
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProline derived guanidine catalysts forge extensive H-bonded architectures: a solution and solid state study†
Zahraa S. Al-Taieab,
Simon R. Anettsb,
Jeppe Christensencd,
Simon J. Coles d,
Peter N. Hortond,
Daniel M. Evansb,
Leigh F. Jonese,
Frank F. J. de Kleijneb,
Shaun M. Ledbetterb,
Yassin T. H. Mehdarb,
Patrick J. Murphy*b and
Jack A. Wilsonb
d,
Peter N. Hortond,
Daniel M. Evansb,
Leigh F. Jonese,
Frank F. J. de Kleijneb,
Shaun M. Ledbetterb,
Yassin T. H. Mehdarb,
Patrick J. Murphy*b and
Jack A. Wilsonb
aDepartment of Chemistry, College of Science, Al-Nahrain University, Baghdad, Iraq
bSchool of Natural Sciences (Chemistry), Bangor University, Bangor, Gwynedd LL57 2UW, UK. E-mail: paddy@bangor.ac.uk; Tel: +44 (0)1248 382392
cDiamond Light Source, Didcot, OX12 0DE, UK
dEPSRC National Crystallography Service, School of Chemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
eSchool of Biological, Physical and Forensic Sciences, University of Wolverhampton, WV1 1LY, UK
First published on 11th June 2020
Abstract
The preparation of a range of amino acid derived guanidine organocatalysts is reported together with their application to the Michael addition of 2-hydroxy-1,4-napthoquinone to β-nitrostyrene, achieving a maximum ee of 56%. Some insight into the mechanism was sought by using X-ray crystallography and a detailed study of the intra- and intermolecular hydrogen bonding is reported.
Introduction
We recently reported1 the Michael addition of 2-hydroxy-1,4-napthoquinone 1 to β-nitrostyrene 2 catalyzed by the tetracyclic guanidinium salts 3 in combination with L-proline 4. It was observed that the two substrates which were unreactive on combination in THF were slowly converted to the product 5 with a T1/2 of 465 h. The reaction was also catalyzed slowly in the presence of L-proline 4 with a T1/2 of 579 h in THF. However if the two catalysts 3 and L-proline were used in combination the reaction proceeded with a T1/2 of 80 h. Unfortunately no appreciable enantioselectivity was observed in any of the catalysed reactions which might be attributed to the site of reaction being too far removed from the point of asymmetric induction within the proposed intermediate 6 (Scheme 1). | ||
| Scheme 1 Condition (a) β-nitrostyrene 2, 3 (0.05 equiv.), L-proline 4 (0.05 equiv.), CH2Cl2, rt, 6 days 85%. | ||
The catalysis of the process by the guanidinium salt 3 in conjunction with L-proline 4 was of interest, as proline and proline derivatives have a long history of use in organocatalysis.2 It is also known that several guanidine and guanidinium derivatives have proved effective as organocatalysts. We have reported the use of 3 and related in phase transfer alkylations and epoxidations with high ee's and its free base as a catalyst for Michael additions albeit in low ee.3 Similarly Yu et al.4 reported that guanidine 7 was an efficient catalyst for the Michael reaction of β-ketoesters to 2-nitrostyrene 2 with ee's of up to 95%. Terada et al. reported a series of novel axially chiral guanidines 8 as highly efficient Brønsted base catalysts to promote Michael reactions in high ee's.5 Similarly, Najera et al. reported the benzimidazole catalyst 9 gave enantioselectivities in the Michael additions of malonates to nitrostyrene 2 with selectivity of up to 96% ee, in the presence of TFA as a co-catalyst.6 These authors also reported the use of the 2-aminobenzimidazole-derived organocatalyst 10 for the efficient room temperature asymmetric conjugate addition of 1,3-diketones, β-ketoesters, and malonates to maleimides in up to 97% ee.7 An interesting catalyst containing both guanidine and thiourea moieties 11 was reported by Shubina et al.8 who reported the Michael additions of acetylacetone to β-nitrostyrene 2 in a low 25% ee. They reported that this poor selectivity was thought to arise from a high conformational activity of the guanidine–thiourea complex formed with the enolised acetylacetone, leading to several different transition states which are all of very similar energy (Fig. 1).8
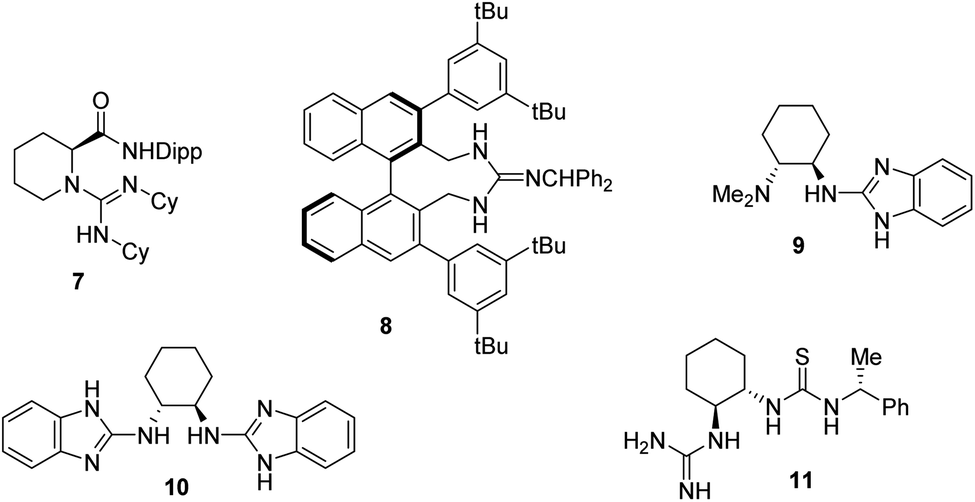 | ||
| Fig. 1 Guanidine organocatalysts. Dipp = 2,6-diisopropylphenyl. | ||
We were interested in preparing a series of guanidines in an attempt to improve and extending the range of guanidines in organocatalysis. We initially decided to focus on the preparation of N-alkylated proline derived catalysts and were inspired by the work of Altenbach et al. who reported9 that the benzimidazole-proline derivative 12 was an effective catalyst in the Hajose–Parrishe–Edere–Sauere–Wiechert aldol reaction of 1,3-ketone 13, leading to 14 in high yields and ee. Similarly, a report by Bhowmick et al. detailed10 the structurally very simple hydrazine derivative of L-proline 15 which was a viable organocatalyst for the aldol reaction of ketones 16 with aldehydes 17, leading to the aldol products 18 in 32–99% ee. Similalrly the low molecular weight cyclohexanediamine derived thiourea catalyst 19, prepared11 by Dixon et al., was employed in intramolecular Michael additions of α,β-unsaturated esters. For example they reported the transformation of 20 into 21 in 86% yield and 94% ee using 19 as the catalyst (Scheme 2).
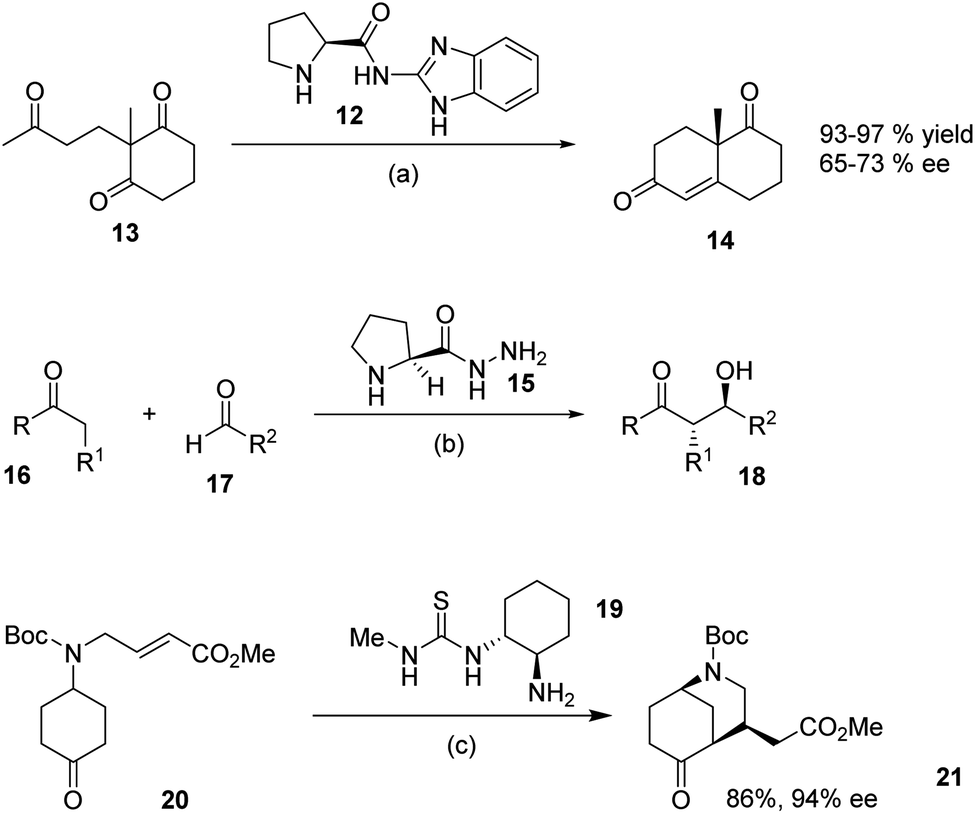 | ||
| Scheme 2 Recent organocatalytic reactions. Conditions: (a) 12 (0.05 mmol), TFA (0.05 mmol), THF, DMSO, MeOH or H2O, rt. 24 h. (b) 15 (0.1 equiv.), PTSA, (0.05 equiv.) in H2O; R = alkyl, R1 = H, alkyl, R2 = aryl. (c) 19 (0.05 equiv.), PhCO2H (0.025 equiv.), CH2Cl2, 50 °C, 48 h. | ||
We envisaged the preparation of a range of guanidines 22, based on a proline scaffold, which are both N-substituted and covalently bonded to a guanidine moiety via an amide bond. This concept was to allow ease of flexibility at these two sites and we hoped the catalysts might be “tunable” by modifying the proline nitrogen to influence steric interactions. Similarly, modification of the groups on the guanidine could also offer some electronic control of its hydrogen bonding ability and basicity (Fig. 2). Our initial aim was thus to prepare a range of guanidine catalysts.
