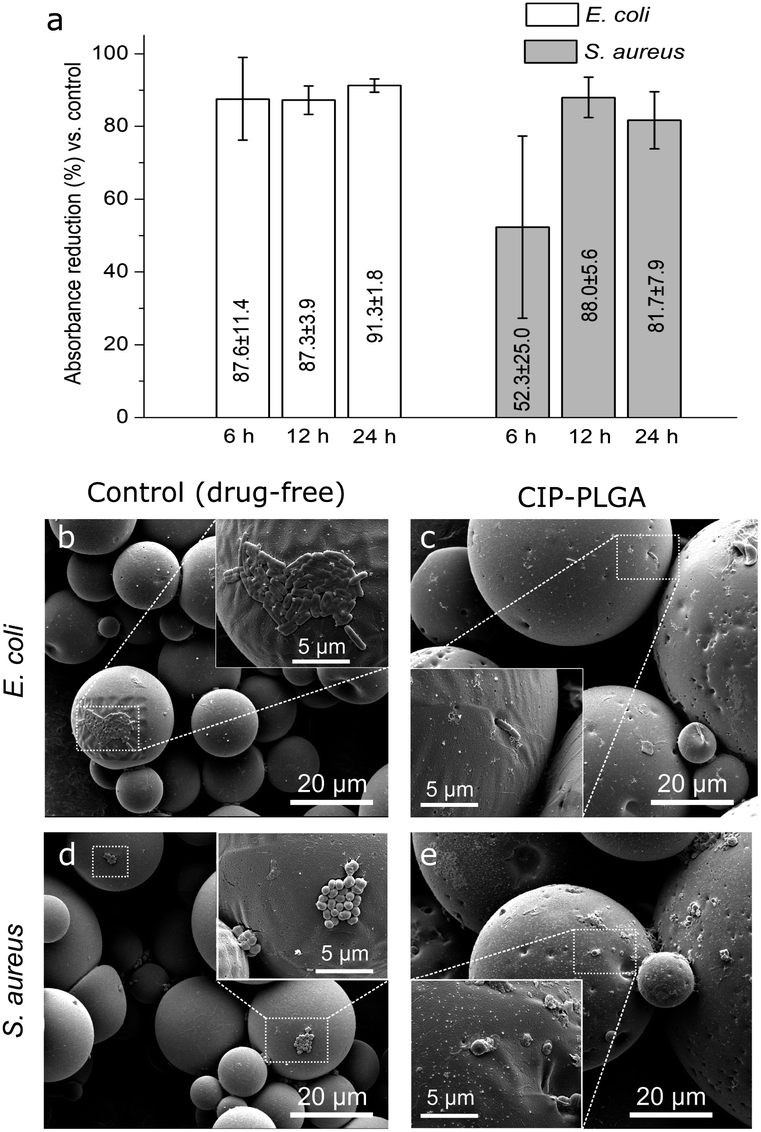 | ||
| Fig. 7 (a) Absorbance reduction of HA-Ti discs carrying CIP-PLGA relative to control (discs carrying drug-free microspheres) after coculture with bacteria (Alamar blue assay; mean ± standard deviation, n = 3); (b–e) scanning electron micrographs of discs after coculture with bacteria for 12 h. | ||
Immobilization of triclosan-loaded PLGA microspheres on PMMA
The above mechanism suggests that, the method may be also applied to other materials able to interact with PVA. PMMA was reported to form carbonyl–hydroxyl hydrogen bonding with alcohols such as PVA.29,37 Additionally, PVA was also observed to adsorb on PMMA irreversibly.30 Based on these observations and its clinical application as craniofacial bone implants,3 PMMA appeared to be an ideal material for further studies.Moreover, to examine the suitability of the method for drugs with different characteristics (e.g., solubility), an oil-soluble antibiotic, triclosan, was selected as the model drug. Triclosan is currently used as coatings on anti-infective sutures.37 Controlled triclosan release systems were also studied for orthopaedics.38
The PMMA discs (Fig. 8a, substrate) were flat with some grinding marks. Drug-free PLGA microspheres were stably immobilized on PMMA discs by vacuum drying (Fig. 9), with 89% retained after 12 d. Comparatively, without vacuum drying, only 28% were retained after 10 min. Good microsphere retention was also observed on discs prepared from a commercial PMMA bone cement (Versabond, Smith & Nephew, Memphis, TN, USA; not shown).
