DOI:
10.1039/C9RA07470H
(Paper)
RSC Adv., 2020,
10, 1219-1225
Rapid and selective electrochemical transformation of ammonia to N2 by substoichiometric TiO2-based electrochemical system†
Received
16th September 2019
, Accepted 19th December 2019
First published on 8th January 2020
Abstract
In this study, we have developed a continuous-flow electrochemical system towards the rapid and selective conversion of ammonia to N2, based on a tubular substoichiometric titanium dioxide (Ti4O7) anode and a Pd–Cu co-modified Ni foam (Pd–Cu/NF) cathode, both of which are indispensable. Under the action of a suitable anode potential, the Ti4O7 anode enables the conversion of Cl− to chloride radicals (Cl˙), which could selectively react with ammonia to produce N2. The anodic byproducts, e.g. NO3−, were further reduced to N2 at the Pd–Cu/NF cathode. EPR and scavenger experiments confirmed the dominant role of Cl˙ in ammonia conversion. Complete transformation of 30 mg L−1 ammonia could be obtained over 40 min of continuous operation under optimal conditions. The proposed electrochemical system also exhibits enhanced oxidation kinetics compared to conventional batch systems. This study provides new insights into the rational design of a high-performance electrochemical system to address the challenging issue of ammonia pollution.
1. Introduction
The excessive discharge of ammonia wastewater accelerates the eutrophication of water bodies and causes the serious water pollution we are currently facing.1–3 To address this issue, various radical-based catalytic processes have been extensively studied.4–6 The hydroxyl radical (OH˙) and sulfate radical (SO4−˙) are two types of highly potent radicals that have been widely applied. However, another important but often neglected type of radical, chloride radicals (Cl˙), have received only limited attention. These Cl˙ are selective oxidants (E0 = 2.4 V vs. NHE) that react with electron-rich groups like ammonia through single electron oxidation, H-abstraction, and addition to unsaturated C–C bonds.7,8 We have previously developed a flow-through electrochemical system enabling the sustainable production of Cl˙ and further converting ammonia to N2.9 Unfortunately, the poor stability of the SnO2-modified carbon nanotube anode under elevated anode potentials significantly limits its wide application. In separate study, Zhang et al. employed a coupled Ti/IrO2–RuO2 anode and a Ti cathode and also achieved conversion of ammonia into N2 within a conventional batch reactor.10 They believed that the produced active chlorine was responsible for the oxidation of ammonia. But the treatment kinetics were far from satisfactory. It is, therefore, highly desirable to develop efficient, environmentally friendly and affordable electrochemical systems for ammonia decontamination.
In recent years, Magnéli phase titanium sub-oxides have been considered as promising electrode materials for electrochemical applications, due to their increased electrical conductivity, chemical stability, strong corrosion resistance and limited production cost.11 These substoichiometric titanium oxides are commercially referred to as Ebonex®, with the generic formula of TinO2n−1 where n is an integer between 4 and 10.12 Magnéli phase Ti4O7 possesses the highest electrical conductivity (e.g., 1500 S cm−1) as well as comparable or even higher oxidative performance compared with other state-of-the-art electrode materials.13 These intriguing characteristics indicate that Ti4O7 may serve as an alternative electrode material for various environmental applications.
Herein, an electrochemical system composed of a Ti4O7 tubular anode and a Pd–Cu co-modified nickel foam (Pd–Cu/NF) cathode was rationally designed. Both are indispensable. The Ti4O7 electrode exhibits high stability under high potentials (e.g., 3.0 V vs. Ag/AgCl) and may oxidize Cl− into chlorine radicals (Cl˙). These radicals can rapidly and selectively convert ammonia into N2 and some by-products (e.g., NO3−). In addition, the macro-porous Pd–Cu/NF cathode can effectively reduce NO3− and other anodic by-products to N2.14 Various advanced characterization techniques were employed to get detailed morphological and compositional information about these electrode materials. The effects of several key parameters on ammonia conversion were also studied. To the best of our knowledge, no reports are available on the rapid and selective electrochemical transformation of ammonia by a Cl˙-mediated Ti4O7 electrode. This study provides an efficient and economical method for the treatment of ammonia wastewater.
2. Materials and methods
2.1 Chemicals and materials
Ti4O7 electrodes with an inner diameter of 20 mm were purchased from Vector Corrosion Technologies (Ontario, Canada) and were cut into typical lengths of 5 cm before use. Carbon rods with a diameter of 4.0 mm were provided by Shenbei Changfeng Carbon Rod Co., Ltd (Shanghai, China). Nickel foam (NF, 0.5 mm thickness) was purchased from Lizhiyuan Electronics Co., Ltd (China). Sulfuric acid (H2SO4, 96–98%), acetone (≥99.5%), palladium(II) chloride (PdCl2, ≥98%), copper(II) sulfate pentahydrate (CuSO4·5H2O, ≥99%), hydrochloric acid (HCl, 36–38%), sodium hydroxide (NaOH, ≥96%), nitric acid (HNO3, ≥90%), ammonium sulfate ((NH4)2SO4, ≥99%) and sodium chloride (NaCl, ≥99.5%) were purchased from Sinopharm Chemical Reagent Co., Ltd. (China). All chemicals were reagent-grade and all solutions were made from DI water with a resistivity of 18.25 MΩ cm.
2.2 Analytical methods
The surface morphology and crystallinity of the Ti4O7 electrode was examined by a field emission scanning electron microscope (FESEM, Model No.: Hitachi S-4800, Japan) and an X-ray diffractometer (XRD, Model No.: Rigaku D/max-2550/PC), respectively. X-ray photoelectron spectroscopy was obtained from a Thermo Fisher Scientific Escalab 250Xi X-ray Photoelectron Spectrometer (XPS) under high vacuum (1 × 10−9 torr). The total organic carbon of real water samples was measured with a multi N/C 2100 TOC Analyzer (Analytik Jena AG, Germany). An MS5000 spectrometer (Freiberg Instruments Inc., Germany) was employed to obtain the electron paramagnetic resonance (EPR) spectra using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as a radical trap. Ammonia concentration was measured by the Nessler reagent method15 using a TU-1810 UV-vis spectrophotometer (Yixin Instruments Equipment Co., Ltd., Shenzhen) at a wavelength of 420 nm. The amount of N2 produced was calculated from the total nitrogen (30 mg L−1 ammonia) minus the measured concentration of ammonia and nitrate. NO3− and NO2− concentrations were determined with a DionexTM ICS-5000 ion chromatography system (IC, Thermo Fisher, USA). NB concentration was quantified by a waters 3000 high performance liquid chromatography system (UHPLC, USA) coupled with a C18 column at 262 nm.
2.3 Fabrication of the Pd–Cu/NF cathode
The fabrication of a Pd–Cu/NF electrode followed a previously reported electrodeposition method.14,16 Briefly, the NF support was cleaned by sequential sonication in H2SO4, acetone, and DI water to remove the surface oxidized layer before use. The electrolyte solution consisted of 2 mM PdCl2, 4 mM CuSO4·5H2O and 0.1 M HCl. The electrodeposition experiment was conducted in a typical three-electrode system using an NF working electrode, a Pt counter electrode, and a saturated Ag/AgCl electrode reference. The deposition potential was controlled at −1.0 V vs. Ag/AgCl for 30 min using a CHI 760E electrochemical workstation.
2.4 Electrochemical experiments
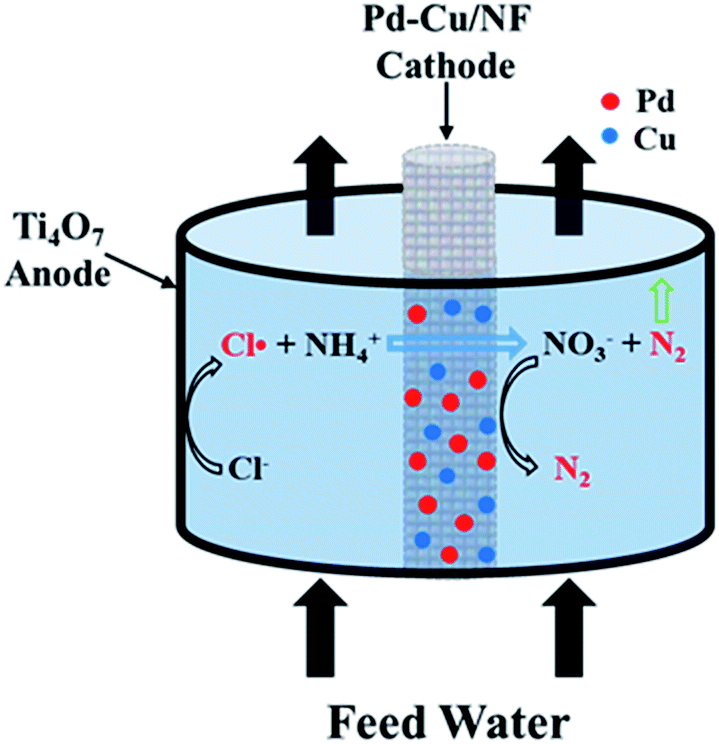
The ammonia oxidation experiment was performed in flow-by mode (Fig. S1†) using a Ti4O7 tube and a Pd–Cu/NF or a carbon rod cathode. The cathode was arranged at the center of the Ti4O7 tube and sealed with silicone rubber to avoid any leakage (Fig. 1). In a typical experiment, 30 mL of 30 mg L−1 (NH4)2SO4 solution with certain NaCl electrolyte was passed through the reactor and returned. The flow rate (2–6 mL min−1) was controlled by an ISM833C-230V peristaltic pump (Cole-Parmer, USA). Besides the flow circulation, no other mixing was provided in the reactor. The cell potential (1.5–3.2 V vs. Ag/AgCl) was provided by a CHI 760E electrochemical workstation. Cyclic voltammetry (CV) experiments were performed using a three-electrode system with a Ti4O7 working electrode, a saturated Ag/AgCl electrode reference electrode and a carbon rod counter electrode. The scan rate was set at 20 mV s−1 using 0.12 M NaCl or 0.12 M Na2SO4 electrolyte.
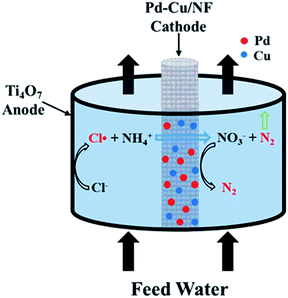 |
| | Fig. 1 Schematic of the proposed electrochemical system. | |
3. Results and discussion
The surface morphology of the Ti4O7 tubular electrode was firstly characterized by FESEM. The results showed that the internal surface of the Ti4O7 tubular electrode presented a typical interconnected porous structure (Fig. S2a†). The average pore size was determined to be 2.9 ± 0.62 μm (Fig. S2b†). The macropores provide sufficient electroactive sites and are beneficial for the mass transport of target molecules.17 XRD patterns of the tubular electrode agree well with the Magnéli phase Ti4O7 standard pattern18 (Fig. S3†). This confirmed that the main component of the anode was Ti4O7. The XPS technique clearly demonstrated the presence of characteristic peaks of O 1s, Ti 2p and C 1s, with a superficial elemental ratio of 53.4% O, 24.3% Ti and 22.3% C (Fig. S4a†). Two Ti 2p characteristic peaks centred at binding energies of 459 eV (2p3/2) and 464.6 eV (2p1/2) can be assigned to Ti4+ and those observed at 456.2 eV (2p3/2) and 461.9 eV (2p1/2) can be assigned to Ti3+ (Fig. S4b†).19,20 The cathodic Pd–Cu/NF exhibited a cross-linked grid structure with certain visible particle agglomerations on the NF surface (Fig. S5†). NF showed three characteristic XRD peaks centred at 44.5°, 51.8°, and 76.4°. A typical Pd peak was observed at 40.1° (Fig. S6†).21 However, no characteristic diffraction of Cu was observed. This may be due to the low crystallinity of the copper phase and the peaks centred at 42° and 48° of the Cu3Pd phase being masked by the strong peak of Ni at 44.5°.22 The presence of Pd and Cu elements on NF can also be verified by the XPS technique. The binding energies of the spin–orbit coupling of the metal (Pd0) were at 340.5 and 335.2 eV, respectively. Another pair of Pd2+ signals was observed at 342.2 and 337.0 eV (Fig. S7a†). The Cu 2p spectrum centred at 931.8, 934.4, and 942.7 eV, is attributed to the presence of Cu0, Cu+, and Cu2+, respectively14 (Fig. S7b†).
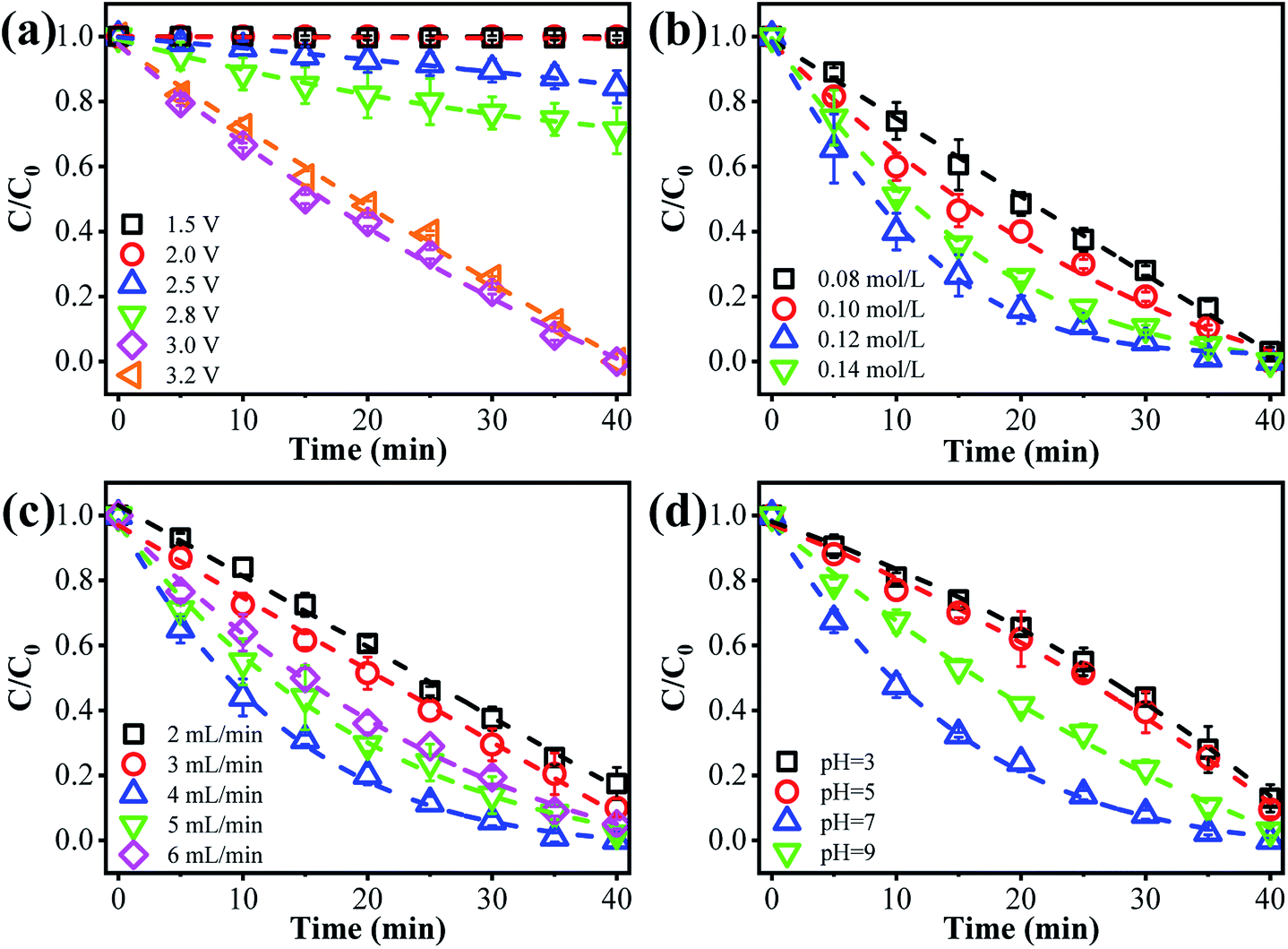
The anodic potential is an essential factor affecting the ammonia conversion performance. As can be seen from Fig. 2a, only negligible ammonia conversion was observed at an anode potential of 1.5 or 2.0 V vs. Ag/AgCl (<3%). Once the anode potential increased to 2.5 V vs. Ag/AgCl, a decreased concentration of ammonia was observed, indicating that an ammonia oxidation reaction had occurred. This may be explained by the occurrence of mono-electron chlorine oxidation (E0 = 2.4 V vs. NHE) under these conditions (eqn (1)).23 The as-produced Cl˙ may selectively react with ammonia to produce NO3− or N2. The ammonia removal performance increased with the anode potential, and complete ammonia transformation within 40 min was observed at an applied anode potential of 3.0 V vs. Ag/AgCl. This indicates that the ammonia conversion efficiency could be improved by increasing the applied voltage. Further increasing the anode potential to 3.2 V vs. Ag/AgCl did not further accelerate the ammonia oxidation kinetics, which may be due to the occurrence of other side reactions, like water splitting. This may produce gas bubbles, e.g. O2, at the anode surface, blocking the surface-active sites. To alleviate such negative consequences, we adopted an upflow configuration of the reactor. An increased flow rate (e.g., 4 mL min−1) also facilitates the rapid sweeping out of bubbles.24,25 Therefore, 3.0 V vs. Ag/AgCl was selected as optimal for subsequent investigations.
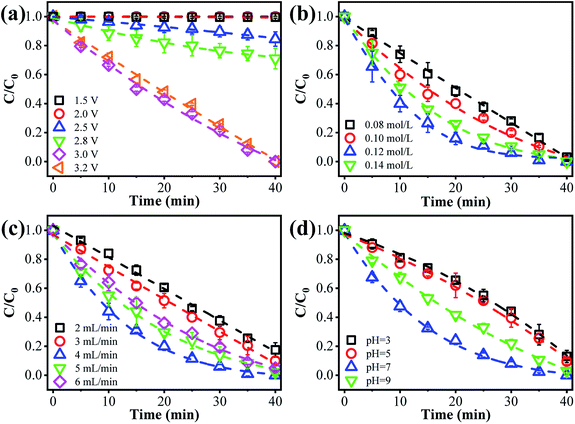 |
| | Fig. 2 The effect of different operational factors on ammonia removal: impact of (a) anode potential, (b) [Cl−], (c) flow rate, and (d) solution pH. | |
The [Cl−] was another key parameter impacting the ammonia conversion.26 As can be seen from Fig. 2b, the ammonia conversion rate increased with increasing [Cl−] from 0.08 M to 0.12 M. This may be attributed to the production of more chloride radicals with increased [Cl−]. However, further increasing the [Cl−] to 0.14 M caused a slight decline in ammonia oxidation efficiency. Excessive [Cl−] may cause unnecessary side reactions (eqn (2) and (3)) leading to the consumption of Cl˙ as well as a decreased ammonia conversion efficiency.
| |
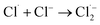 | (2) |
| |
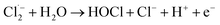 | (3) |
The flow rate is another important factor.27,28 As presented in Fig. 2c, the ammonia conversion efficiency increased with flow rate from 2 to 4 mL min−1. This can be explained by the fact that these ammonia ions have more chances to come into contact with these active sites at an increased flow rate in the recirculated filtration mode. Whereas at a flow rate of 6 mL min−1, the ammonia oxidation efficiency showed a decreasing trend. This may be ascribed to the decreased residence time within the reactor (or insufficient contact with the reaction sites) and only an incomplete ammonia oxidation was achieved. We thus chose an optimal flow rate of 4 mL min−1 for subsequent study, considering the “trade-off” effect between flow rate and residence time of ammonia.
The impact of solution pH on ammonia conversion was further studied under optimized experimental conditions. Interestingly, as shown in Fig. 2d, the best ammonia conversion performance was obtained at neutral pH. For example, over 40 min of continuous filtration reaction, complete ammonia conversion was obtained at pH 7. Whereas only 87%, 90% and 96% were achieved at pH values of 3, 5, and 9, respectively. This indicates that the designed electrochemical system has great prospects for the treatment of environmental water matrixes. The pH-dependent equilibrium between NH3 and NH4+ is responsible for the increase in ammonia conversion at neutral pH. According to a previous report,29 ammonia in aqueous solution can exist in either un-ionized form (NH3) or ionized form (NH4+). In these two forms of ammonia, the un-ionized one is more susceptible to oxidation and more abundant under neutral pH conditions. Similar results were previously reported by Zhu et al.,30 who found that NH3–N was completely removed in a neutral solution but hardly removed in an acidic solution. Conversely, Ji et al.31 reported a highly selective method for transforming ammonia to N2, based on a novel solar-driven photoelectrochemical coupled chlorine radical reaction system, in which the ammonia removal efficiency was enhanced when the solution acidity increased from pH 6 to 2.
The impact of different operation systems on the conversion of ammonia was further investigated. As presented in Fig. S8,† compared with a conventional batch reactor, the present continuous-flow system demonstrated obviously enhanced kinetics (>99.9% vs. 62.3%). This could be due to the convection-enhanced mass transport within the proposed electrochemical system, while diffusion alone dominates the mass transport within the batch system. Moreover, the as-produced gas bubbles may easily block the surface-active sites within the batch reactor, further damaging the overall performance.
The Cl˙ can be produced by direct oxidation by Ti4O7 or through the instantaneous adduct of Cl− with OH˙.32 While other side reactions may also consume Cl˙, such as Cl˙ reacting with Cl− to form 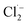 and further reacting to produce HOCl/OCl− (eqn (2)–(4)). It is known that OH˙, Cl˙ and HClO are strong oxidants that can react with NH4+.16 In particular, the Cl˙ could react selectively with NH4+ (kCl˙ = 5 × 1013 M−1 s−1)33 with a much higher reaction rate compared with that of OH˙ (kOH˙ = 3 × 108 M−1 s−1).34 These Cl˙ can selectively convert NH4+ into N2, NO3− or NO2− (eqn (5)–(7)). However, the contribution of ammonia conversion from other reactive chlorine species cannot be eliminated, which will be discussed later.
and further reacting to produce HOCl/OCl− (eqn (2)–(4)). It is known that OH˙, Cl˙ and HClO are strong oxidants that can react with NH4+.16 In particular, the Cl˙ could react selectively with NH4+ (kCl˙ = 5 × 1013 M−1 s−1)33 with a much higher reaction rate compared with that of OH˙ (kOH˙ = 3 × 108 M−1 s−1).34 These Cl˙ can selectively convert NH4+ into N2, NO3− or NO2− (eqn (5)–(7)). However, the contribution of ammonia conversion from other reactive chlorine species cannot be eliminated, which will be discussed later.
| | |
2NH4+ + 6Cl˙ → N2 + 6Cl− + 8H+
| (5) |
| | |
NH4+ + 8Cl˙ + 3H2O → NO3− + 8Cl− + 10H+
| (6) |
| | |
NH4+ + 6Cl˙ + 2H2O → NO2− + 6Cl− + 8H+
| (7) |
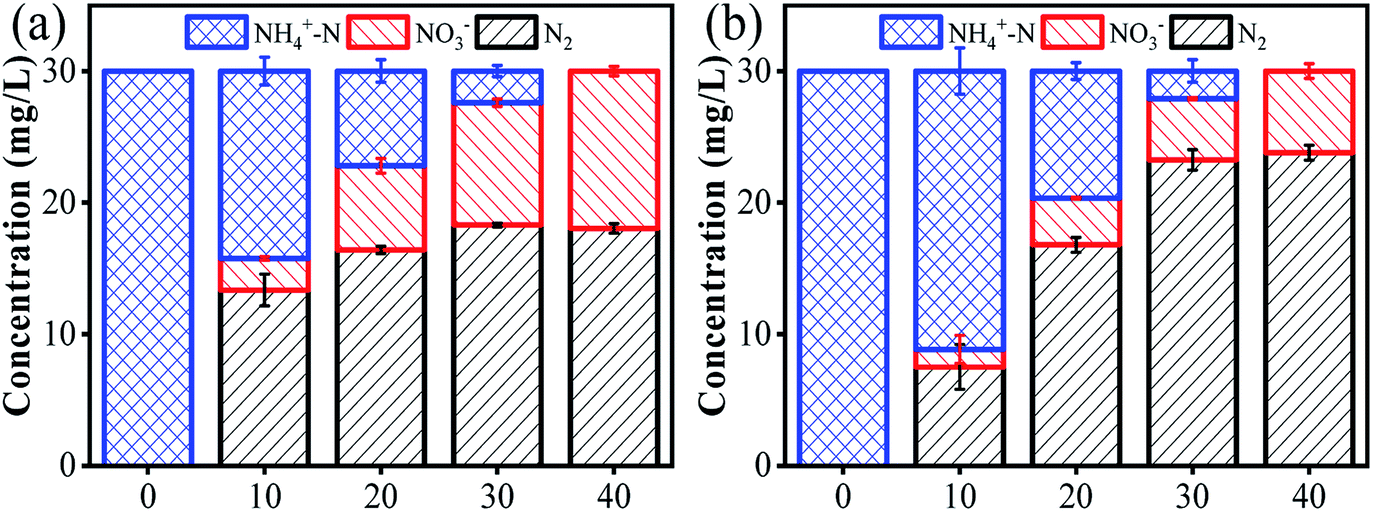
The changes in ammonia conversion products over time were investigated under an optimized anode potential of 3.0 V vs. Ag/AgCl, [Cl−] of 0.12 M, flow rate of 4 mL min−1 and pH of 7. As shown in Fig. 3a, NO3− and N2 content increased gradually with time to 12 mg L−1 and 18 mg L−1, respectively, over 40 min when using a carbon rod cathode. NO2− was not detected during this process, possibly due to the strong oxidative ability of Ti4O7. However, the ideal product of the present system is N2, rather than NO3−. Since NO3− is negatively charged, it has a strong electrostatic repulsion from the cathode surface. Moreover, these NO3− ions can also be converted back to NH4+ (eqn (8)).35
| | |
NO3− + 7H2O + 8e− → NH4+ + 10OH−
| (8) |
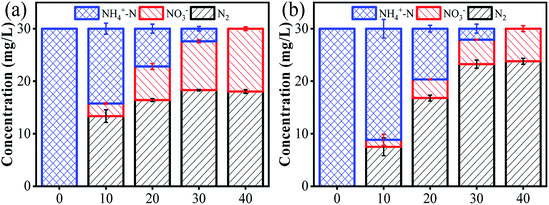 |
| | Fig. 3 The conversion of ammonia in 40 min by employing different cathode materials: (a) carbon rod, and (b) Pd–Cu/NF. Reaction conditions: anode potential of 3.0 V vs. Ag/AgCl, [Cl−] of 0.12 M, flow rate of 4 mL min−1, and pH of 7. | |
To address this issue, a Pd–Cu/NF cathode was further employed to replace the carbon rod cathode.16 NF, with excellent electrical conductivity and a three-dimensional network, is considered to be a promising cathode material.14 Moreover, Pd and Cu have been proven to be effective promoters for NO3− reduction reaction to N2.36 In a recent study, a Pd–Cu/NF cathode demonstrated an evidently enhanced N2 yield compared with a Pd/NF or a Cu/NF cathode in an SnO2-CNT based electrochemical system.14 Importantly, the as-fabricated Pd–Cu/NF surface is positively charged. Hence, the negatively-charged NO3− can be easily absorbed onto the cathode surface via electrostatic attraction. Therefore, another set of experiments was performed using the Pd–Cu/NF cathode coupled with the Ti4O7 anode. The results showed that ammonia was gradually converted to NO3− and N2 over time. Compared with the carbon rod cathode, the NO3− removal rate increased by 50% over 40 min of reaction when using the Pd–Cu/NF cathode (Fig. 3b). In addition, Pd and Cu coatings are also essential. The results indicate that similar ammonia removal rates can be obtained with Pd/NF or Cu/NF cathodes. However, the presence of Pd or Cu alone only partially contributed to the conversion of NO3− to N2 (Fig. S9†). This confirmed the capacitive characteristics of the Pd–Cu/NF electrode, which can rapidly adsorb NO3− onto the cathode via electrostatic attraction. NO3− can be reduced to NO2− after adsorption on the Cu site, and then further reduced to N2 on the Pd site (eqn (9)–(12)).
| | |
NO3− + Cu ↔ Cu − NO3ads−
| (9) |
| | |
Cu − NO3ads− + H2O + 2e− → NO2− + Cu + 2OH−
| (10) |
| | |
NO2− + Pd ↔ Pd − NO2ads−
| (11) |
| | |
2Pd − NO2ads− + 4H2O + 6e− → N2 + 2Pd + 8OH−
| (12) |
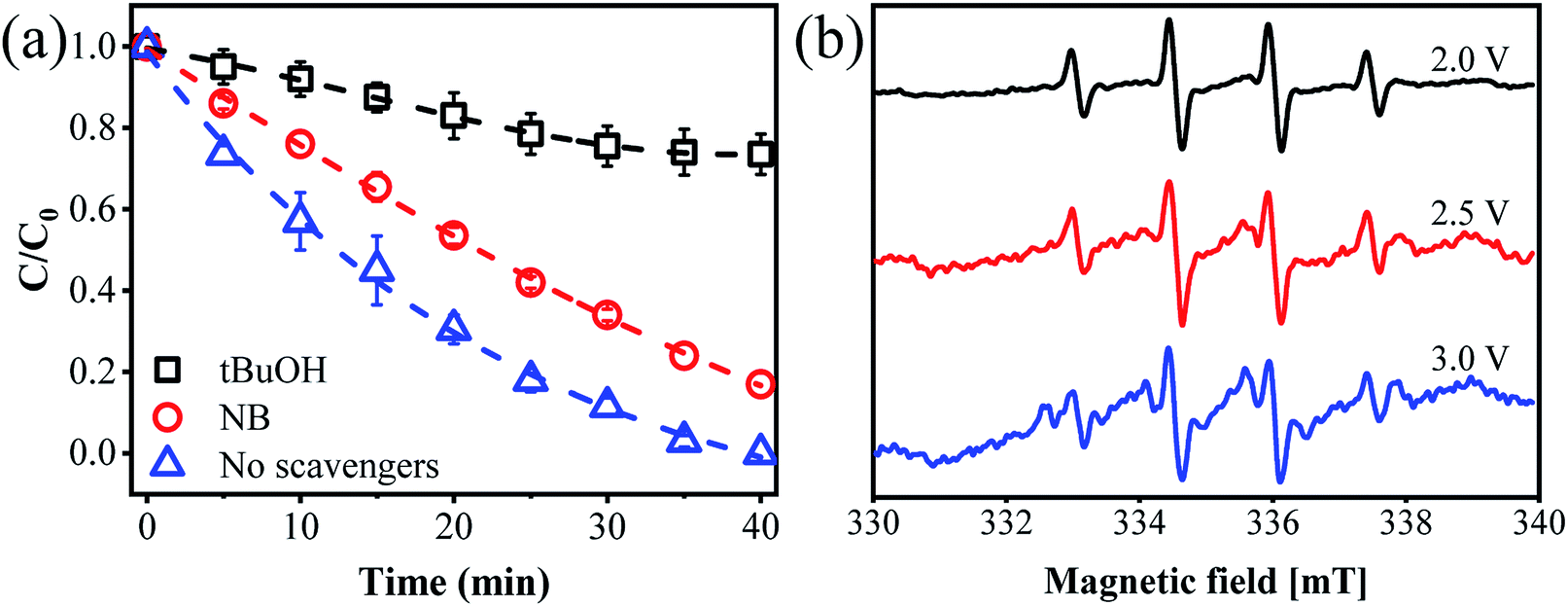
To distinguish the dominant active species on ammonia conversion, we spiked a few scavengers into the ammonia solution before flowing it into the reactor. We chose nitrobenzene (NB) as an OH˙ quenching agent with a k of 3.9 × 109 M−1 s−1 and tert-butanol (tBuOH) as a joint quenching agent of Cl˙ and OH˙ with k values of 1.9 × 109 and 6 × 108 M−1 s−1, respectively.37 As shown in Fig. 4a, complete ammonia conversion was observed within 40 min of reaction in the absence of any scavengers. However, the ammonia conversion decreased by 74% in the presence of tBuOH within the same period. This suggests that the ammonia removal process indeed involved radicals and that HOCl plays only a minor role. To quantitatively determine the amount of [OH˙], we used an indirect method by adding 1 mM NB into the reaction medium due to their high affinity. According to eqn (13), [OH˙] is determined to be 1.06 × 10−11 M with a KNB of 0.04133 min−1 (Fig. S10†). While the ammonia conversion decreased by only 17% when NB was added, proving that Cl˙ may play a dominant role in the process of ammonia oxidation. It is of note that other radicals such as NO3˙ may also exist in the solution and contribute to ammonia conversion. However, the reaction rate constant of NO3˙ with tBuOH (k = 5.7 × 104 M−1 s−1)38 was 5 orders of magnitude lower than that of Cl˙. It is thus reasonable to believe that the contribution from NO3˙ is negligible.
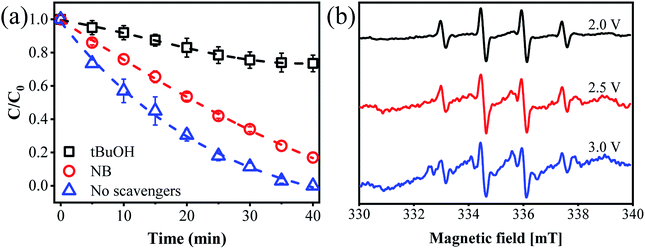 |
| | Fig. 4 (a) Effect of different scavengers on ammonia removal. (b) EPR spectra with DMPO observed from the flow-by experiment at different applied anode potentials. | |
The production of Cl˙ and OH˙ during electrochemical filtration was further determined by the EPR technique. As presented in Fig. 4b, the typical 4-peak spectrum of OH˙ with an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 was detected with DMPO as a spin trapping agent.39 However, an alternative 11-peak spectrum was detected at an increased anode potential of 2.5 or 3.0 V vs. Ag/AgCl. This 11-peak spectrum can be divided into a 4-peak (DMPO-OH˙) signal and another 7-peak signal. According to previous studies, these 7-peak signals may be ascribed to DMPO-Cl, which are enhanced with an applied potential up to 3.0 V vs. Ag/AgCl40 (Fig. S11†). Similar results were previously reported by Li et al.14 This confirmed the formation of OH˙ and Cl˙ within the present electrochemical flow-by system. In addition, we performed another control experiment using Na2SO4 to replace NaCl. As expected, only negligible ammonia conversion (<5%) was observed, regardless of the anode potentials (Fig. S12†), further indicating the essential role of Cl−. The CV spectrum in 0.12 M of NaCl and Na2SO4 solution also indicates that an oxidation reaction occurred at an anode potential of 2.92 V vs. Ag/AgCl using an NaCl electrolyte, as evidenced by the obvious oxidative peak. This oxidative peak could be due to the ammonia oxidation reaction in the presence of Cl˙, while no visible redox peak was observed when using the Na2SO4 electrolyte (Fig. S13†). This indirectly verifies the essential role of Cl˙ for ammonia transformation in this reaction system.
:2:2:1 was detected with DMPO as a spin trapping agent.39 However, an alternative 11-peak spectrum was detected at an increased anode potential of 2.5 or 3.0 V vs. Ag/AgCl. This 11-peak spectrum can be divided into a 4-peak (DMPO-OH˙) signal and another 7-peak signal. According to previous studies, these 7-peak signals may be ascribed to DMPO-Cl, which are enhanced with an applied potential up to 3.0 V vs. Ag/AgCl40 (Fig. S11†). Similar results were previously reported by Li et al.14 This confirmed the formation of OH˙ and Cl˙ within the present electrochemical flow-by system. In addition, we performed another control experiment using Na2SO4 to replace NaCl. As expected, only negligible ammonia conversion (<5%) was observed, regardless of the anode potentials (Fig. S12†), further indicating the essential role of Cl−. The CV spectrum in 0.12 M of NaCl and Na2SO4 solution also indicates that an oxidation reaction occurred at an anode potential of 2.92 V vs. Ag/AgCl using an NaCl electrolyte, as evidenced by the obvious oxidative peak. This oxidative peak could be due to the ammonia oxidation reaction in the presence of Cl˙, while no visible redox peak was observed when using the Na2SO4 electrolyte (Fig. S13†). This indirectly verifies the essential role of Cl˙ for ammonia transformation in this reaction system.
To examine the potential applications of the proposed system, ammonia conversion with different background solutions was investigated. To do this, 30 mg L−1 of (NH4)2SO4 were added to local tap and lake water samples before flowing into the electrochemical system. A similar conversion trend for ammonia was observed in all cases (Fig. S14†). Complete ammonia conversion could still be obtained in tap water with a background total organic carbon (TOC) of 1.8 mg L−1. In addition, the removal of ammonia was about 97% when challenged with lake water with a background TOC of 93.2 mg L−1. The effect of selected common anions (e.g., carbonate, phosphate and silicate) on ammonia conversion was further investigated under optimal operating conditions (Fig. S15†). The results showed that such inhibition was negligible in the presence of carbonate at varying concentrations. Silicate and phosphate posed a slight inhibitory effect on ammonia conversion. When the concentration increased from 5 to 20 mM, the ammonia conversion efficiency decreased from 100% to 86.5% and 82.3%, respectively. This indicates that the complex composition of natural water and the presence of some competitive ions produces only negligible inhibition of ammonia conversion. This result can be explained by the fact that the Cl˙ react selectively with other electron-rich groups (e.g., NH4+). The stability of Ti4O7 was further evaluated by performing four consecutive cycles of ammonia removal experiments in DI-H2O conditions. The results showed an average 98.7 ± 2.2% ammonia removal efficiency, indicating the excellent anodic stability of the Ti4O7 electrode (Fig. S16†). These positive outcomes indicate that the proposed electrochemical system is a potential technology for removing ammonia from various water environments. It should be noted that the toxicity of Cl˙ is beyond the scope of this work. We would like to leave this key question for further exploration.
4. Conclusions
In summary, a continuous-flow electrochemical system was developed towards effective and exhaustive ammonia removal. The proposed electrochemical system employs a highly stable tubular Magnéli phase Ti4O7 anode coupled with a Pd–Cu co-modified NF cathode. The as-produced Cl˙ can react selectively with NH4+ to produce N2 as it flows through the electrochemical system. This flow-by electrochemical system also demonstrated enhanced oxidative kinetics compared with a conventional batch system.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Natural Science Foundation of Shanghai, China (No. 18ZR1401000), the Fundamental Research Funds for the Central Universities (No. 2232019G-11), and the Shanghai Pujiang Program (No. 18PJ1400400).
References
- S. Y. Weon, S. I. Lee and B. Koopman, Environ. Technol., 2004, 25, 1211–1219 CrossRef CAS PubMed.
- L. Chmielarz and M. Jablonska, RSC Adv., 2015, 5, 43408–43431 RSC.
- Z. J. Li, X. P. Zhang, H. F. Dong, X. C. Zhang, H. S. Gao, S. J. Zhang, J. W. Li and C. M. Wang, RSC Adv., 2015, 5, 81362–81370 RSC.
- W. J. Tang, Y. Zhang, J. Bai, J. H. Li, J. C. Wang, L. S. Li, T. S. Zhou, S. Chen, M. Rahim and B. X. Zhou, Sep. Purif. Technol., 2020, 234, 9 CrossRef.
- J. Lee, H. Park and W. Choi, Environ. Sci. Technol., 2002, 36, 5462–5468 CrossRef CAS PubMed.
- X. R. Zhang, W. G. Li, E. R. Blatchley, X. J. Wang and P. F. Ren, Water Res., 2015, 68, 804–811 CrossRef CAS PubMed.
- Y. Y. Xiang, J. Y. Fang and C. Shang, Water Res., 2016, 90, 301–308 CrossRef CAS PubMed.
- Y. Yang, J. J. Pignatello, J. Ma and W. A. Mitch, Environ. Sci. Technol., 2014, 48, 2344–2351 CrossRef CAS PubMed.
- F. Li, X. Peng, Y. B. Liu, J. C. Mei, L. W. Sun, C. S. Shen, C. Y. Ma, M. H. Huang, Z. W. Wang and W. G. Sand, Chemosphere, 2019, 229, 383–391 CrossRef CAS PubMed.
- C. Y. Zhang, D. He, J. X. Ma and T. D. Waite, Water Res., 2018, 145, 220–230 CrossRef CAS PubMed.
- F. C. Walsh and R. G. A. Wills, Electrochim. Acta, 2010, 55, 6342–6351 CrossRef CAS.
- S. Nayak and B. P. Chaplin, Electrochim. Acta, 2018, 263, 299–310 CrossRef CAS.
- C. Trellu, C. Coetsier, J. C. Rouch, R. Esmilaire, M. Cretin and C. Causserand, Water Res., 2017, 131, 310–319 CrossRef PubMed.
- F. Li, X. Peng, Y. B. Liu, J. Mei, L. Sun, C. Shen, C. Ma, M. Huang, Z. Wang and W. Sand, Chemosphere, 2019, 229, 383–391 CrossRef CAS PubMed.
- K. Fang, H. Gong, W. Y. He, F. Peng, C. H. He and K. J. Wang, Chem. Eng. J., 2018, 348, 301–309 CrossRef CAS.
- Y. Zhang, J. Li, J. Bai, Z. Shen, L. Li, L. Xia, S. Chen and B. Zhou, Environ. Sci. Technol., 2018, 52, 1413–1420 CrossRef CAS PubMed.
- L. J. J. Janssen and L. Koene, Chem. Eng. J., 2002, 85, 137–146 CrossRef CAS.
- L. Guo, Y. Jing and B. P. Chaplin, Environ. Sci. Technol., 2016, 50, 1428–1436 CrossRef CAS PubMed.
- X. Y. Tao, J. G. Wang, Z. G. Ying, Q. Cai, G. Zheng, Y. Gan, H. Huang, Y. Xia, C. Liang, W. Zhang and Y. Cui, Nano Lett., 2014, 14, 5288–5294 CrossRef CAS PubMed.
- X. X. Li, A. L. Zhu, W. Qu, H. Wang, R. Hui, L. Zhang and J. Zhang, Electrochim. Acta, 2010, 55, 5891–5898 CrossRef CAS.
- Z. Q. Zhang, Y. P. Xu, W. X. Shi, W. Wang, R. J. Zhang, X. Bao, B. Zhang, L. Li and F. Y. Cui, Chem. Eng. J., 2016, 290, 201–208 CrossRef CAS.
- Y. Yoshinaga, T. Akita, I. Mikami and T. Okuhara, J. Catal., 2002, 207, 37–45 CrossRef CAS.
- C. D. Vecitis, M. H. Schnoor, M. S. Rahaman, J. D. Schiffman and M. Elimelech, Environ. Sci. Technol., 2011, 45, 3672–3679 CrossRef CAS.
- P. Gayen, C. Chen, J. T. Abiade and B. P. Chaplin, Environ. Sci. Technol., 2018, 52, 12675–12684 CrossRef CAS PubMed.
- T. X. H. Le, H. Haflich, A. D. Shah and B. P. Chaplin, Environ. Sci. Technol. Lett., 2019, 6, 504–510 CrossRef CAS.
- J. Ding, Q. L. Zhao, J. Q. Jiang, L. Wei, K. Wang, Y. Zhang, W. Hou and H. Yu, Environ. Sci. Pollut. Res., 2017, 24, 5152–5158 CrossRef CAS PubMed.
- K. W. Kim, Y. J. Kim, I. T. Kim, G. I. Park and E. H. Lee, Electrochim. Acta, 2005, 50, 4356–4364 CrossRef CAS.
- Y. B. Liu, P. Wu, F. Q. Liu, F. Li, X. An, J. Liu, Z. Wang, C. Shen and W. Sand, Environ. Sci. Technol., 2019, 53, 1527–1535 CrossRef CAS PubMed.
- S. H. Lin and C. L. Wu, Water Res., 1996, 30, 715–721 CrossRef CAS.
- X. Zhu, J. Ni and P. Lai, Water Res., 2009, 43, 4347–4355 CrossRef CAS PubMed.
- Y. Z. Ji, J. Bai, J. H. Li, T. Luo, L. Qiao, Q. Zeng and B. Zhou, Water Res., 2017, 125, 512–519 CrossRef CAS PubMed.
- B. P. Chaplin, Acc. Chem. Res., 2019, 52, 596–604 CrossRef CAS PubMed.
- Y. D. Gao, I. M. Alecu, P. C. Hsieh, B. P. Morgan, P. Marshall and L. N. Krasnoperov, J. Phys. Chem. A, 2006, 110, 6844–6850 CrossRef CAS.
- L. Jing, B. Chen, D. Y. Wen, J. S. Zheng and B. Y. Zhang, Environ. Sci. Pollut. Res., 2018, 25, 2691–2701 CrossRef CAS PubMed.
- X. R. Zhang, W. G. Li, E. R. Blatchley III, X. Wang and P. Ren, Water Res., 2015, 68, 804–811 CrossRef CAS PubMed.
- Y. Wang, J. H. Qu and H. J. Liu, Chin. Chem. Lett., 2006, 17, 61–64 CAS.
- M. J. Watts and K. G. Linden, Water Res., 2007, 41, 2871–2878 CrossRef CAS PubMed.
- S. P. Mezyk, T. D. Cullen, K. A. Rickman and B. J. Mincher, Int. J. Chem. Kinet., 2017, 49, 635–642 CrossRef CAS.
- X. R. Wang, Y. Li, L. P. Tang, W. Gan, W. Zhou, Y. F. Zhao and D. S. Bai, Chin. Chem. Lett., 2017, 28, 394–399 CrossRef CAS.
- T. Li, Y. Jiang, X. Q. An, H. Liu, C. Hu and J. Qu, Water Res., 2016, 102, 421–427 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07470h |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Jiancheng Meia,
Chensi Shenab,
Manhong Huangab,
Ming Yangc,
Zhiwei Wangbd,
Wolfgang Sandae and
Fang Li*ab
*ab,
Jiancheng Meia,
Chensi Shenab,
Manhong Huangab,
Ming Yangc,
Zhiwei Wangbd,
Wolfgang Sandae and
Fang Li*ab
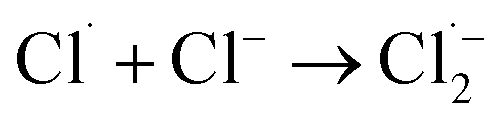

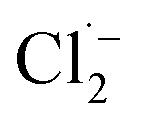 and further reacting to produce HOCl/OCl− (eqn (2)–(4)). It is known that OH˙, Cl˙ and HClO are strong oxidants that can react with NH4+.16 In particular, the Cl˙ could react selectively with NH4+ (kCl˙ = 5 × 1013 M−1 s−1)33 with a much higher reaction rate compared with that of OH˙ (kOH˙ = 3 × 108 M−1 s−1).34 These Cl˙ can selectively convert NH4+ into N2, NO3− or NO2− (eqn (5)–(7)). However, the contribution of ammonia conversion from other reactive chlorine species cannot be eliminated, which will be discussed later.
and further reacting to produce HOCl/OCl− (eqn (2)–(4)). It is known that OH˙, Cl˙ and HClO are strong oxidants that can react with NH4+.16 In particular, the Cl˙ could react selectively with NH4+ (kCl˙ = 5 × 1013 M−1 s−1)33 with a much higher reaction rate compared with that of OH˙ (kOH˙ = 3 × 108 M−1 s−1).34 These Cl˙ can selectively convert NH4+ into N2, NO3− or NO2− (eqn (5)–(7)). However, the contribution of ammonia conversion from other reactive chlorine species cannot be eliminated, which will be discussed later.