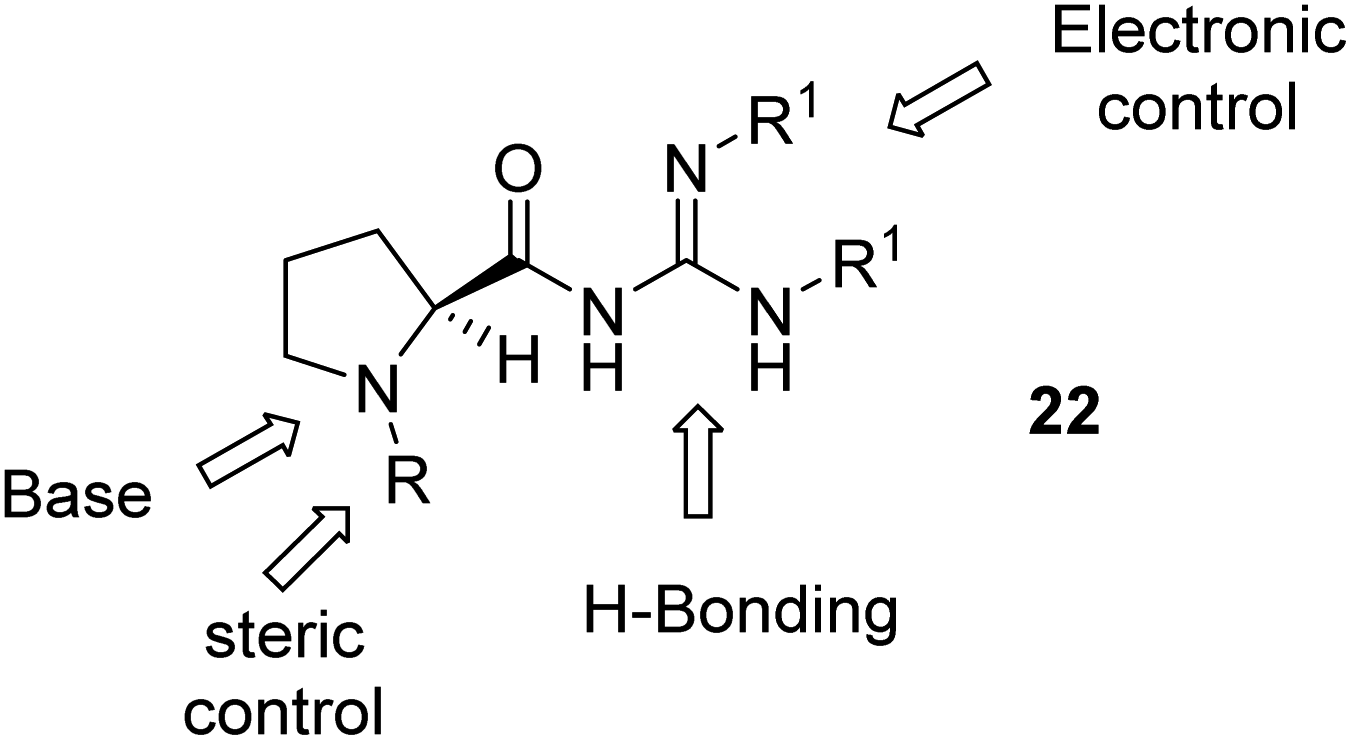 | ||
| Fig. 2 Generic catalyst structure 22. R = alkyl, R1 = alkyl, aryl, acyl. | ||
Preparation of the studied catalysts
The catalysts initially investigated were prepared by the coupling of the N-alkylated-L-proline 23a–d derivatives with alkyl, acyl or aryl substituted guanidines (see ESI†) using CDI as a coupling reagent in DMF (Scheme 3). This reagent was employed, as several other reagent combinations that were tested (DCC/NHS, DCC/HOBT, HBTU, MeOCOCl/NEt3), were found to be ineffective and gave considerable problems on attempted purification. We initially attempted the preparation of the benzoyl substituted catalyst 24 and the crude compound was obtained, however on attempted purification it underwent hydrolysis on contact with moisture to give the parent benzoyl guanidine. This was somewhat worrying as if the other guanidines are prone to hydrolysis this might limit the use of these compounds. We next attempted the coupling of N-Cbz-guanidine and were pleased to obtain the compounds 25a–d in good yields after chromatography. We similarly prepared the diphenyl substituted catalysts 26a–d and found that the yields were good for the isopropyl- and cyclohexyl-substituted compounds whilst the methyl and benzyl were poor. The observation that coupling of N-methyl-L-proline 23a under mixed-anhydride and other peptide coupling methods is problematic has been made by Lygo and Moore12 who did not offer a clear insight as to why this was so, however our work might offer a possible explanation. The phenyl-substituted catalysts 27a-b were also prepared in reasonable yield using method B in which the guanidine is generated in situ from a guanidinium salt and sodium hydride. We next attempted the preparation of the potentially more basic guanidines 28a–d, from 1,1-dimethyl guanidine, using method B. On analysis of the crude reaction mixture of this reaction by proton NMR, the reactions appeared to be successful as the products were present. However, on work up we found that the products were highly polar and difficult to separate from the by-products of the reaction. In particular, the N-methyl substituted catalyst 28a had a high water solubility/instability and was not isolatable if an aqueous work up was employed. In the case of the N-benzyl substituted 28b, a low yield of the desired product was obtained as was a low yield of the dimeric compound 28e. For reasons to be discussed later, the focus of catalyst preparation switched to the N-methyl substituted prolines and we next prepared the substituted guanidines 29 (N-Boc), 30 (N-Me, N′-Boc) and 31 (N-Me, N′-Cbz) by method A in good to excellent yields. We also attempted the preparation of the 1,1-dimethyl substituted Cbz 32 and Boc 33 guanidines and found that there was no indication of the formation of these compound, as on work up only recovered guanidine starting material was observed. This would suggest that either the guanidines were not reactive enough to displace the imidazole in the coupling intermediate or that the products 32 and 33 were hydrolysed during work up. We also prepared the benzimidazole derived prolines 34a–d in good yields using coupling method A with again the lowest yield being observed for the N-methyl substituted 34a. Similarly we prepared the methyl substituted benzimidazoles 35 and 36 in 41% and 45% yield respectively. We also prepared the benzothiazole 37a catalyst in 74% yield using method A, however the benzoxazole 37b was not accessible by this method which might be due to the low nucleophilicity of 2-aminobenzoxazole. Similarly the N-methyl 38a and the N-benzyl-imidazole catalyst 38b were prepared using the standard method in 97% and 68% yield respectively. We also attempted to prepare other heterocyclic derived catalysts with limited success. The pyridine derived catalyst 39a was prepared using the CDI coupling method but the yield obtained (18%) was poor. Unfortunately, the pyrimidine 39b and pyrazine 39c derivatives could not be prepared using these conditions. We next attempted the preparation of the bis-L-proline substituted guanidines 40a–d (method C) and found that the catalyst 40b–d were formed in good to excellent yields. It was also observed that catalyst 40a appeared to be formed under the conditions of the reaction as evidenced by proton and carbon NMR, however the product was lost on aqueous work up or on chromatography. This appeared to be due to a combination of factors including high water solubility and decomposition on silica gel and in water. The hydrazine derived N-methyl-L-proline catalysts 41c-d were also prepared in good yields by esterification of N-methyl-L-proline 23a with methanolic HCl followed by reaction with the corresponding hydrazine. | ||
| Scheme 3 Synthesis of the studied catalysts; conditions (A) 23a-d or 42a-b, CDI, DMF, rt, 24h. (B) (i) 23a-d or 42a-b, CDI, DMF, rt, 24h, (ii) guanidine. HX (X = Cl, HCO3, NO3), NEt3. (C) (i) 23a-d, CDI, DMF, rt, 24h, (ii) guanidine hydrochloride (0.5 equiv.), NaH (0.6 equiv.) DMF, rt, 24h. (D) (i) 23a, AcCl, MeOH, 0 °C 1h, (ii) reflux, 12h, (iii) NH2NHR1, rt, 24h. R = Me, Bn, iPr, Cy; R1 = H, Ph, Boc, Cbz. | ||
We also prepared a range of N,N-dimethyl alanine substituted guanidines (43a–47a) and also N,N-dimethyl phenylalanine substituted guanidines (43b–47b) using the previously employed CDI coupling method (Scheme 3). The Cbz-substituted 43a-b and Boc-substituted guanidines 44a-b were easily prepared in generally high yields. In the case of 44a, the slightly lower yield might be due to problems associated with the aqueous work up as significant losses were observed. The phenyl-substituted guanidines 45a-b were also prepared using the CDI coupling method with phenylguanidinium nitrate and hydrochloride, however compound 45a was a very polar material and proved impossible to purify from polar organic and inorganic by products. Compound 45b was less problematic and was purified by chromatography to give a moderate yield (46%). The benzimidazole substituted guanidines 46a and 46b were similarly prepared using this method in 92% and 42% yields respectively. The preparation of the symmetric disubstituted catalysts 47a and 47b was also attempted and again the L-alanine derived 47a could not be obtained in a pure form which again appeared to be due to high aqueous solubility/instability. Pleasingly the phenylalanine derived compound 47b was obtained in 77% yield under identical conditions after chromatography.
Michael addition reaction of 2-hydroxy-1,4-napthoquinone 1 with β-nitrostyrene 2
With these catalysts in hand we investigated the Michael addition reaction of 2-hydroxy-1,4-napthoquinone 1 with β-nitrostyrene 2 which in our hands had proven to be a robust reaction with little evidence of competing side reactions (Scheme 4a–c).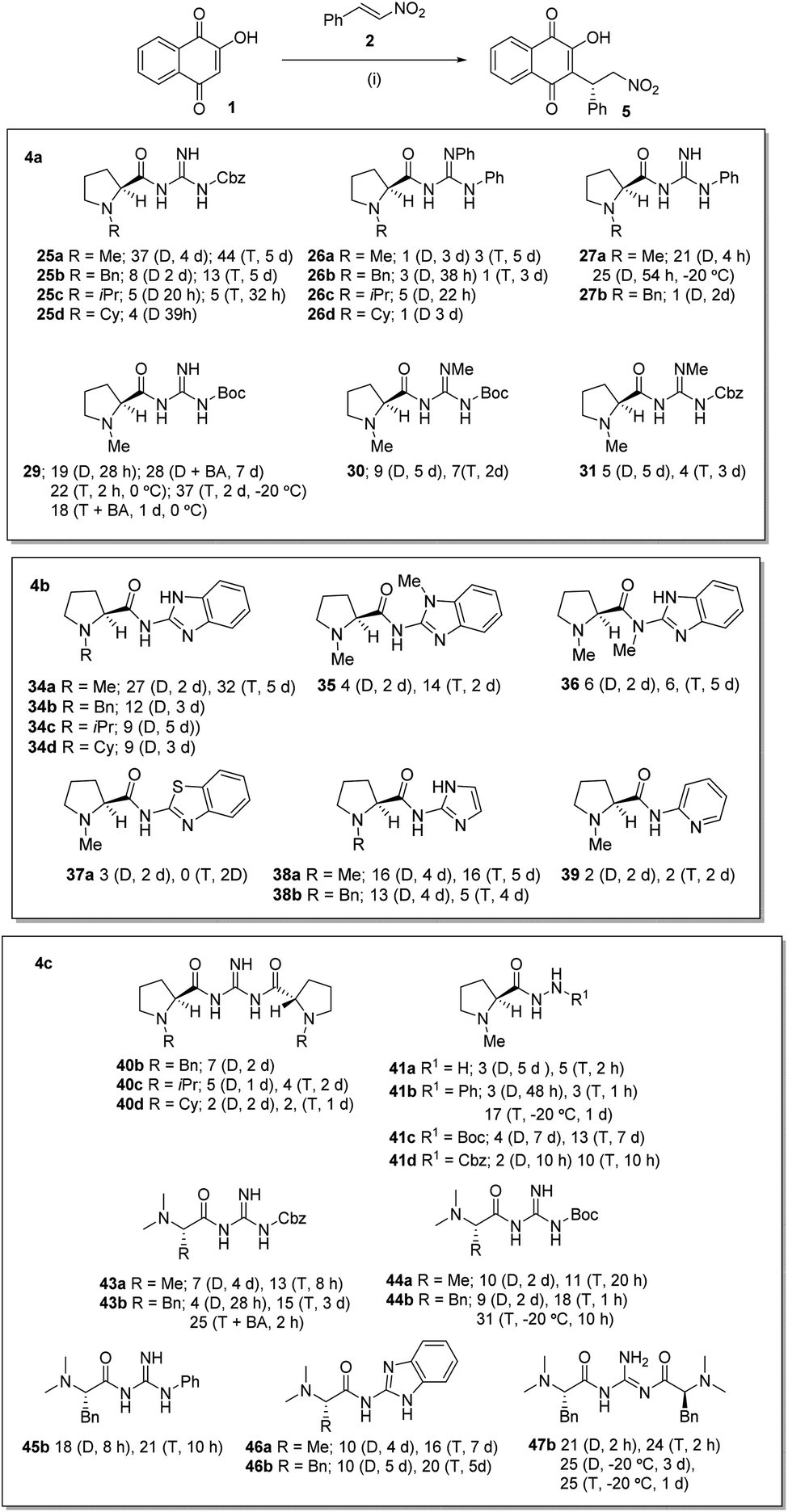 | ||
| Scheme 4 (a–c): Catalyzed Michael reaction between 1 and 2: conditions (i) catalyst (0.1–0.04 equiv.), solvent (0.03–0.08 M), −78 to 0 °C 7–8 h, then 0 °C-rt. Results are given as ee (solvent, time); D = dichloromethane, T = toluene. | ||
The catalysts 25a–d, were initially investigated (Scheme 4a) and were pleased to see that all of these guanidines were effective catalysts leading to good conversions over 1–5 d as evidenced by NMR sampling of the reaction mixtures. We were pleased to observe some level of asymmetric induction (18–44% ee) with all the catalyst with 25a being the most effective. The highest ee was obtained using toluene as solvent, giving the product in 44% ee over 5 days and in a 75% isolated yield. Various solvent systems (CH2Cl2, MeCN, PhMe, THF, EtOAc and benzene) were studied, with the best results being obtained in dichloromethane and toluene. Using toluene presented some solubility problems associated with the starting material 1, which was overcome by performing the reaction at higher dilution. It was also apparent that the catalytic reactions using 25a–d were generally slow with 88–100 h being required for a reasonable conversion. It was also apparent that whilst all these compounds catalyzed the reaction, there was a steady decrease in ee as the size of the L-proline N-substituent increased from Bn to iPr to Cy. The diphenyl-substituted catalysts 26a–d were investigated next (Scheme 4a) and whilst these compounds were found to catalyze the reaction at a similar rate to 25a–d, the ee's of the product were consistently very low (1 to 5% ee) and at this point in the investigation we had no clear explanation for this. We next investigated the N-alkylated-benzimidazole prolines 34a–d (Scheme 4b) and the results obtained with these catalysts were similar to those reported for 25a–d. It was found that the N-methyl catalyst 34a gave the highest levels of asymmetric induction with a 32% ee in toluene and a 27% ee in dichloromethane, however again there was a steady decrease in ee as the size of the proline N-substituent increased from Bn to iPr to Cy. We concluded from these studies that the size of the N-substituent on the proline is critical and that larger substituents appeared to detrimental to the ee of the reaction. We next investigated the mono-phenyl substituted catalysts 27a and 27b (Scheme 4a) and again the N-methyl substituted catalyst 27a gave the best asymmetric induction. Interestingly and in contrast to the previous catalysts, the reaction time using 27a in dichloromethane was shorter, leading to a 91% yield of 5 over 4 h with an ee of 21%. We repeated this reaction at −20 °C and obtained a slightly improved 25% ee over 54 h. This result was in stark contrast to the previous catalysts and we had no clear explanation for this at this point in the investigation. Again increasing the size of the N-substituent to a Bn group was detrimental to the ee as catalyst 27b gave no appreciable levels of asymmetric induction in any of the solvents studied. We also investigated the Boc-substituted guanidine 29 (Scheme 4a) which gave a 19% ee in dichloromethane over 28 h and on the addition of benzoic acid as a co-catalyst the ee improved to 28%. Interestingly the reaction with 29 in toluene at 0 °C was rapid when compared to previous catalysts leading to an 89% yield of the product in 22% ee over 2 h. Repeating this experiment at −20 °C for 24 h gave an improved 37% ee but a lower yield (37%). Repeating the reaction in the presence of benzoic acid in toluene over 24 h gave a diminished ee of 18% and a much lower yield (38%).