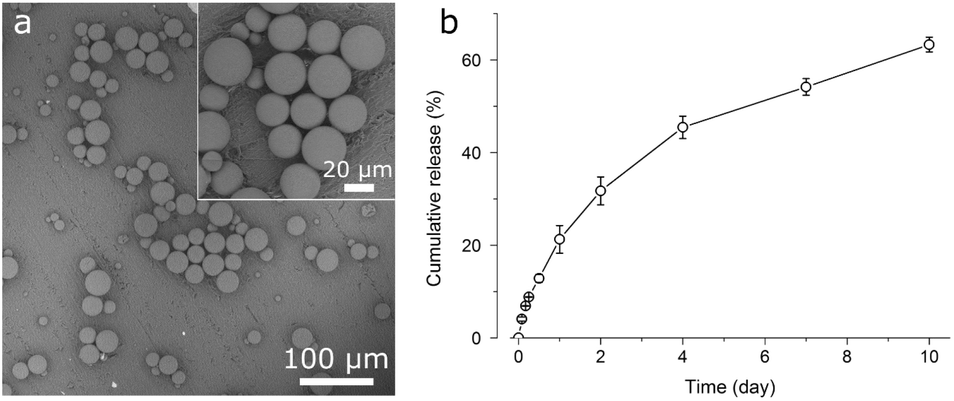 | ||
| Fig. 8 (a) Backscattered scanning electron micrographs of triclosan-PLGA microspheres immobilized by vacuum drying on a PMMA disc; (b) cumulative in vitro triclosan release from triclosan-PLGA microspheres (mean ± standard deviation, n = 3). | ||
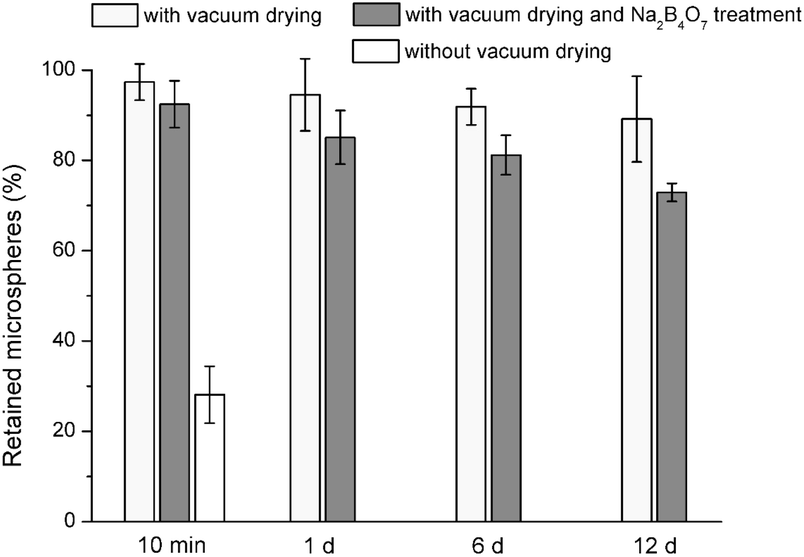 | ||
| Fig. 9 Proportions of drug-free PLGA microspheres retained on PMMA discs after immersion for various periods in PBS (mean ± standard deviation, n = 3); microspheres were immobilized with or without vacuum drying, or were pretreated with sodium borate solution and subsequently immobilized with vacuum drying. | ||
Similarly, treatment with sodium borate reduced the retention of microspheres (immobilized by vacuum drying) moderately (73% retained after 12 d). This suggested that –OH group-mediated binding was not the predominant contributor to their immobilization on PMMA, and other forces have contributed significantly. The exact mechanisms are unclear, but two possibilities may be considered. First, the van der Waals forces between PLGA and PMMA should have contributed. Second, molecular segments in polymers are (to different degrees) mobile even at room temperature;39 this motion may have caused local interpenetration-entanglement of polymer chains between microspheres and PMMA, thereby leading to their adhesion to the latter.40 Additionally, the PMMA surface was rougher than HA-Ti and this may have created a greater mechanical interlock. However, other possibilities are also open.
Triclosan-PLGA microspheres (Fig. 8a) were 5–37 μm in size (mean: 21 ± 9 μm). In PBS (Fig. 7b), they released triclosan for >10 d in a sustained manner (64% released after 10 d). This pattern was likely related to the low water solubility of triclosan as well as the non-porous nature of the microspheres. The microspheres were immobilized on PMMA discs and cocultured with the two bacteria. Discs carrying triclosan-PLGA formed inhibition zones against both (Fig. 10), whereas those carrying drug-free ones gave none.
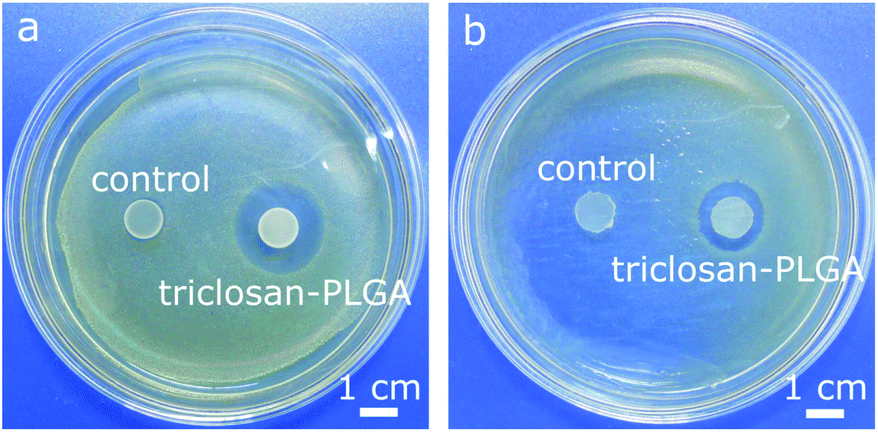 | ||
| Fig. 10 Photographs of PMMA discs carrying immobilized drug-free (control) and triclosan-PLGA microspheres after coculture with (a) S. aureus and (b) E. coli for 24 h. | ||
Quantitative test found that, between 6–12 h, the discs carrying triclosan-PLGA reduced the absorbance by 94.1–94.6% (for E. coli) and ∼100.0% (for S. aureus) relative to controls (Fig. 11), confirming the bactericidal property of the immobilized discs.
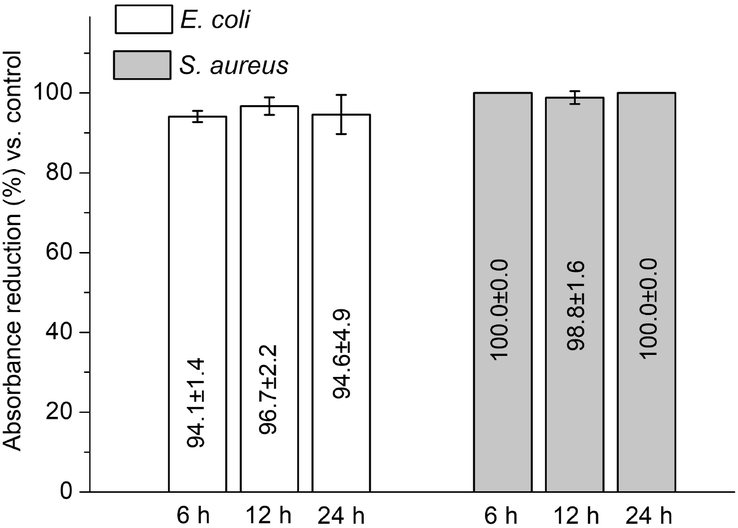 | ||
| Fig. 11 Absorbance reduction of PMMA discs carrying triclosan-PLGA microspheres relative to control (discs carrying drug-free microspheres) after coculture with bacteria (Alamar blue assay; mean ± standard deviation, n = 3). | ||
This study shows that, the vacuum-drying treatment can immobilize PLGA microspheres on different biomaterials without requiring special or potentially adverse conditions, although likely involving different mechanisms for different surfaces. This offers a simple and versatile method for achieving local antibiotic release from bone implant surfaces. Further studies are required to better characterize microsphere-biomaterial interactions at molecular levels and provide basic understanding for rational development of multi-functional bone implants.