In an attempt to increase the basicity of the guanidine and to modify the intramolecular hydrogen-bonding (vide infra) in the catalysts, the N-methylated-N′-Boc catalyst 30 and the N-methylated-N′-Cbz catalyst 31 (Scheme 4a) were investigated. These catalysts both gave poor ee's and slower reaction times and yields in both toluene and dichloromethane when compared to catalysts 25a and 29. These results might indicate that increasing the basicity of the guanidine does not help the reaction process or that interfering with the hydrogen-bonding pattern at this position is detrimental. These results were mirrored in the reactions of the N-methylated benzimidazole catalysts 35 and 36 (Scheme 4b) when compared to 34a. These compounds were found to catalyze the reaction but again yields and ee's were lower (6–14% ee) and reaction times generally slower. Two catalysts were prepared in which the guanidine was replaced by a benzothiazole 37a and a pyridine 39a and again these catalyzed the reaction but gave poor ee's and slow reaction times. The imidazole catalysts 38a and 38b (Scheme 4b) were both effective catalysts with 38a leading to a 5 in 16% ee in dichloromethane or toluene, whilst 39a gave lower ee's of 13% and 5% ee in the respective solvents.
We next investigated the C2-catalysts 40b–d (Scheme 4c) and were disappointed to find that whilst catalyzing the reactions effectively the ee's of the products were very low (1–7%) in either toluene or dichloromethane. Interestingly adding benzoic acid to the reaction decreased the conversion time but no increase in ee was apparent (see ESI†). The hydrazine derived catalysts 41a–d (Scheme 4c) were investigated next and it was found that unsubstituted hydrazine 41a gave slow reaction times and poor ee's. By contrast phenyl-substituted hydrazine 41b gave a more rapid reaction in toluene whilst the reaction was again slow in dichloromethane. Unfortunately, the ee's of these reactions were again very low (3% ee), however repeating the reaction of 41b in toluene at −20 °C gave an improved ee of 17% but over a longer time (1 d). The Boc-substituted hydrazine 41c gave much slower reaction times but an improved 13% ee with toluene as reaction solvent. This slower reaction time might be due to an electronic effect as reactions of the Cbz-substituted hydrazine 41d were more rapid (10 h) but ee's were slightly lower (10% ee).
Moving on from the L-proline derived catalysts we examined those derived from L-alanine and L-phenylalanine. The N-Cbz-protected guanidines 43a and 43b (Scheme 4c) were investigated initially and these were found to be effective catalysts. Compound 43a gave an ee of 7% in dichloromethane over 4 d and 13% in toluene over 20 h, whilst 43b gave 4% ee in dichloromethane over 28 h and 15% ee in toluene over 3 d. The latter result was repeated using benzoic acid as a co-catalyst and this led to a faster conversion and an improvement in ee to 25%. Following this the N-Boc-protected guanidines 44a and 44b (Scheme 4c) were investigated initially and again were found to be effective catalysts. Compound 44a gave an ee of 10% in dichloromethane over 2 d and 11% in toluene over 20 h, whilst 44b gave 9% in dichloromethane over 2 d and an 18% ee in toluene over 1 h. The latter result was repeated at 0 °C over 10 h and gave product 5 in 31% ee. These results seem to suggest the bulkier benzyl group found in the L-phenylalanine catalysts gives a more effective catalyst. Moving to the phenyl-substituted catalyst 45b (Scheme 4c), similar results were found with an ee of 18% in dichloromethane over 8 h and 21% in toluene over 10 h. The benzimidazole catalysts 46a and 46b (Scheme 4c) were similarly investigated and overall were found to be slower that the previously investigated cases. Thus 46a gave an ee of 10% in dichloromethane over 4 d and 16% in toluene over 7 d, whilst 46b gave 10% ee in dichloromethane over 5 d and 20% ee in toluene over 5 d. Finally the C2-catalyst 47b gave similar results with an ee of 21% in dichloromethane over 2 h and 24% ee in toluene over 2 h. These reactions were repeated at −20 °C and ee of 25% in dichloromethane was obtained over 3 d and a 25% ee in toluene over 1 d.
Our overall conclusions from this work appear to be that the N-methyl-L-proline derived catalysts give better ee's in the reactions and increasing the substituent in general has a detrimental effect on the ee. The best groups on the guanidine of these catalysts appears to be those with Cbz-, Boc-, phenyl and benzimidazole substituents, whilst diphenyl and methyl substituted variants of the Cbz-, Boc- and benzimidazole catalysts led to lower ee's. Throughout this work the best solvent for this reaction appears to be toluene (and other aromatic solvents; see ESI†) with dichloromethane also being effective, however acetonitrile and other solvents were not successful. Cooling of the more rapid reactions appears to increase the ee of the product however, yields are lower and reaction times are prolonged. Throughout this work several catalysts seemed to increase the rate of reaction leading to completion or near completion in 1–4 h instead of the more typical 2–7 days, particularly catalysts 27a, 29 + BA, 40c + BA, 40d + BA, 41b, 43b + BA, 44b and 47b. The exact reason for this was unclear and we thus decided to investigate the catalysts further.
Crystallographic and racemization studies
From the beginnings of this project we were interested in the crystallographic nature of our catalysts as it was felt that this information might lead to an insight into the efficiency (or not) of the systems employed. The initial crystallographic work of the previous student on this project gave some insight into the H-bonding patterns observed in the N-methyl-L-proline catalysts 25a, 26a, 27a and 34a catalysts. Three of these catalysts 25a, 26a and 34a, had a distinctive H-bonding pattern (Fig. 3), which consisted of a strong intramolecular H-bond between the amide protons (NH) and the pyrrolidine nitrogen (bond a) and a complementary NH-bond between the guanidine NH and the amide carbonyl (bond b). This pattern holds the amide at a trans-configuration (E-) and in compound 25a we refer to this as E-abd as the carbamate has an H-bond to the guanidine NH2. In the case of 26a and 34a where no carbamate is present the E-ab pattern is found. We reasoned that the H-bonds found in these structures might still be present whilst they are in solution and this explain why these bases are slow to catalyze the Michael reaction. Some NMR evidence is also available which demonstrated that the 13C NMR data for compound 26a shows distinctive individual signals for each phenyl suggesting that rapid interconversion between these two phenyls is not occurring on the NMR timescale. Some further evidence for this is found in the structure of the guanidine 27a (Fig. 3), which differs in its H-bonding pattern in that the pyrrolidine amine (N1) is not intramolecularly H-bonded and only the carbonyl–NH bond (bond b; N3(H1)⋯O1 = 2.02 Å) is present as the amide is in the form of a N-methyleneamide; this is termed a Free-b type system. Whilst this evidence is tenuous, this difference in H-bonding might explain the increased reaction rate observed in the Michael addition reaction for compound 27a (proceeded in a 91% yield over 4 h), which was rapid when compared to the timescale of the other catalysts (typically took 4–5 days to reach completion). Another interesting feature is that the N4-phenyl group found in catalyst 26a appears to eclipse the nitrogen N1. Catalysts 26a–d gave very poor ee's in their reactions to form 5 and this eclipsing of the N might be a contributing factor (Fig. 3).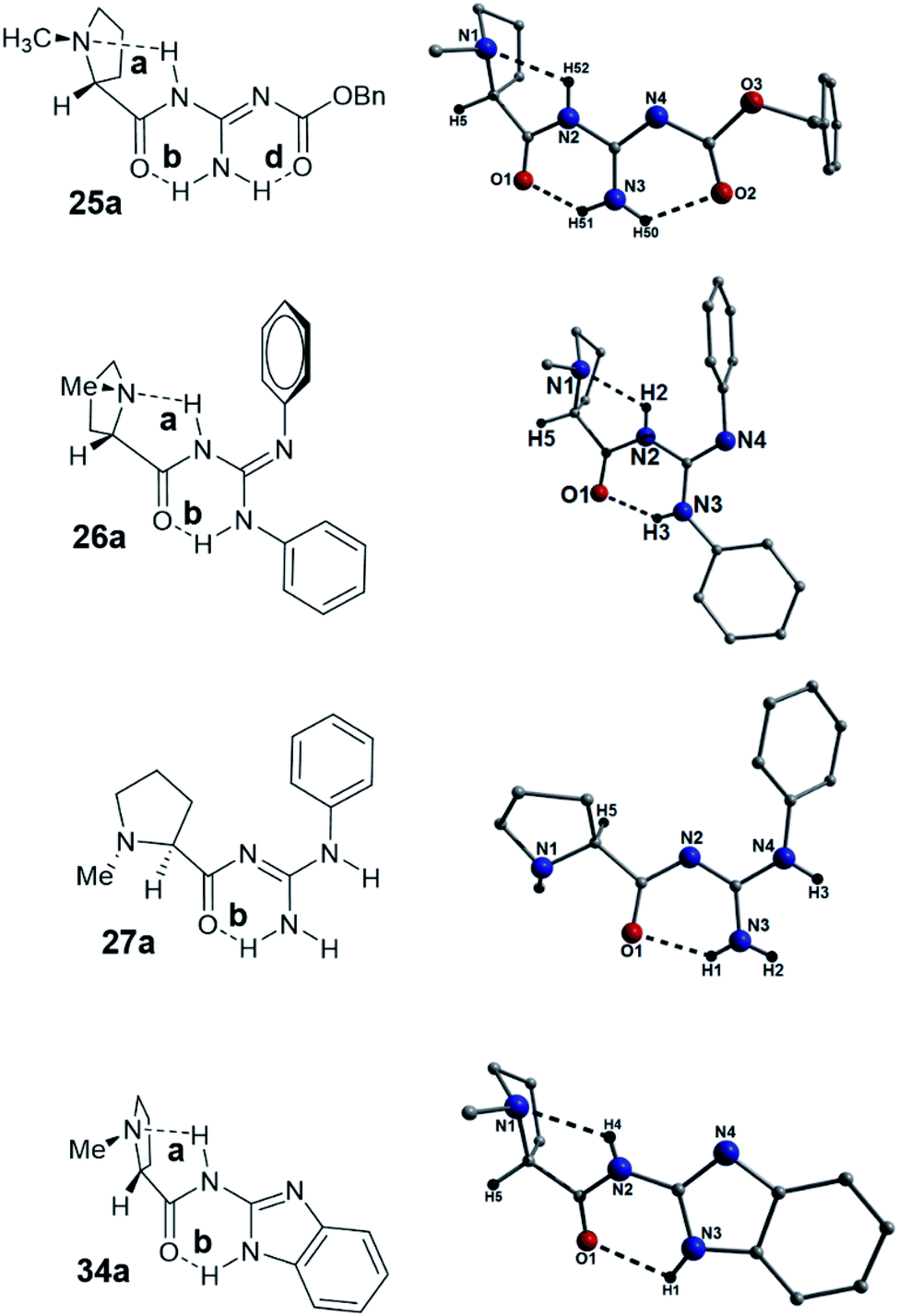 | ||
| Fig. 3 ChemDraw representations (left) of catalysts 25a, 26a, 27a and 34a along with their corresponding crystal structures (right). Colour code as used throughout this work: C (grey), N (blue), O (red) and H (black). Dashed lines represent all intramolecular H-bonding interactions (N2(H52)⋯N1 = 2.26 Å, N3(H51)⋯O1 = 1.87 Å and N3(H50)⋯O2 = 2.00 Å in 25a; N2(H2)⋯N1 = 2.15 Å and N3(H3)⋯O1 = 2.01 Å in 26a; N3(H1)⋯O1 = 2.02 Å in 27a; N2(H4)⋯N1 = 2.21 Å and N3(H1)⋯O1 = 2.35 Å in 34a. | ||
However, the most surprising and somewhat troubling result from our crystallographic studies was observed when examining the structure of 26a which was found to crystallize as a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (S:R) mixture of epimers within the unit cell (Fig. 4). Upon comparison with a simulated spectrum, the pXRD collection on 26a was consistent with this observation (Fig. S25†). Similar racemization was observed in the crystal structures of compounds 41c and 43a. More specifically, the species 41c was found to produce two types of crystal (crystals 1 and 2) within the same batch having crystallized in the Pna21 (41c-crystal 1) and P21 (41c-crystal 2) space groups. Crystal 1 is racemic in nature (Fig. 4), while all 41c units in crystal 2 exhibit the S configuration (Fig. S22†). Interestingly, the simulated powder patterns obtained from crystal 1 and crystal 2 are virtually identical. Although they have almost identical unit cell dimensions (Table S5† and Fig. S26†), this also means there is significant correlation in how the species pack within a unit cell. Likewise, the bulk powder sample of 41c also corresponds well to both simulated patterns. Unfortunately, the near identical nature of their simulated patterns severely hampers any attempts at obtaining a crystal 1/crystal 2 ratio in 41c. The catalyst 43a also crystallized as a racemate (Fig. S23†).
:1 (S:R) mixture of epimers within the unit cell (Fig. 4). Upon comparison with a simulated spectrum, the pXRD collection on 26a was consistent with this observation (Fig. S25†). Similar racemization was observed in the crystal structures of compounds 41c and 43a. More specifically, the species 41c was found to produce two types of crystal (crystals 1 and 2) within the same batch having crystallized in the Pna21 (41c-crystal 1) and P21 (41c-crystal 2) space groups. Crystal 1 is racemic in nature (Fig. 4), while all 41c units in crystal 2 exhibit the S configuration (Fig. S22†). Interestingly, the simulated powder patterns obtained from crystal 1 and crystal 2 are virtually identical. Although they have almost identical unit cell dimensions (Table S5† and Fig. S26†), this also means there is significant correlation in how the species pack within a unit cell. Likewise, the bulk powder sample of 41c also corresponds well to both simulated patterns. Unfortunately, the near identical nature of their simulated patterns severely hampers any attempts at obtaining a crystal 1/crystal 2 ratio in 41c. The catalyst 43a also crystallized as a racemate (Fig. S23†).
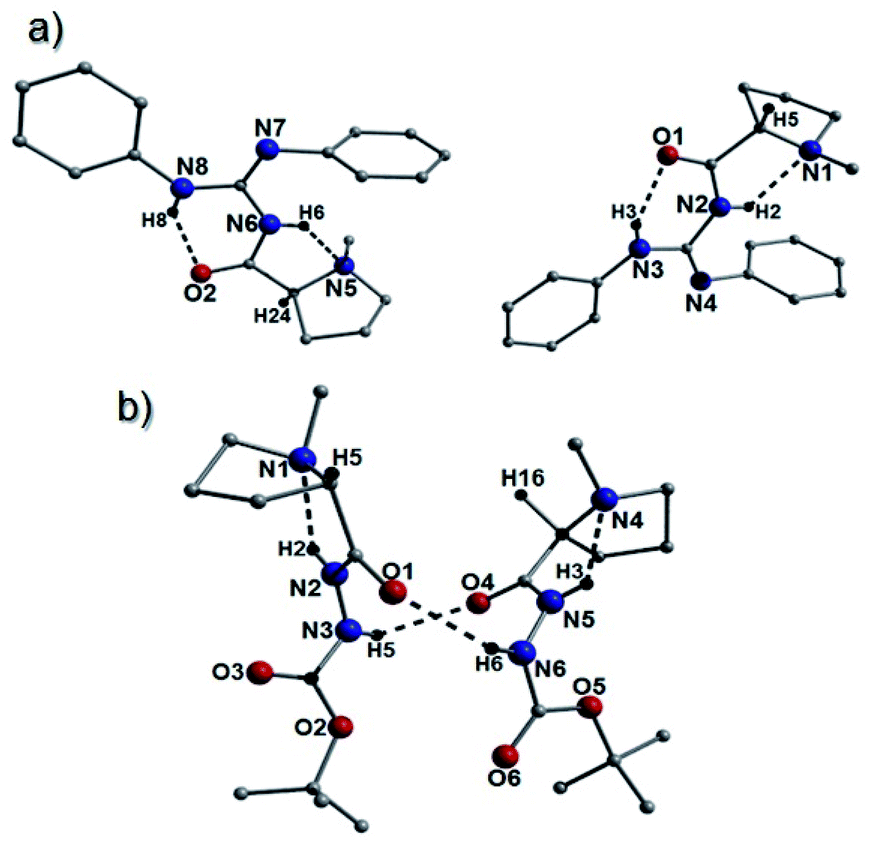 | ||
| Fig. 4 (a) Crystal structure highlighting the two organic moieties (S-left and R-right) comprising the asymmetric unit in 26a. The chiral protons are labelled H24 and H5. The dashed lines represent the intramolecular H-bonding interactions: N8(H8)⋯O2 = 2.03 Å; N6(H6)⋯N5 = 2.07 Å; N3(H3)⋯O1 = 2.01 Å and N2(H2)⋯N1 = 2.15 Å. Note: the disorder observed at both proline moieties is not shown. Catalyst 26a crystallises as an 2:1 S:R epimeric mixture. (b) The asymmetric unit in 41c (crystal 1). Dashed lines represent the intermolecular H-bonding (O1⋯(H6)N6 = 2.25 Å, N3(H5)⋯O4 = 2.18 Å) and intramolecular H-bonding (N2(H2)⋯N1A = 2.37 Å and N5(H3)⋯N4 = 2.20 Å). | ||
This was a surprising observation, as the L-proline used in the synthesis was of the reported optical rotation, as was the N-methylproline 23a, which was prepared using a literature procedure.13 The epimerization of several of the catalysts was investigated by NMR in deuterated methanol under neutral, weakly basic (NEt3) and acidic (PhCO2H) conditions and whilst decomposition (hydrolysis) was observed over prolonged time-periods (>5 days), no evidence of deuterium incorporation at the proline chiral center was observed. We thus investigated the nature of the most successful catalyst 25a by HPLC and found it to also be a 3:1 mixture of epimers by HPLC (by comparison with a separately prepared racemic sample). For compound 25a, the PXRD patterns show observable differences. Realistically the only differences between the 86% ee and 100% ee patterns, is the sharpness of the peaks in the 100% ee (Fig. S28†). However they are both totally different from the racemic mixture, suggesting that the racemate has a completely different type of packing to the pure chiral form.
We concluded from this that the epimerisation must be occurring at the CDI coupling stage. We thus repeated the preparation of 25a by coupling 23a with N-Cbz-guanidine which was activated using CDI in DMF over different times. Analysis of the products by HPLC and optical rotation data was performed and the traces compared with an independently prepared racemic and enantiomerically pure sample of 25a (obtained by repeated recrystallization) (Scheme 5, Table 1).
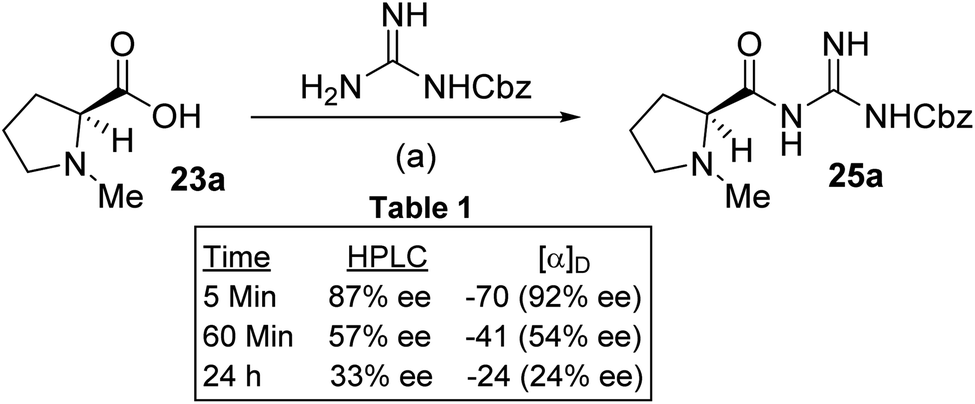 | ||
| Scheme 5 (a) (i) 23a, CDI, DMF, 5 min, 1 h or/24 h, rt; (ii) N-Cbz-guanidine, rt 24h; 47–53% yield. | ||
It was apparent from these experiments that even under short activation times racemization was occurring very rapidly. The only possible explanation of this is that the intermediate imidazole amide 48 was undergoing base catalyzed epimerization, via enolate 49. The likely base for this process is the amide 48 itself and in the cases where relatively strong guanidine bases are used in the coupling, their presence might exacerbate this process (Scheme 6).
 | ||
| Scheme 6 Proposed mechanism for the racemization of the proline catalyst 25a. (a) (i) CDI, DMF, (ii) N-Cbz-guanidine. | ||
We thus repeated the preparation of the Michael adduct 5 using enantiomerically pure 25a and were able to increase the ee of 5 to 56% ee over 48 h reaction in toluene. Similarly catalyst 29 was prepared using a short coupling time and repeated recrystallized to constant optical rotation and this again gave an increases ee for 5 to 41% (from 22%) over 10 h in toluene. These two compounds represent the best catalysts studied to date within this work.
Comparison of crystal H-bonding patterns and solution chemistry
We still wished to rationalize the reactions from the standpoint of the crystal structures and to this end, investigated the structures of several of the catalysts and were able to obtain crystallographic data on compounds 25a, 26a, 26c, 26d, 27a, 29, 30, 31, 34a, 34b, 34c, 38b, 39, 40b, 40d, 41b, 41c, 43a, 43b and 44b. As expected prior to their crystallization (and shown in Tables S1–S5†), the majority of our catalysts crystallized in Sohncke type space groups which include: P1 (30), P21 (25a, 26c, 26d, 29, 34a, 38, 40d, 41c (crystal 2) and 43b), P212121 (27a and 40b), C2221 (39) and P65 (34b). On close inspection and as highlighted in Fig. 3–12 as well as throughout the ESI (Fig. S1–S24†), intramolecular hydrogen bonding is commonplace within each catalyst. More specifically, the majority of interactions occur between guanidine protons, neighboring carbonyl oxygen and proline nitrogen acceptor atoms (e.g. N7(H54)⋯O5 = 1.83 Å and (N6(H55)⋯N5 = 2.26 Å, respectively; Fig. 5). | ||
| Fig. 5 ChemDraw (top) and crystal structure (middle) representations of 25a. (Bottom): The two asymmetric units in 25a. The dashes lines represent the intramolecular interactions within each moiety. These distances are: N2(H52)⋯N1 = 2.26 Å; N3(H50)⋯O2 = 2.00 Å; N3(H51)⋯O1 = 1.87 Å; N6(H55)⋯N5 = 2.26 Å; N7(H53)⋯O4 = 2.03 Å; N7(H54)⋯O5 = 1.83 Å. The longer contacts O2⋯H9A(C9) = 2.36 Å and O5⋯H24B(C24) = 2.33 Å are not shown. | ||
We hoped to use the X-ray structures obtained to give some insight into the possible H-bonding patterns found in these catalysts in solution. We initially compared the X-ray structures of the compounds 25a, 29, 30 and 31. Compounds 25a and 29 have no methyl substituent on the guanidine and 25a gave two H-bonds between the amide carbonyls and two of the N–H's of the guanidine (bond b; N3(H51)⋯O1 = 1.87 Å and bond d; N3(H50)⋯O2 = 2.00 Å). Additionally a H-bond between the proline nitrogen atoms (N1) and the amide NH (bond a; N2(H52)⋯N1 = 2.26 Å) was present and this compound was thus an E-abd type (Fig. 5).
The analogous Boc-protected compound 29 was prepared in order to study changes in steric factors near the guanidine and surprisingly it had a different H-bonding pattern (E-abc) to that of 25a. Catalyst 29 had two H-bonds between the amide and the carbamate carbonyls and two of the N–H's of the guanidine (bond b; N3(H3)⋯O1 = 1.94 Å and bond c; N2(H1)⋯O2 = 2.09 Å). The H-bond between the proline nitrogen atom (N1) and the amide NH (bond a; N2(H1)⋯N1 = 2.10 Å) was also present (Fig. 6).
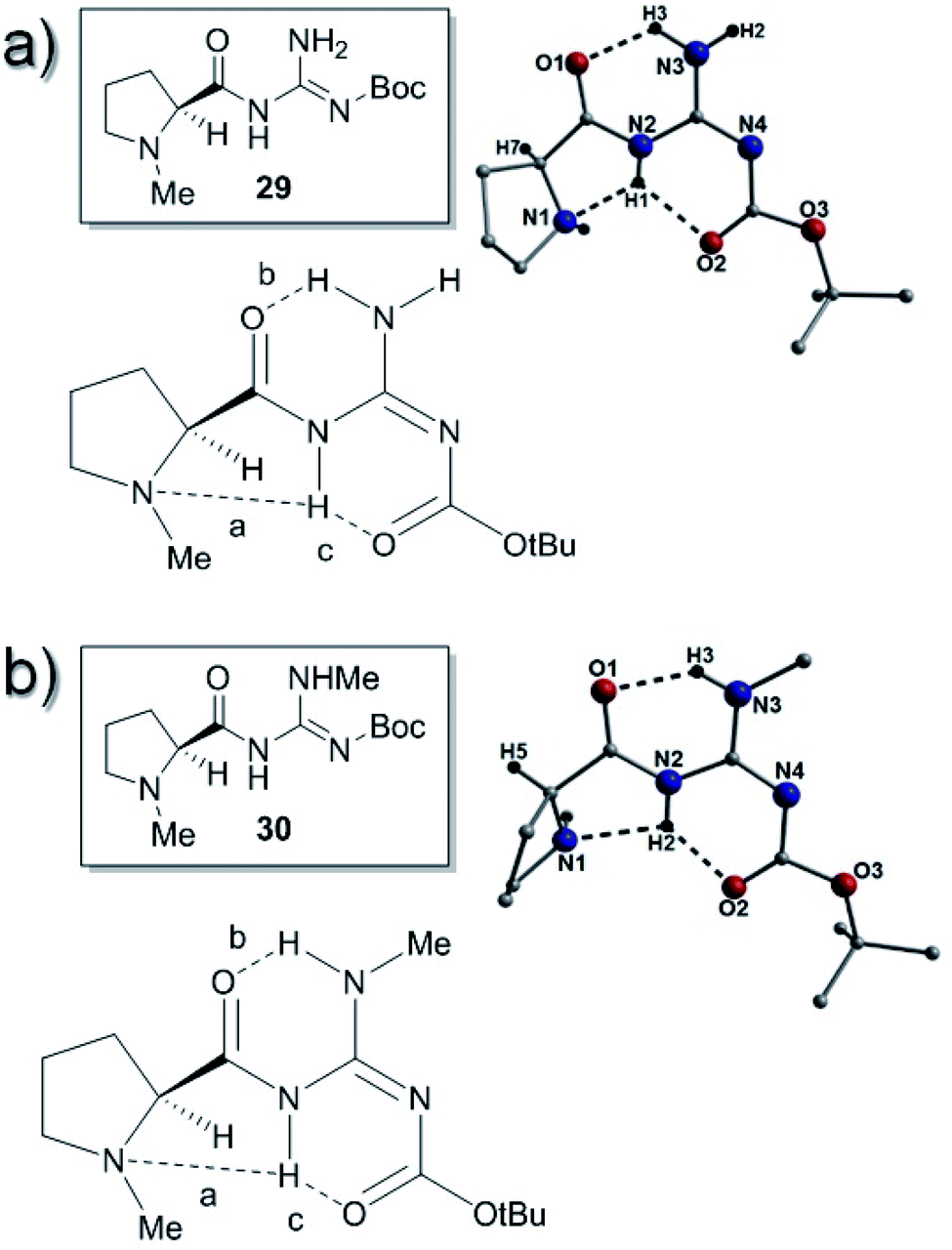 | ||
| Fig. 6 (a) ChemDraw, H-bonding and crystal structure representations of 29. The intramolecular H-bonding distances (dashed lines) are: N2(H1)⋯N1 = 2.10 Å, N2(H1)⋯O2 = 2.09 Å and N3(H3)⋯O1 = 1.94 Å. The chiral proton is labelled H7. Note: there are two units of 29 in the asymmetric unit and therefore only one is shown here. (b) ChemDraw, H-bonding and crystal structure of 30. The intramolecular H-bonding distances (dashed lines) are 2.37 Å (N2(H2)⋯N1), 1.92 Å (N3(H3)⋯O1) and 1.92 Å (N2(H2)⋯O2). Note: there are two complete molecules in the asymmetric unit and so only one is shown here. | ||
Compounds 30 and 31 were prepared in order to disrupt the intramolecular hydrogen bonding patterns in the catalysts. Catalyst 30 gave a structure with an identical H-bonding pattern (E-abc) to catalyst 29 as it had two H-bonds between the amide carbonyls and two of the N–H's of the guanidine (bond b; N3(H3)⋯O1 = 1.92 Å and bond c; N2(H2)⋯O2 = 1.92 Å). The H-bond between the proline nitrogen atom (N1) and the amide NH (bond a; N2(H2)⋯N1 = 2.37 Å) was also present (Fig. 6).
On close inspection of the X-ray crystal structure of compound 31, more evidence of the proposed racemization process was observed as the unit-cell was shown to comprise a 2:1 mixture of S:R enantiomers. The powder X-ray diffraction spectrum obtained from 31 was found to compare favourably with the corresponding simulated spectrum generated by single crystal data (ESI, Fig. S29†). Despite this the S-enantiomer had an identical E-abc arrangement to 29 with two H-bonds between the amide carbonyls and two of the N–H's of the guanidine (bond b; N7(H34)⋯O4 = 1.90 Å and bond c; N6(H33)⋯O5 = 1.95 Å). The H-bond between the proline nitrogen atom (N5) and the amide NH (bond a; N6(H33)⋯N5 = 2.44 Å) was also still present (Fig. 7a).
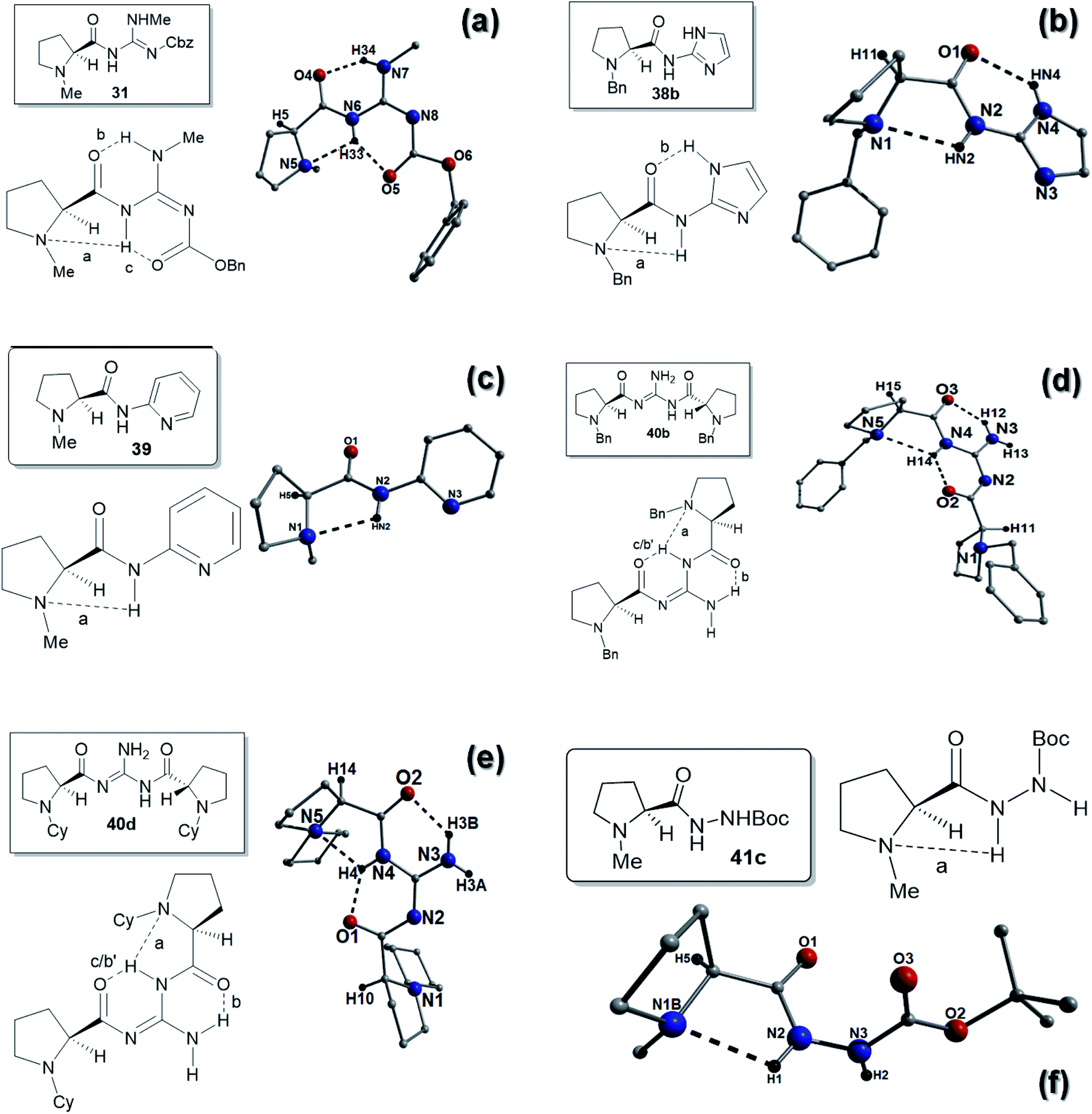 | ||
| Fig. 7 ChemDraw and crystal structure representations of 31 (a), 38b (b), 39 (c), 40b (d), 40d (e) and 41c (f). All intramolecular H-bonding distances are represented as dashes lines. These distances are: (a) N6(H33)⋯N5 = 2.44 Å, N6(H33)⋯O5 = 1.95 Å, N7(H34)⋯O4 = 1.90 Å. (b) N2(HN2)⋯N1 = 2.36 Å, N4(HN4)⋯O1 = 2.24 Å. (c) N2(HN2)⋯N1 = 2.29 Å. (d) N4(H14)⋯N5 = 2.44 Å, N4(H14)⋯O2 = 1.99 Å, N3(H12)⋯O3 = 1.99 Å. (e) N4(H4)⋯N5 = 2.28 Å, N3(H3B)⋯O2 = 2.06 Å, N4(H4)⋯O1 = 1.93 Å. (f) N2(H1)⋯N1B = 2.32 Å. Note: 31 crystallises as an 2:1 S:R epimer mixture. The S-enantiomer is shown here. There are two types of crystals in the bulk material of 41c (labelled crystal 1 and crystal 2). Crystal 1 is racemic in nature and all 41c units in crystal 2 exhibit the S configuration. The S-enantiomer extracted from crystal 1 is shown in (f). | ||
It was observed that both catalyst 30 and 31 gave very slow reaction times and very low ee's in all the solvents studied and might indicate that substituting the hydrogens on the guanidine leads to a lowering of ee, possibly because we are blocking a site for H-bonding interactions. Contrasting these results to catalyst 29, which gave very rapid reaction times in toluene, the lack of a methyl group might enable the guanidine N3–H2 and N4 positions to form a bidentate interaction with the quinone 1 or the nitro-group of β-nitrostyrene 2 and promote the reaction.
We were unable to obtain any X-ray data on the methyl-substituted benzimidazoles 35 and 36, however we were able to obtain a structure on the imidazole compound 38b (Fig. 7b). Compound 38b had an E-ab hydrogen bonding pattern and possessed a H-bond between the proline nitrogen and the amide HN bond (bond a; N2(HN2)⋯N1 = 2.36 Å) and a H-bond between the imidazole NH and the amide carbonyl (bond b; N4(HN4)⋯01 = 2.24 Å). The ee's for the reaction of this catalyst were low, which might be due to the effect of the N-benzyl group as the corresponding N-methyl catalyst 38a gave slightly better ee's. The lower ee's might indicate that the benzimidazole ring plays a role in the better ee's achieved with the catalyst 34a.
The pyridine catalyst 39 had a single H-bond (E-a) between the proline nitrogen and the amide HN bond (bond a; N2(HN2)⋯N1 = 2.29 Å). Reaction times were reasonable for this catalyst (48 h) which might reflect the basicity of the pyridine, however no appreciable ee's were observed in the reactions (Fig. 7c). The two C2-symmetric catalysts 40b and 40d and were found to possess similar hydrogen bonding patterns (see Fig. 7d and e). In compound 40b, one amide NH was H-bonding to the proline nitrogen (bond a; N4(H14)⋯N5 = 2.44 Å) and this hydrogen was also H-bonding to the other amide carbonyl (bond c/b′: N4(H14)⋯O2 = 1.99 Å). The amide carbonyl is also H-bonding to the guanidine NH2 (bond b; N3(H12)⋯O3 = 1.99 Å). The other proline nitrogen is free of intramolecular H-bonds so this H-bonding interaction overall is an E-abc/Free-b arrangement (Fig. 7d). Similarly an identical H-bonding patterns was observed for 40d in that one amide NH was H-bonding to the proline nitrogen (bond a; N4(H4)⋯N5 = 2.28 Å) and this hydrogen was also H-bonding to the other amide carbonyl (bond c/b′: N4(H4)⋯O1 = 1.93 Å). The amide carbonyl is also H-bonding to the guanidine NH2 (bond b; N3(H3B)⋯O2 = 2.06 Å). Again the other proline nitrogen is free of intramolecular H-bonds so this H-bonding interaction is also an E-abc/Free-b arrangement (Fig. 7e).
In both 40b and 40d it is apparent that the proline nitrogen (labelled N5 in both cases) is not involved in intramolecular hydrogen-bonding and this might leave it free for base catalysed reactions. This observation might explain why several of the reactions of these catalysts took relatively short times to complete (24–48 h, Scheme 4c and appendix IV). Unfortunately it seems apparent that the relatively strong intramolecular H-bonds of the guanidine NH's and the carbonyls might preclude the formation of the bidentate H-bonding pattern we had hoped for. This, together with the known problems associated with bulky nitrogen substituent groups may have led to the poor ee's.
As previously stated the co-crystallisation of two polymorphs of 41c (crystal 1: 1:1 racemic mixture and crystal 2: S-configuration only) indicated that the reaction has proceeded with partial racemization and that the 1:1 mixture has crystallized from a partially racemic mixture together with the S-enantiomer. Despite this a similar H-bond between the proline nitrogen and the amide HN bond (bond a; N2(H2)⋯N1 = 2.23 Å) was observed (E-a type) on examination of the S-enantiomer. This was shorter than that found in other catalysts, which might explain the slow reaction times for this compound compared to the other hydrazine catalyst (Fig. 7f).
The X-ray structure was also obtained for the phenyl hydrazine catalyst 41b and was found to crystallize as a 2:1 mixture of the S- and R-enantiomers again indicating partial racemization or preferential crystallization. The powder XRD data obtained from a polycrystalline sample of 41b suggests that its single crystal data is representative of the bulk sample (Fig. S30†). The H-bond pattern is similar to the majority of the guanidine catalysts in that it is an E-amide-ab′ type arrangement. As such, there is a H-bond between the nitrogen and the amide NH bond (bond a; N2(H2)⋯N3 = 2.29 Å) and a long H-bond between the amide carbonyl and the other NH of the hydrazine (bond b′; N1(H1)⋯O1 = 2.68 Å). This catalyst gave a very short Michael reaction time (1 h in toluene, xylene and benzene) and may be explained by the presence of the hydrazine PhNH, which will be more basic than the examples where this NH is a carbamate. Low ee's (2–3%) were obtained using this catalyst, however, on cooling the ee in toluene was improved to 17% (Fig. 8).
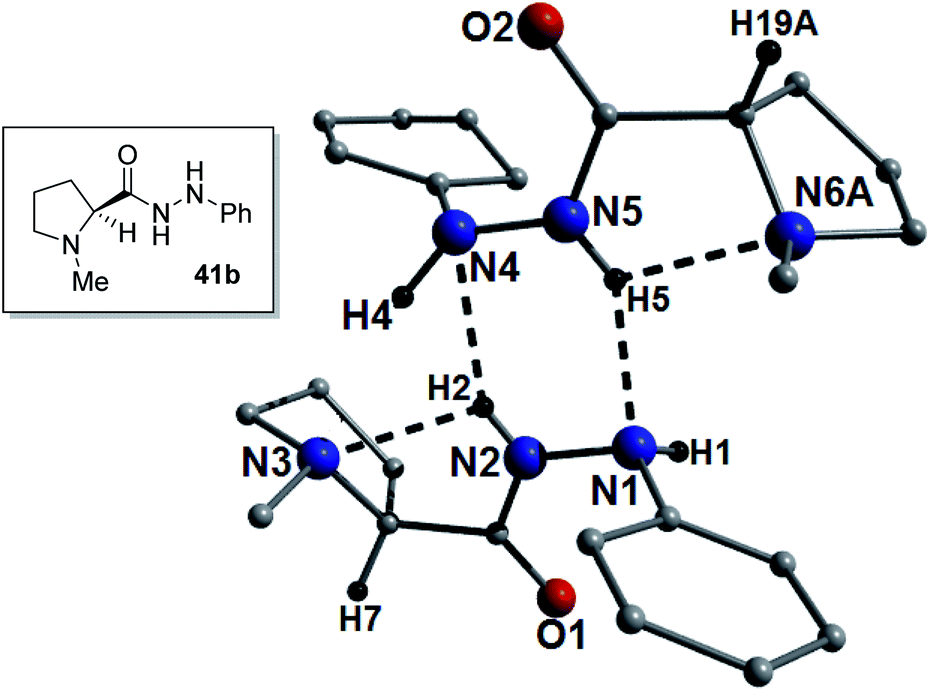 | ||
| Fig. 8 ChemDraw (inset) and crystal structure representations of 41b. The dashed lines represent intra- and intermolecular H-bonding interactions with distances: N2(H2)⋯N3 = 2.29 Å, N5(H5)⋯N6A = 2.30 Å, N2(H2)⋯N4 = 2.26 Å and N5(H5)⋯N1 = 2.18 Å. Note: the A label denotes a disordered (R:S) proline unit (only A is shown here). For clarity the N1(H1)⋯O1 intramolecular interaction is not shown here. | ||
X-Ray structures of the dimethyl-L-alanine catalyst 43a and the dimethyl-L-phenylalanine catalysts 44a and 44b were also obtained. Again, unfortunately, the dimethyl-L-alanine catalyst 43a crystallized as a racemic mixture (single crystal and pXRD corroborate batch uniformity; ESI, Fig. S31†), however the S-enantiomer gave an E-abc type with H-bonds between the N–H of the amide and the dimethylamine together with an H-bond between a guanidine NH and the amide carbonyl (bond a; N3B(HN3)⋯N4 = 2.30 Å and bond b; N2(HN2A)⋯O3B = 2.05 Å). A third H-bond between the amide NH and the carbonyl of the Cbz groups was also observed (bond c; N3B(HN3)⋯O2 = 1.96 Å), which was the shortest H-bond of the three (Fig. 9).
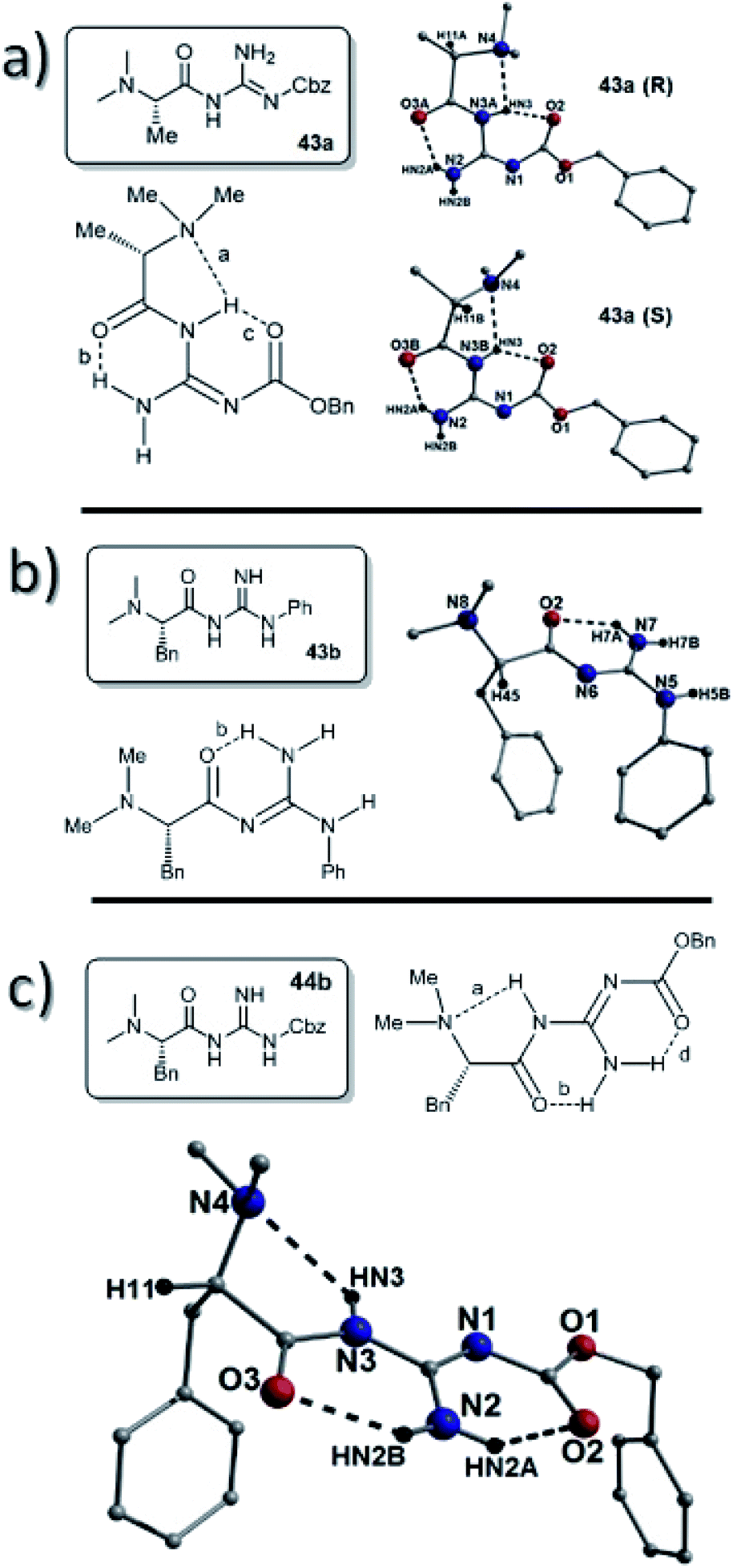 | ||
| Fig. 9 (a) The ChemDraw representation and crystal structures of the R- and S-enantiomers observed in the racemic compound 43a. Intramolecular H-bonding shown using dashed lines at distances: N2(NH2A)⋯O3A = 2.06 Å, N3A(HN3)⋯N4 = 2.30 Å and N3A(HN3)⋯O2 = 1.96 Å (in R) and N2(HN2A)⋯O3B = 2.05 Å, N3B(HN3)⋯2.30 Å and N3B(HN3)⋯O2 = 1.96 Å (in S). (b) ChemDraw and crystal of catalyst 43b. The dashed lines represent intramolecular H-bonding interactions (N7(H7A)⋯O2 = 1.98 Å). Note: there are four molecules of 43b in the asymmetric unit. A solitary unit is shown here. (c) Crystal structure obtained from 44b. The dashed lines represent intramolecular H-bonding at distances: N2(HN2B)⋯O3 = 2.03 Å, N2(HN2A)⋯O2 = 1.98 Å and N3(HN3)⋯N4 = 2.48 Å. | ||
The structure of the dimethyl-L-phenylalanine-phenyl guanidine catalyst 43b was obtained and this had a significantly different Free-b type H-bonding pattern. In this structure the amide was present as an N-methyleneformamide which lacks an N–H bond. The only intramolecular H-bond was between the guanidine NH2 and the amide carbonyl (bond b; N7(H7A)⋯O2 = 1.98 Å) (Fig. 12).
The dimethyl-L-phenylalanine catalyst 44b gave no signs of racemization in the crystal. This compound displayed an E-abd H-bonding pattern with a hydrogen bond between the N–H of the amide and the dimethylamine together with a H-bond between a guanidine NH and the amide carbonyl (bond a; N3(HN3)⋯N4 = 2.48 Å and bond b; N2(HN2B)⋯O3 = 2.03 Å). A third H-bond was again observed, this time between the guanidine NH2 and the carbonyl of the Cbz group (bond c; N2(HN2A)⋯O2 = 1.98 Å), which again was the shortest H-bond of the three (Fig. 9).
The Michael reaction using the dimethyl-L-alanine catalyst 43a gave poor ee's (7–13%), over relatively long reaction times (18–96 h). This might be a reflection on the degree of racemisation or possibly the level of H bonding. The dimethyl-L-phenylalanine catalyst 44b gave better ee's (4–25%) but again generally relatively long reaction times. In contrast, the dimethyl-L-phenylalanine phenyl guanidine catalyst 43b gave rapid reaction times (8–10 h) in all solvents and the ee's (18–21%) were high when compared to the other two catalysts. This might possibly be due to the more basic nature of the guanidine, which is substituted with only one electron withdrawing carbonyl containing group. These observations might suggest that the presence of the carbamate protecting groups is detrimental to the efficiency of the reaction and does not lead to a high ee product (as previously postulated).
Whilst it is difficult to draw conclusions from the X-ray data, it is obvious the presence of racemization is a major concern in the potential for these catalysts. Despite this there is some evidence that the more substituted catalysts are less efficient and that a maximum of two substituents seems to be the most promising. Some interesting observations were also made regarding intermolecular H-bonding interactions and this is discussed below.
Intermolecular interactions
In terms of intermolecular interactions and as exampled by the species 25a, 40b and 43b, the individual organic moieties in a number of our catalysts self-assemble in the solid-state as planar sheet arrays through complementary H-bonding interactions as highlighted in Fig. 10 (25a), Fig. 11 (40b) and Fig. 12 (44b). In all these examples, the H-bond connections are forged through guanidine protons and neighboring carbonyl oxygen, alcohol oxygen and/or guanidine nitrogen acceptor atoms (i.e. N2(H52)⋯O1′ = 2.58 Å and N4⋯(H51′)N3′ = 2.63 Å in Fig. 10). For example, the catalyst 25a comprises two organic crystallographically unique moieties within its asymmetric unit and partake in side-on C–H⋯π intermolecular interactions between proline protons and juxtaposed benzoate aromatic rings (e.g. C17(H17B)⋯[C10–C15] = 3.01 Å and C2(H2B)⋯[C25–C30] = 3.20 Å) to form an interdigitated rectangular dimeric assembly. Likewise, intermolecular complementary H-bonding between the individual guanidine units connect these dimeric units along the a unit cell direction to form tubular sheet-like arrays throughout the crystal structure in 25a (Fig. 10).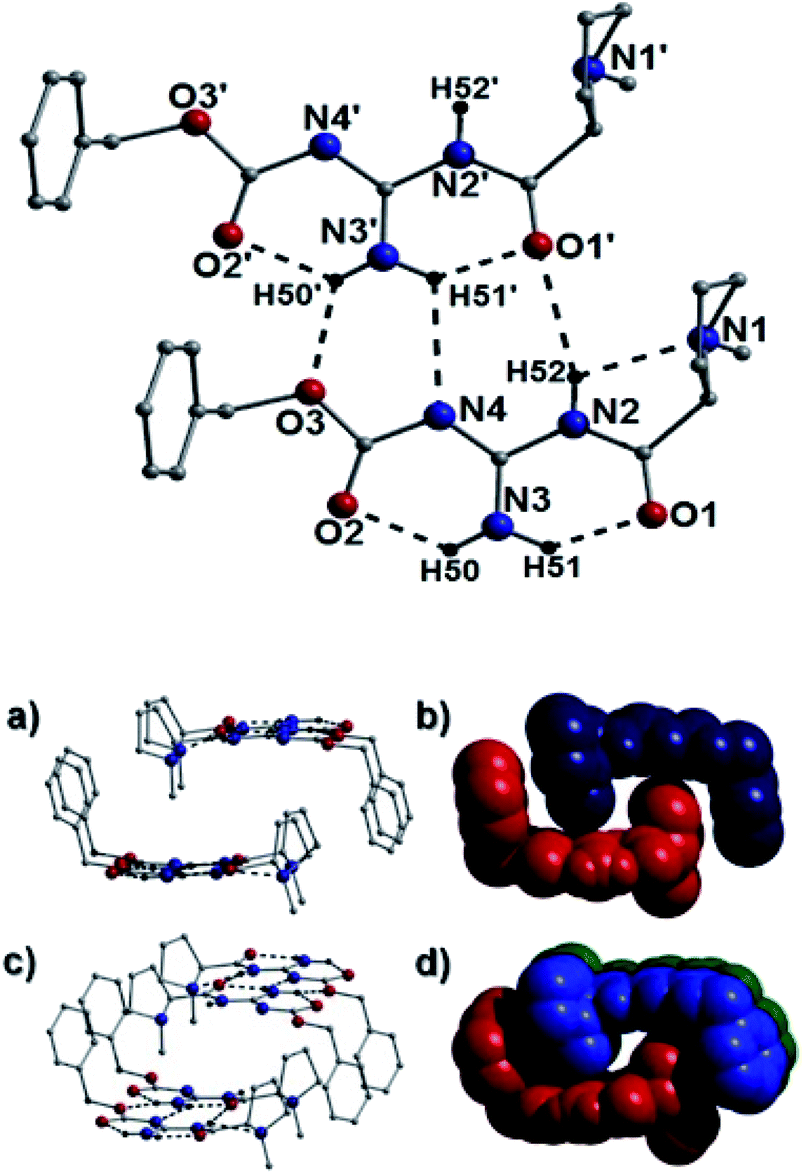 | ||
| Fig. 10 (Top): Intermolecular H-bonding arrangement (dashed lines) between two crystallographically equivalent 25a units. Selected distances: O3⋯(H50′)N3′ = 2.20 Å; N4⋯(H51′)N3′ = 2.63 Å and N2(H52)⋯O1′ = 2.58 Å. Note: intramolecular H-bonding also shown (dashed lines). (Bottom): Regular (a and c) and space-fill (b and d) views of the packing arrangement observed in 25a. Each unit (in b and d) is represented by a different colour to highlight the space efficient interdigitation of the U-shaped organic units in 25a. | ||
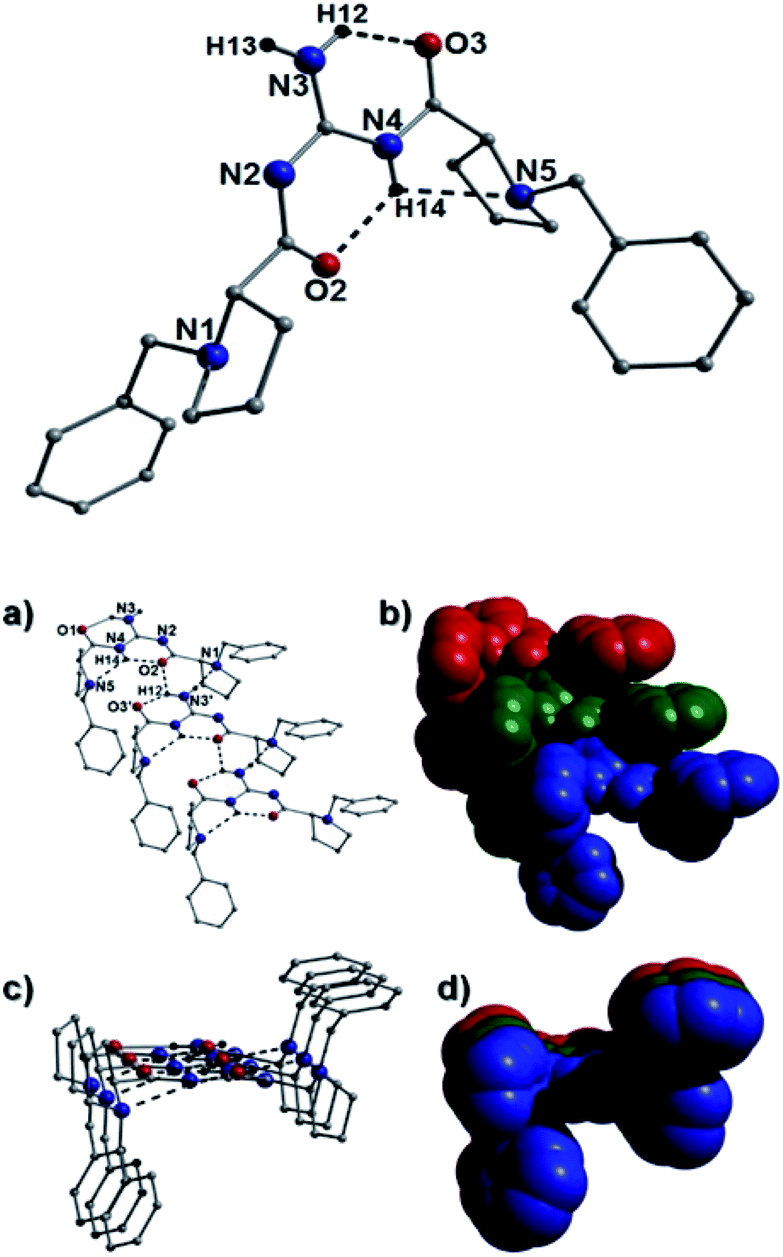 | ||
| Fig. 11 (Top): The crystal structure of 40b. The dashed lines represent the intramolecular hydrogen bonding interactions: N4(H14)⋯O2 = 1.99 Å; N3(H12)⋯O3 = 1.99 Å and N4(H14)⋯N5 = 2.44 Å. (Bottom): A section of the H-bonded (dashed lines) chains in 40b as viewed off-set (a) and parallel (c) to the plane of the H-bonded guanidine moieties along with their space-fill representations (b and d respectively). The individual guanidine units are colour coded. The intermolecular H-bonded distances are: N1⋯(H13′)C13′ = 2.06 Å (not shown); O2⋯(H12′)N3′ = 2.40 Å and C17(H17A)⋯O3′ = 2.80 Å. | ||
 | ||
| Fig. 12 (Top left): Crystal structure of 44b highlighting (a) the intramolecular H-bonding interactions in 44b (dashed lines) (N3(HN3)⋯N4 = 2.48 Å; N2(HN2A)⋯O2 = 1.98 Å and N2(HN2B)⋯O3 = 2.03 Å) and (b and c) the superimposable U-shaped 1D H-bonded rows of 44b units propagating along the a direction of the unit cell. Selected intermolecular H-bonding interactions: N3(HN3)⋯O3′ = 2.54 Å; N1⋯(HN2B′)N2′ = 2.49 Å and O1⋯(HN2A′)N2′ = 2.22 Å. (d) is a space-fill representation of the 1D rows in 44b. Each colour represents a single organic unit. (Top right): Crystal structure of the dimeric asymmetric unit 40d as viewed perpendicular (a) and parallel (c) to the plane of the H-bonded (dashed lines) guanidine moieties along with their colour coded space-fill representations (b and d, respectively). Intramolecular H-bonded distances: N3(H3B)⋯O2 = 2.06 Å; N4(H4)⋯O1 = 1.93 Å; N8(H8D)⋯O3 = 2.05 Å; N7(H7)⋯O4 = 1.95 Å; N7(H7)⋯N6 = 2.31 Å and N4(H4)⋯N5 = 2.28 Å. Intermolecular H-bonded distances: N8(H8C)⋯N2 = 2.11 Å; N3(H3A)⋯N9 = 2.09 Å; N3(H3B)⋯N10 = 2.89 Å and N8(H8D)⋯N1 = 2.96 Å. (Bottom left): (a) A H-bonded helical chain of individual units of 34b propagating along the c unit cell direction. The dashed lines represent the intermolecular H-bonding interactions in 34b (N2(H20)⋯O1′ = 2.26 Å and N4⋯H19′(N3′) = 2.02 Å). (b) The asymmetric unit in 34b highlighting (dashed lines) the intramolecular H-bonding interactions (N2(H20)⋯N1 = 2.33 Å and N3(H19)⋯O1 = 2.36 Å). (c) A single H-bonded helical chain in 34b as viewed along the c direction of the unit cell. (Bottom right): Crystallographic packing of 34b as viewed along the c direction of the unit cell. | ||
Akin to the connectivity arrangements in 25a, the chair-shaped organic units in 40b produce superimposable H-bonded sheets along the a unit cell direction (Fig. 11). The intermolecular hydrogen bonding interactions are forged between guanidine protons and neighbouring carbonyl oxygen acceptor atoms (e.g. N3(H12)⋯O2′ = 2.40 Å). The individual H-bonded rows in 40b connect along the bc plane of the unit cell through C–H⋯π interactions between proline protons and neighbouring aromatic rings at distances of (for instance): 3.83 Å (C15(H15)⋯[C20–C25]), 3.46 Å (C15(H16B)⋯[C20–C25]) and 3.44 Å (C17(H17B)⋯[C20–C25]). In a similar fashion, the individual U-shaped organic units in 44b connect along the a cell direction via numerous intermolecular interactions to form a H-bonded half pipe array as highlighted in Fig. 12.
In a similar vein, the catalysts 29, 30 and 40d arrange into H-bonded dimers and are presumably too sterically hindered by their tert-butyl (29 and 30) and cyclohexane (40d) groups to forge planar sheet topologies as previously described. For instance, the catalyst 40d crystallises in the monoclinic P21 space group (screw axis along b cell direction) and exhibits two organic moieties in the asymmetric unit. Indeed these U-shaped guanidine moieties H-bond to one another via their guanidine protons (H3A and H8C) to form dimeric units that forge a U-shaped half-pipe motif as highlighted in Fig. 12.
Interestingly, the benzimidazole containing proline derived catalyst 34b crystallises in the hexagonal P65 space group whose translational symmetry is manifested through the formation of H-bonded helical chains propagating along the c direction of the unit cell (Fig. 12-bottom left). These chains organise themselves into a space efficient brickwork motif to give the packing arrangement shown in Fig. 12 (bottom right). Moreover, the structurally related catalyst 34c is the only member to encapsulate a solvent molecule during crystallisation. More specifically, a crystallographically unique water of crystallisation lies at H-bonding distance from the organic moiety in 34c. This molecule is held in position through H-bonding with a guanidine proton (H2N) at a distance of 2.15 Å (N2(H2N)⋯O2) and a guanidine N acceptor atom of an adjacent organic moiety at a distance of 2.09 Å (N4⋯(H20)O2) (Fig. S14 and S15†). As a result, these waters of crystallisation as molecular cement in connecting the individual brick units in 34c as they propagate along the a unit cell direction (Fig. S16†).
Conclusions
The work presented here has illustrated that amino acid derived guanidine have some potential as asymmetric organocatalysts however several problems have arisen. Crystallographic investigation of the catalysts has led to the discovery of quite prevalent racemization occurring during their formation under CDI activated coupling. Obviously, this is a major drawback to their use as catalysts and they are not competitive in terms of producing the targeted asymmetric products. Our investigation in the solid state demonstrated extensive intra- and intermolecular H-bonding abilities of the proline, guanidine and/or amide functional groups within these organic moieties. These interactions lead to the production of elaborate and aesthetically pleasing hydrogen bonded extended network architectures. Consideration of the intramolecular H-bonding gave a loose correlation with catalytic efficiency and it inferred that these interactions remain in place in solution. A consideration of these structural interactions might lead the way to development of new catalysts. Indeed our current work addresses this together with new methodology to produce related catalysts without the problems associated with racemisation.Experimental
General procedures
Unless otherwise noted, reactions were stirred and monitored by TLC. TLC plates were visualized using iodine, phosphomolybdic acid or under UV light. All anhydrous reactions were conducted under a static argon atmosphere using oven dried glassware that had previously been cooled under a constant stream of nitrogen. Reagents and starting materials were purchased from commercial suppliers and used without further purification unless otherwise noted. All anhydrous solvents used in reactions were distilled over either sodium wire and benzophenone (THF/DE) or calcium hydride (DCM), and used either immediately or stored over molecular sieves prior to use. Flash column chromatography was performed on Davisil® silica gel (35–70 microns) with the eluent specified in each case, TLC was conducted on precoated E. Merck silica gel 60 F254 glass plates. Melting points were determined using a Gallenkamp MF370 instrument and are uncorrected. 1H and 13C NMR spectra were recorded on a Bruker Avance 400 or 500 spectrometer with an internal deuterium lock at ambient temperature at 400 or 500 MHz with internal references of δH 7.26 and δC 77.016 ppm for CDCl3, δH 3.31 and δC 49.0 ppm for CD3OD and δH 2.54 ppm and δH 39.52 ppm for DMSO. All mass spectra were performed at the EPSRC National Mass Spectrometry Service Centre based in Swansea. Low resolution Chemical Ionisation (CI) and Electrospray Ionisation (ESI) mass spectra were recorded on a Micromass Quattro II spectrometer and high resolution mass spectra were recorded on either a Finnigan MAT 900 XLT or a Finnigan MAT 95 XP. Infrared samples were prepared as thin films or solutions using sodium chloride plates or as KBr discs; spectra were recorded on a Bruker Tensor 37 FT-IR. Optical rotations were determines on an ADP400 polarimeter. The N-alkylated prolines were prepared by literature methods and the guanidines and heterocycles used in the study were prepared by literature methods or are commercially available, full details of these preparations are given in the ESI.† All PXRD samples were collected on a Rigaku MiniFlex 600 (CuKα radiation (40.0 kV, 15.0 mA), over a 2θ range of 5 to 40°.General methods for the preparation of the catalysts 21–41 and 43–47
Spectroscopic data
251, 2804, 1665, 1571, 1525, 1493 cm−1; MS (CI) m/z 160.1 (100%), 269.1 (40%, [M − H]+) 293.1 (55% [M + Na]+); HRMS (CI) m/z found 293.1376, C15H18N4ONa ([M + Na]+) requires 293.1373.General method for the reaction of 2-hydroxy-1,4-naphthoquinone 93 with β-nitrostyrene 52
2-Hydroxy-1,4-napthoquinone 1 (100 mg, 0.574 mmol) and the required catalyst (0.04–0.1 equiv.) were dissolved in the required solvent and cooled to the required temperature (−20 to 0 °C, see Table 1). β-Nitrostyrene 2 (128.5 mg, 0.861 mmol, 1.5 equiv.) was then added and the mixture stirred for the required time and temperature. Reaction progress was determined by sampling and H NMR analysis. On completion the solvent was evaporated to give a deep red residue which was purified by column chromatography eluting firstly with 2–4% EA in petrol to remove excess 2 then followed with DCM to give the product 5 as a yellow solid. Illustrative example from catalyst 25a; 56% ee, [α]25D −16.0 (acetone, c = 1.46); lit. [α]17D −44.8 (acetone, c = 1.0),14 lit. [α]25D −34.0 (acetone, c = 1.46).15 Enantiomeric excess were determined either on a Chiralcel AS-H (250 × 4.6 mm, mobile phase 96% hexane, 4% isopropanol, 0.1% TFA, 1.5 mL min−1 at 40 °C, detecting at 254 nm; R enantiomer 23.5 min, S enantiomer 26.2 min) or a Phenomenex Lux Amylose-1 (250 × 4.6 mm, mobile phase 70% hexane, 30% isopropanol, 0.5 mL min−1 at 40 °C, detecting at 254 nm; R enantiomer 13.2 min, S enantiomer 14.3 min).X-ray diffraction studies
Catalysts 25a, 34c, 40b and 40d, were collected on an Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E+ Super Bright molybdenum rotating anode generator with HF Varimax optics (100 μm focus). The cell determination and data collection were carried out using the CrystalClear-SM Expert package (Rigaku, 2012) data reduction cell refinement and absorption correction were carried out using CrysAlisPro software (Rigaku OD, 2015).16Catalysts 30, 34a, 39, 41b, 41c-crystal 1, 43a and 44b were collected on an Rigaku AFC11 goniometer equipped with an Hypix 6000 detector mounted at the window of an 007HF copper rotating anode generator with HF Varimax optics (150 μm focus). Cell determination data collection, data reduction cell refinement and absorption correction were carried out using CrysAlisPro software (Rigaku OD, 2015).16
Catalysts 26a, 26c, 26d, 27a, 29, 31, 34b, 38b, 41c-crystal 2 and 43b were collected at the synchrotron Diamond Light Source (beamtime I19)17 using a Dectris Pilatus 2M detector (CCDC numbers: 1952629–1952649). Data collection was carried out using the beamline control software, data reduction cell refinement and absorption correction were carried out using XIA2.18 All structures were initially solved and refined using SHELXT and SHELXL-201419 within OLEX-2.20
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC (DE, EP/J01821X/1), the ERASMUS and SOCRATES schemes (FFJDK), BEACON (ERDF; DE, PJM), the Iraqi government (ZSSA-T) and the government of Saudi Arabia (YTHM) for funding and support. This work was carried out with the support of the Diamond Light Source, instrument I19 (proposal MT15762). We also thank the EPSRC National Crystallography Service at the University of Southampton and the National Mass Spectrometry Facility at Swansea for their excellent work.References
- M. T. Allingham, E. L. Bennett, D. H. Davies, P. M. Harper, A. Howard-Jones, Y. T. H. Mehdar, P. J. Murphy, D. A. Thomas, P. W. R. Caulkett, D. Potter, C. M. Lam and A. C. O'Donoghue, Tetrahedron, 2016, 72, 496–503 CrossRef CAS.
- S. Bhowmick, A. Mondal, A. Ghosh and K. C. Bhowmick, Tetrahedron: Asymmetry, 2015, 26, 1215–1244 CrossRef CAS.
- P. J. Murphy, M. St. Allingham and A. J. L. Howard-Jones, Tetrahedron, 2003, 44, 8677–8680 CrossRef.
- Z. Yu, X. Liu, L. Zhou, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2009, 48, 5195–5198 CrossRef CAS PubMed.
- M. Terada, T. Ikehara and H. Ube, J. Am. Chem. Soc., 2007, 129, 14112–14113 CrossRef CAS PubMed.
- D. Almasi, D. A. Alsono, E. Gomez-Bengoa and C. Najera, J. Org. Chem., 2009, 74, 6163–6168 CrossRef CAS PubMed.
- E. Gomez-Torres, D. A. Alonso, E. Gomez-Bengoa and C. Najera, Org. Lett., 2011, 13, 6106–6109 CrossRef CAS PubMed.
- T. E. Shubina, M. Freund, S. Schenker, T. Clark and S. B. Tsogoeva, Beilstein J. Org. Chem., 2012, 8, 1485–1498 CrossRef CAS.
- G. Tang, Ü. Gün and H. Altenbach, Tetrahedron, 2012, 68, 10230–10235 CrossRef CAS.
- S. Bhowmick, S. S. Kunte and K. C. Bhowmick, RSC Adv., 2014, 4, 24311–24315 RSC.
- A. D. G. Yamagata and D. J. Dixon, Org. Lett., 2017, 19, 1894–1897 CrossRef PubMed.
- G. Moore, Novel phosphonium salts and bifunctional organocatalysts in asymmetric synthesis, PhD thesis, University of Nottingham, 2013.
- Z. Han, R. Wang, Y. Zhou and L. Liu, Eur. J. Org. Chem., 2005, 934–938 CrossRef CAS.
- S. B. Woo and D. Y. Kim, Beilstein J. Org. Chem., 2012, 8, 699–704 CrossRef CAS PubMed.
- W. Yang and D. Du, Adv. Synth. Catal., 2011, 353, 1241–1246 CrossRef CAS.
- O. D. Rigaku, CrysAlis PRO, Rigaku Oxford Diffraction Ltd, Yarnton, England, 2015 Search PubMed.
- (a) D. R. Allan, H. Nowell, S. A. Barnett, M. R. Warren, A. Wilcox, J. Christensen, L. K. Saunders, A. Peach, M. T. Hooper, L. Zaja, S. Patel, L. Cahill, R. Marshall, S. Trimnell, A. J. Foster, T. Bates, S. Lay, M. A. Williams, P. V. Hathaway, G. Winter, M. Gerstel and R. W. Wooley, Crystals, 2017, 7(11), 336 CrossRef; (b) H. Nowell, S. A. Barnett, K. E. Christensen, S. J. Teat and D. R. Allan, J. Synchrotron Radiat., 2012, 19, 435–441 CrossRef CAS PubMed.
- G. Winter, J. Appl. Crystallogr., 2010, 43, 186–190 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. J. Puschmann, Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1952629–1952649. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra07508a |
| This journal is © The Royal Society of Chemistry 2020 |