Conclusion
PLGA microspheres prepared using PVA as the emulsifier were immobilized on two commonly used orthopaedic biomaterials (HA-coated Ti, PMMA) by a vacuum-drying method. The immobilized microspheres allowed the release of ciprofloxacin and triclosan to kill adjacent S. aureus and E. coli. The immobilization on HA-coated Ti was attributable to the interaction between HA and the –OH groups of residual PVA on the microsphere surface. The immobilization on PMMA appeared to arise from more interactions that need to be further investigated. This simple method is applicable for multiple biomaterials and offers a potential technique for developing anti-infective bone implant surfaces.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by National Natural Science Foundation of China (81071456), Science & Technology Department of Sichuan Province (2016JY0123, 2018JY0100, 2018SZYZF0012), and Startup Program of Affiliated Hospital of Southwest Medical University (19038). Valuable technical comments from two anonymous reviewers are appreciated.References
- L. I. Havelin, L. B. Engesæter, B. Espehaug, O. Furnes, S. A. Lie and S. E. Vollset, Acta Orthop. Scand., 2000, 71, 337–353 CrossRef CAS
.
- J. Ding, J. Zhang, J. Li, D. Li, C. Xiao, H. Xiao, H. Yang, X. Zhuang and X. Chen, Prog. Polym. Sci., 2019, 90, 1–34 CrossRef CAS
- A. Wolff, G. Santiago, M. Belzberg, C. Huggins, M. Lim, J. Weingart, W. Anderson, A. Coon, J. Huang, H. Brem and C. D. Gordon, J. Craniofac. Surg., 2018, 29, 887–894 CrossRef PubMed
- H. O. Gbejuade, A. M. Lovering and J. C. Webb, Acta Orthop., 2015, 86, 147–158 CrossRef PubMed
- S. Isefuku, C. J. Joyner and A. H. Simpson, J. Orthop. Trauma, 2003, 17, 212–216 CrossRef PubMed
- E. M. Hetrick and M. H. Schoenfisch, Chem. Soc. Rev., 2006, 35, 780–789 RSC
- D. A. Puleo and A. Nanci, Biomater, 1999, 20, 2311–2321 CrossRef CAS PubMed
- M. Stigter, J. Bezemer, K. de Groot and P. Layrolle, J. Contr. Release, 2004, 99, 127–137 CrossRef CAS PubMed
- S. Li, S. Dong, W. Xu, S. Tu, L. Yan, C. Zhao, J. Ding and X. Chen, Adv. Sci., 2018, 5, 1700527 CrossRef PubMed
- X. Zhao, Y. Han, T. Zhu, N. Feng, Y. Sun, Z. Song, S. Li, J. Liu and J. Ding, J. Biomed. Nanotechnol., 2019, 15, 1213–1222 CrossRef CAS PubMed
- G. Schmidmaier, M. Kerstan, P. Schwabe, N. Südkamp and M. Raschke, Injury, 2017, 48, 2235–2241 CrossRef CAS PubMed
- M. van der Elst, C. P. Klein, J. M. de Blieck-Hogervorst, P. Patka and H. J. Haarman, Biomater, 1999, 20, 121–128 CrossRef CAS PubMed
- K. Mulia, G. J. Witkamp, G. J. S. Dawes, L. E. Fratila-Apachitei, I. Apachitei, J. Duszczyk and H. Pellikaan, J. Biomater. Appl., 2011, 25, 401–412 CrossRef CAS PubMed
- S. K. Goel and E. J. Beckman, Polymer, 1993, 34, 1410–1417 CrossRef CAS
- J. S. Son, Y. A. Choi, E. K. Park, T. Y. Kwon, K. H. Kim and K. B. Lee, J. Biomed. Mater. Res. B Appl. Biomater., 2013, 101, 247–257 CrossRef PubMed
- C. Brunot, L. Ponsonnet, C. Lagneau, P. Farge, C. Picart and B. Grosgogeat, Biomater, 2007, 28, 632–640 CrossRef CAS PubMed
- D. M. Liu, Q. Yang and T. Troczynski, Biomater, 2002, 23, 691–698 CrossRef CAS PubMed
- E. S. Darley and A. P. MacGowan, J. Antimicrob. Chemother., 2004, 53, 928–935 CrossRef CAS
- S. W. Sinton, Macromolecules, 1987, 20, 2430–2441 CrossRef CAS
- S. N. Rampersad, Sensors, 2012, 12, 12347–12360 CrossRef CAS PubMed
- S. Kockisch, G. D. Rees, J. Tsibouklis and J. D. Smart, Eur. J. Pharm. Biopharm., 2005, 59, 207–216 CrossRef CAS
- D. Xiao, Q. Liu, D. Wang, T. Xie, T. Guo, K. Duan and J. Weng, Appl. Surf. Sci., 2014, 309, 112–118 CrossRef CAS
- S. K. Sahoo, J. Panyam, S. Prabha and V. Labhasetwar, J. Contr. Release, 2002, 82, 105–114 CrossRef CAS
- S. C. Lee, J. T. Oh, M. H. Jang and S. I. Chung, J. Contr. Release, 1999, 59, 123–132 CrossRef CAS
- K. Merat, J. Chaodamrongsakul, W. Tanthanuch and V. Vao-soongnern, J. Non-Cryst. Solids, 2013, 371, 47–52 CrossRef
- P. Stoyanov, S. Akhter and J. M. White, Surf. Interface Anal., 1990, 15, 509–515 CrossRef CAS
- T. K. Kundu, N. Karak, P. Barik and S. Saha, Int. J. Soft Comput. Eng., 2011, 1, 19–24 Search PubMed
- S. Hajatdoost and J. Yarwood, Appl. Spectrosc., 1996, 50, 558–564 CrossRef CAS
- Y. Zhang, G. Ping, N. Kaji, M. Tokeshi and Y. Baba, Electrophoresis, 2007, 28, 3308–3314 CrossRef CAS PubMed
- S. Mollazadeh, J. Javadpour and A. Khavandi, Ceram. Int., 2007, 33, 1579–1583 CrossRef CAS
- F. Xu, Y. Li, X. Wang, J. Wei and A. Yang, J. Mater. Sci., 2004, 39, 5669–5672 CrossRef CAS
- A. E. Ismanto and J. C. Liu, Compos. Interfac., 2014, 21, 639–650 CrossRef CAS
- S. Malynych, I. Luzinov and G. Chumanov, J. Phys. Chem. B, 2002, 106, 1280–1285 CrossRef CAS
- K. Vasilev, J. Cook and H. J. Griesser, Expet Rev. Med. Dev., 2009, 6, 553–567 CrossRef PubMed
- D. Q. Xiao, F. Yang, Q. Zhao, S. Chen, F. Shi, X. Xiang, L. Deng, X. Sun, J. Weng and G. Feng, RSC Adv., 2018, 8, 29526–29534 RSC
- X. Cao, M. Jiang and T. Yu, Makromol. Chem., 1989, 190, 117–128 CrossRef CAS
- W. K. Chang, S. Srinivasa, R. Morton and A. G. Hill, Ann. Surg., 2012, 255, 854–859 CrossRef PubMed
- B. D. W. Richards, R. Bayston and W. Ashraff, J. Bone Joint Surg., 2012, 94, 58 CrossRef PubMed
- A. Vesel and M. Mozetic, Vacuum, 2012, 86, 634–637 CrossRef CAS
- N. A. Peppas and J. J. Sahlin, Biomater, 1996, 17, 1553–1561 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08246h |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |