DOI:
10.1039/C9RA07380A
(Paper)
RSC Adv., 2020,
10, 376-386
Functionalization of hydrophobic surfaces with antimicrobial peptides immobilized on a bio-interfactant layer†
Received
13th September 2019
, Accepted 16th December 2019
First published on 2nd January 2020
Abstract
The design of functionalized polymer surfaces using bioactive compounds has grown rapidly over the past decade within many industries including biomedical, textile, microelectronics, bioprocessing and food packaging sectors. Polymer surfaces such as polystyrene (PS) must be treated using surface activation processes prior to the attachment of bioactive compounds. In this study, a new peptide immobilization strategy onto hydrocarbonaceus polymer surfaces is presented. A bio-interfactant layer made up of a tailored combination of laccase from trametes versicolor enzyme and maltodextrin is applied to immobilize peptides. Using this strategy, immobilization of the bio-inspired peptide KLWWMIRRWG-bromophenylalanine-3,4-dihydroxyphenylalanine-G and KLWWMIRRWG-bromophenylalanine-G on polystyrene (PS) was achieved. The interacting laccase layers allows to immobilize antimicrobial peptides avoiding the chemical modification of the peptide with a spacer and providing some freedom that facilitates different orientations. These are not strongly dominated by the substrate as it is the case on hydrophobic surfaces; maintaining the antimicrobial activity. Films exhibited depletion efficiency with respect to the growth of Escherichia coli bacteria and did not show cytotoxicity for fibroblast L929. This environmentally friendly antimicrobial surface treatment is both simple and fast, and employs aqueous solutions. Furthermore, the method can be extended to three-dimensional scaffolds as well as rough and patterned substrates.
Introduction
Biomedical devices have become an essential component of the human healthcare system. Due to their low cost, widespread availability, and versatile mechanical properties, industrial polymers such as polystyrene (PS), polypropylene and polyethylene are often used in the manufacture of medical products.1 However, these materials do not present significant antibacterial properties. In addition, their surfaces must be treated using surface activation processes prior to the attachment of bioactive compounds.2 Despite considerable research and development into biomedical devices and implants significant risk of infections related to biomedical devices and implants persists. The development of an implant coating having broad-spectrum antimicrobial activity is challenging due to the fast rate at which bacteria evolve resistance to antibiotics. An important goal within the biomaterials field is the development of surface functionalization methods that inhibit bacterial surface attachment and growth which can postpone or prevent infections and device failure.3 Past work has made progressing antibacterial surface design utilizing strategies such as surfaces functionalized with antimicrobial agents, low-adhesion surfaces (e.g. superhydrophobic interfaces), and polymer matrices that release antibiotics.4,5 A promising approach to overcome this challenge is to utilize naturally occurring antimicrobial peptides or synthetic analogues against bacterial infections. One strategy that has shown significant targeting potential is the use of antimicrobial peptides (AMPs), which exhibits a number of advantageous properties: effectiveness against a broad range of bacteria including antibiotic-resistant strains, low-toxicity to mammalian cells, small molecular size and high stability.6,7 Furthermore, advantages of immobilized peptides comprise long-term stability. AMPs are produced by the immune systems of animals, insects and plants and are secreted when infections occur.8–10 Consisting of approximately 12 to 100 amino acids, AMPs are amphiphilic molecules having a high content of cationic and hydrophobic amino acids providing side chains responsible for the antibacterial activity.11 Arising from electrostatic interactions between positively charged amino acids and the negatively charged bacterial cell membrane, AMPs work by compromising membrane integrity and transport permeability.12–15 Physical adsorption and covalent bonding strategies have been studied for AMPs immobilization on surfaces. Layer-by-layer deposition is a common physical immobilization technique applied to hydrophilic materials such as metal oxides, mainly due to electrostatic interactions16,17 or the adding of a specific surface binding recognition sequence to the peptide.18–20 This method may be applied on hydrophobic polymer surfaces after applying a process to increase the concentration of functional groups such as hydroxyl and carbonyl groups, and next covalent attachment using click chemistry.21 Another viable method is solid phase peptide synthesis on polymer resins in which the protected amino acids are incorporated by assembling the peptide sequence from its C- to its N-terminus; however, these methods are time consuming and costly.21–23 Although peptides with hydrophobic amino acids may adsorb strongly enough on hydrophobic surfaces to form a stable film under controlled conditions. The peptides when are amphiphilic likely adsorb with their hydrophobic segments facing hydrophobic surfaces and the restricted orientation of the peptide might cause a reduction in its antimicrobial properties.24 For instance, Hilpert et al. reported and improved peptide antimicrobial properties when adsorbed at an orientation such that their hydrophobic segments are away from the interface; facing the bacteria membrane.22 Formation of stable laccase/maltodextrin layers on highly oriented pyrolytic graphite (HOPG) was reported in Corrales et al.25 Laccase molecules adsorb irreversible on this hydrophobic surface in short contact times; forming layers that can serve as binding sites for the interaction and attachment of other biomolecules. In this study, we present a new surface immobilization strategy for AMPs with or without −3,4-dihydroxyphenylalanine (DOPA) in their sequence which profits from using laccase as a bio-interfactant.26 The antimicrobial peptide used for immobilization is based on the peptide Tet-124 sequence KLWWMIRRW, previously synthesized by Hilpert et al.22 This peptide showed partial inhibition of Pseudomonas aeruginosa. Applying the adhesion mechanism of mussel foot proteins, we have modified the AMP sequence adding glycine-(4-bromophenylalanine)-(3,4-dihydroxyphenylalanine)-glycine to the original sequence reported, thus obtaining KLWWMIRRW-G-BrPh-DOPA-G. The amino acid DOPA was added in the sequence to improve the peptide adhesion, comparing the stability of both peptides on the surface after long rinsing times, and to favorably be oriented at the interface of these films.27 We have changed the peptide C-terminal such that two glycines flank the DOPA in order to minimize steric hindrance, allowing for more flexibility,28 and to prevent intramolecular crosslinking to neighbouring amino groups due to the potential laccase catalyzed oxidation of the DOPA. 4-Bromophenylalanine (BrPh) was added to the sequence in order to quantitatively determine the peptide adsorption fraction and to differentiate the peptide and the enzyme adsorbates within the layers. The formation of thin films in situ was characterized using quartz crystal microbalance with dissipation (QCM-D), and the resulting adsorbates were studied using X-ray photoelectron spectroscopy (XPS), atomic force microscopy (AFM), and water contact angle. The peptide laccase catalyzed oxidation on the surface was studied carrying on a selective derivatization reaction for DOPA containing peptides. Antimicrobial properties against Escherichia coli and, moreover, the fibroblast L-929 biocompatibility of the bio-films were tested.
Materials and methods
Commercial polystyrene (PS) and highly pyrolytic oriented graphite (HOPG, quality ZYH from NT-MDT, Russia) freshly cleaved in air were used as substrates. Acetic acid, sodium hydroxide (NaOH), citric acid, sodium acetate and sodium chloride (NaCl) were used as grade reagents (Sigma-Aldrich, Germany). The laccase mixture suspension (LMS) was prepared by mixing laccase-maltodextrin (ASA Spezialenzyme GmbH, Wolfenbüttel, Germany) at 0.1 mg mL−1 in 0.2 M sodium acetate buffer; at pH value of 4.5. Substrates were coated using 100 μL of solution per 1 cm2 of substrate area. For ex situ investigations, LMS was placed directly onto PS or HOPG substrates, and after different times, the aqueous formulation was removed. Samples were then rinsed using water and dried using pressurized air. Stability tests in buffers with different pH values were performed submerging the substrate in the buffer for the corresponding time and then rinsing with water. Modified substrates were then in contact with the 0.1 mg mL−1 peptide solution (50 μL cm−2). Finally, the substrates were rinsed and dried. Samples were stored under aseptic conditions at room temperature.
Solid phase peptide synthesis
Tet-124, Tet-124-G-BrPh-DOPA-G, Tet-124-G-BrPh and HHGHSPTSPQVR were synthesized using a 433A solid phase peptide synthesizer (Applied Biosystems, Farmington, CT, USA) that utilizes the FastMocTM-protocol. Peptide sequences and short names are shown in Table 1. DOPA was introduced into the sequences as (2S)-3-(2,2-dimethyl-1,3-benzodioxol-5-yl)-2-(9H-fluoren-9-ylmethoxycarbonylamino) propanoic acid (Fmoc-DOPA(acetonide)-OH, Merck Nova Biochem, Hohenbrunn, Germany) during the synthesis. Peptides were synthesized at a 0.1 mmol scale using either Fmoc-glycine or Fmoc-tryptophan (Boc) pre-loaded with tritylchloride P resin (TCP). Amino acids beside DOPA were obtained from IRIS Biotech (Marktredwitz, Germany) and Fmoc-DOPA (acetonide)-OH from Merck Nova Biochem. TCP resins with a pre-loading at ≈0.55 mmol g−1 were used (Intavis AG, Heidelberg, Germany). In Fmoc (9-fluorenylmethoxycarbonyl) peptide solid phase synthesis, amino acids bound to a resin were sequentially assembled. First, a resin having the first amino acid sequence was pre-loaded. Amino acids were protected at the N-terminus by the Fmoc group and the carboxylic acid terminus is activated by 1-hydroxybenzotriazol (HOBt) and 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU). 20% Piperidine solution was used to cleave Fmoc groups following each coupling step. Peptide side chain removal was performed using a solution composed of 3600 μL trifluoroacetic acid, 200 μL de-ionized (DI) water and 200 μL triisopropylsilane. After synthesis, resins were treated with the cleavage solution for two hours. After filtering cleavage solutions using a syringe filter (PTFE, 0.45 μm), peptides were collected by precipitation in ice-cold 60 mL tert-butyl methyl ether, centrifugation and solvent transfer to DI water. Following lyophilization, the obtained peptides appeared as white powders.
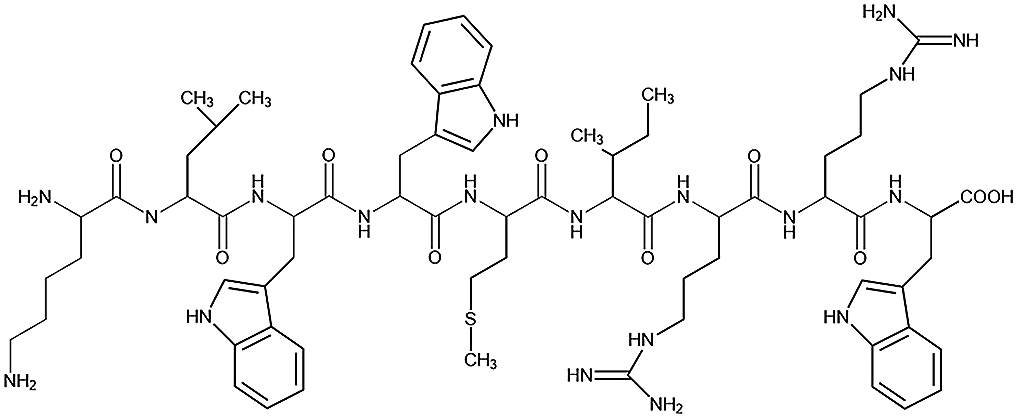
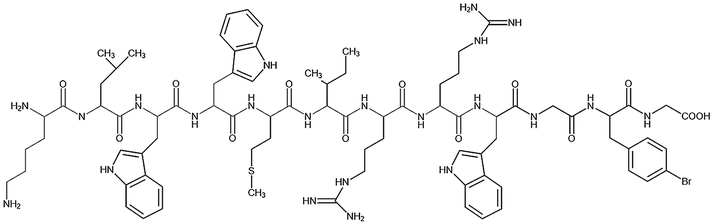
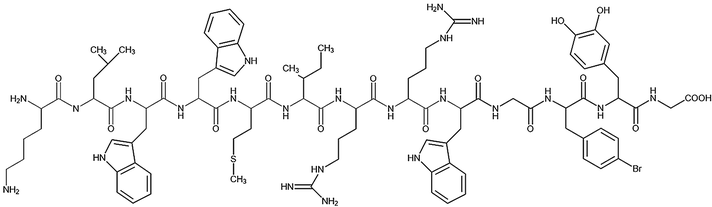
Table 1 Peptide sequences and short names
| Name |
Sequence |
| Tet-124 |
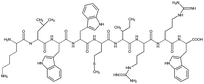 |
| Tet-124-G-BrPh-G |
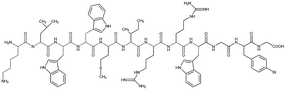 |
| Tet-124-G-BrPh-DOPA-G |
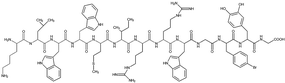 |
Peptides were characterized using a BioCad 700E HPLC system (Applied Biosystems, USA) and Varian HPLC columns (Polaris 5u C18-A, 250 × 46 mm). Peptides were further characterized using a MALDI-ToF-MS Voyager DE-Pro (Applied Biosystems, Foster City, USA) using α-cyano-4-hydroxycinnamic acid as matrix. Purity for all samples was determined to be >95%. Samples were stored at −20 °C.
QCM-D
To evaluate peptide adsorption onto PS, a QCM-D system (Q-Sense Analyzer, Biolin Scientific, Sweden) and PS-coated QCM-D sensors (QSX 310, Q-Sense, Sweden) were used. The crystal sensors were 14 mm diameter, 10 nm layer thickness, and <3 nm surface roughness. The adsorption from the laccase formulation onto the crystal surface contact was monitored for the overtones (n = 1, 3, 5, 7,9,11 and 13) at 25 °C. To remove surface contaminants before use, crystals were placed in a UV-ozone chamber for 15 min. For the measurement, the testing solutions were pumped through the flow cells at a constant flow rate of 100 μL min−1 in the following order: buffer solution for 30 min (baseline), laccase maltodextrin suspension, peptide solution and buffer solution (washing step). Resonance frequency changes Δf determined by adsorbed mass were recorded and analyzed using commercial software (QSoft, supplier Q-Sense, Sweden). The Sauerbrey model was used to calculate the mass, due to the small changes on the dissipation coefficient (ΔD is close to zero). Measurements were repeated at least three times and a representative data is shown.
Contact angle measurements
Apparent water contact angles were measured using a goniometer (OCA15 Plus, Data Physics Instruments, Germany) and applying the sessile drop technique. For each measurement, 10 μL drops were formed using HPLC-grade water (Acros Organics, Germany) and the subsequently reported contact angles were taken as an average of at least three measurements.
XPS
XPS spectra were taken using a Kratos Axis Ultra system (Kratos Analytical Ltd, UK) with a monochromatic Al Kα source operated at a base pressure of 4 × 10−8 Pa, low-energy electrons (<5 eV) for sample neutralization, and a 0° electron take-off angle. One bulk peptide sample was characterized with a Thermo Scientific™ K-Alpha™+ (Thermo Fisher Scientific Inc., US) instrument with attached argon glovebox for the handling of air sensitive samples applying a take-off angle of electrons of 0°, excitation of photoelectrons by monochromatic Al Kα radiation, applying the constant analyzer energy mode (CAE) with a pass energy 40 eV in high resolution spectra and 150 eV in survey spectra, profiting from charge compensation by a dual beam argon/electron source with ultra-low energy beam neutralization. At these conditions, XPS probes the sample at a ≈10 nm depth from the incident surface. For high-resolution spectra, 20 eV and 40 eV analyzer pass energies were used, whereas for survey spectra 160 eV analyzer pass energy was employed. Probe area was elliptical in shape having major and minor axes of 700 μm and 300 μm, respectively. The binding energy values were referenced by positioning the C1s signal for hydrocarbon species of PS substrates at 284.8 eV. Surface concentration values were calculated based on the simplifying geometric model assumption that the sample surface is homogeneously composed. Thus, such concentration values contribute to facilitating a quantitative comparison between distinct surface states.
AFM
AFM images of PS and HOPG surfaces were obtained using a scanning probe microscope (Park systems XE7, Japan) operated in tapping mode. Si-tipped cantilevers (≈5 nm radius) having ≈250 kHz resonance frequencies (force constant k = 0.7 N m−1) were used. PS films were deposited onto silica surfaces by pipetting 1 wt% PS in toluene onto the substrate, 100 μL cm−1,2 and allowed to dry. Contact mode scanning was used to produce the holes in the organic layers. The software XEI Park systems was used for image analysis.
Determination of free catechol moieties
Catechol free moieties were determined using the derivatization protocol described by Sharov et al.29 Solutions of 50, 20, 10, 5 μg mL−1 of Tet-124-G-BrPh-DOPA-G were prepared and next, a solution containing molar excess of 105 fold of benzylamine (BA, Sigma Aldrich Germany) and potassium hexacyanoferrate(III) (K3Fe(CN)6, Roth, Germany) in methanol/water 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 was added during 2 hours under shaking at 37 °C. Based on the QCM-D analysis the approx. adsorbed peptide amount on the laccase surface per cm2 were calculated and a solution of 105 fold molar excess was pipetted on the surface during 2 hours under shaking at 37 °C. Afterwards, the fluorescence of the supernatant solution was measured using a Fluorolog Horiba Scientific TCSPC instrument; 344 nm excitation wavelength and emission spectra between 420 to 580 nm. Under our experimental conditions, the analysis is sensitive for concentrations higher than 1 μg mL−1 and consequently, 8 cm2 PS modified substrates were used.
:9 was added during 2 hours under shaking at 37 °C. Based on the QCM-D analysis the approx. adsorbed peptide amount on the laccase surface per cm2 were calculated and a solution of 105 fold molar excess was pipetted on the surface during 2 hours under shaking at 37 °C. Afterwards, the fluorescence of the supernatant solution was measured using a Fluorolog Horiba Scientific TCSPC instrument; 344 nm excitation wavelength and emission spectra between 420 to 580 nm. Under our experimental conditions, the analysis is sensitive for concentrations higher than 1 μg mL−1 and consequently, 8 cm2 PS modified substrates were used.
Cell culture
L-929 cells (Leibniz Institute DSMZ-German Collection of microorganisms and cell cultures, Braunschweig, Germany, DSMZ no: ACC 2, species murine (mouse) Mus musculus, connective tissue fibroblasts) were seeded in 75 cm2 cell culture flasks at 37 °C, applying 5% CO2 and a humidified atmosphere. The culture medium was prepared by mixing Roswell Park Memorial Institute (RPMI) 1640 medium with L-glutamine (Lonza, Belgium), 10% fetal bovine serum (Biochrom AG, Germany), penicillin (100 μg mL−1), and 100 μg mL−1 streptomycin (Lonza, Belgium) under aseptic conditions. First, the medium was removed from the culture flask, and then cells were washed with 4 mL 0.01 M PBS buffer (Sigma-Aldrich, Germany), prior to adding 1 mL of a 1% trypsin solution in PBS (Lonza, Belgium) for 10 min at 37 °C. Cells were counted by using a cell counter (Cellometer Auto T4 from Nexcelom Bioscience, United States). Finally, a volume corresponding to a quantity of 1 × 106 cells was pipetted into a new culture flask and culture medium was added to a final volume of 20 mL. Prior to cell seeding, test samples were sterilized under UV light for at least 15 min. From the cell suspension, a volume of 1 mL in fresh medium was added on each sample at a seeding density of 1 × 105 cells per mL.
Biocompatibility (WST-1) and cell staining
The test was performed following standard protocols (DIN EN ISO 10993-5 and 10993-12). Samples were sterilized under UV light for 15 minutes, immersed in 1 mL culture medium in a 12-well plate and pre-incubated for 24 hours. At the same time, an aliquot of 100 μL cell suspension was pipetted onto a 96-well plate (CellStar, Sigma-Aldrich) according to experimental planning (5 × 104 cells per well) and pre-incubated for 24 hours. Following pre-incubation, the culture medium was removed from the 96-well plate and replaced with the corresponding extracts from the 12-well plates containing the test samples. Extracts were used diluted with fresh media. For the positive control, >90% HEMA (2-hydroxyethylmethacrylate) was used at concentrations of 100% and 10%, respectively. For the negative control, fresh culture medium was used. After 24 hours of incubation, the liquids were removed from all wells and replaced with WST-1 solution (Roche, Switzerland, 10 μL WST-1 per 1 mL culture medium). After a 2 hour incubation, the absorbance was detected at 450 nm. The results were plotted with GraphPad Prism 5 software.
The cell culture procedure was performed, afterwards the samples were placed in the incubator for 24 hours. After the incubation period, the medium was removed, and the samples were washed two times with 1 mL PBS buffer. After each wash, but before the removal of the washing solution, samples were briefly shaken at 100 rpm. Cells were fixed in 4% paraformaldehyde in PBS at room temperature for 10 to 30 minutes. Next, 0.1% Triton X-100 in PBS was used to permeabilize the cells. Fixed cells were stained with rhodamine phalloidin (Thermo Fisher Scientific, United States) and DAPI (Sigma-Aldrich, Germany).
Minimal inhibition concentration (MIC) of peptides against E. coli
A Escherichia coli (E. coli) suspension at 1 × 106 CFU mL−1 in minimal medium (MM, 1 mL LB in 9 mL PBS) as well as a bacteria suspensions having a additionally different peptide concentrations were incubated on a 96-well plate for 18 hours at 37 °C. 200 μL of each solution was pipetted in a 96-microplate (3 filled wells) and additional 50 μL of LB-medium was pipetted into the filled wells. Over the plate, a breathable film was pasted, and the growth of the bacteria was followed overnight at 37 °C using a microplate multimode reader (Mithras LB 940, Berthold Technologies, Germany), optical density changes at 620 nm. The lowest concentration at which growth was not detected was considered the minimal inhibition concentration (MIC). Average values taken from three independent experiments are reported.
Antibacterial properties
The attachment of E. coli to 1 cm2 neat PS substrates (control) as well as PS substrates modified using LMS and LMS/Tet-124-G-BrPh-DOPA-G was investigated. E. coli colonies obtained from an agar plate were incubated in 5 mL of LB for 16 hours at 37 °C (overnight culture). The bacterial cells were washed by centrifugation and re-suspended using sterile PBS. The pellet was then suspended in minimal medium (MM, 0.1 mL of LB mixed with 9.9 mL of PBS). Centrifugation and washing using MM were repeated three times. The bacterial concentration was tuned to ≈1 × 106 CFU mL−1. Samples were prepared by pipetting 50 μL of bacterial suspension onto the substrates, covered using 1 cm × 1 cm coverslips, and incubated for either 4 hours or 18 hours at 37 °C for the adhesion and antimicrobial test, respectively. Samples were then rinsed using 100 μL of sterile PBS. Bacteria were then detached from the substrates by immersing the samples in an Eppendorf containing PBS, agitation for 3 min using an ultrasonic bath (Sonorex RK100 Bandelin Electronic, Berlin, Germany), and vortex mixing for 1 min. Both solutions, obtained from the first rinsing and detachment procedure, were mixed and 200 μL of the resulting solution were transferred to a 96-well microplate. After adding 50 μL LB to each well, optical density changes were measured at 620 nm during 24-hour at 37 °C. In addition, surfaces, which were rinsed with PBS but did not undergo the detachment procedure using ultrasound, were stained using a LIVE/DEAD BacLight staining Kit (Thermofisher Scientific, Unites States) following the supplier's protocol.
Results and discussion
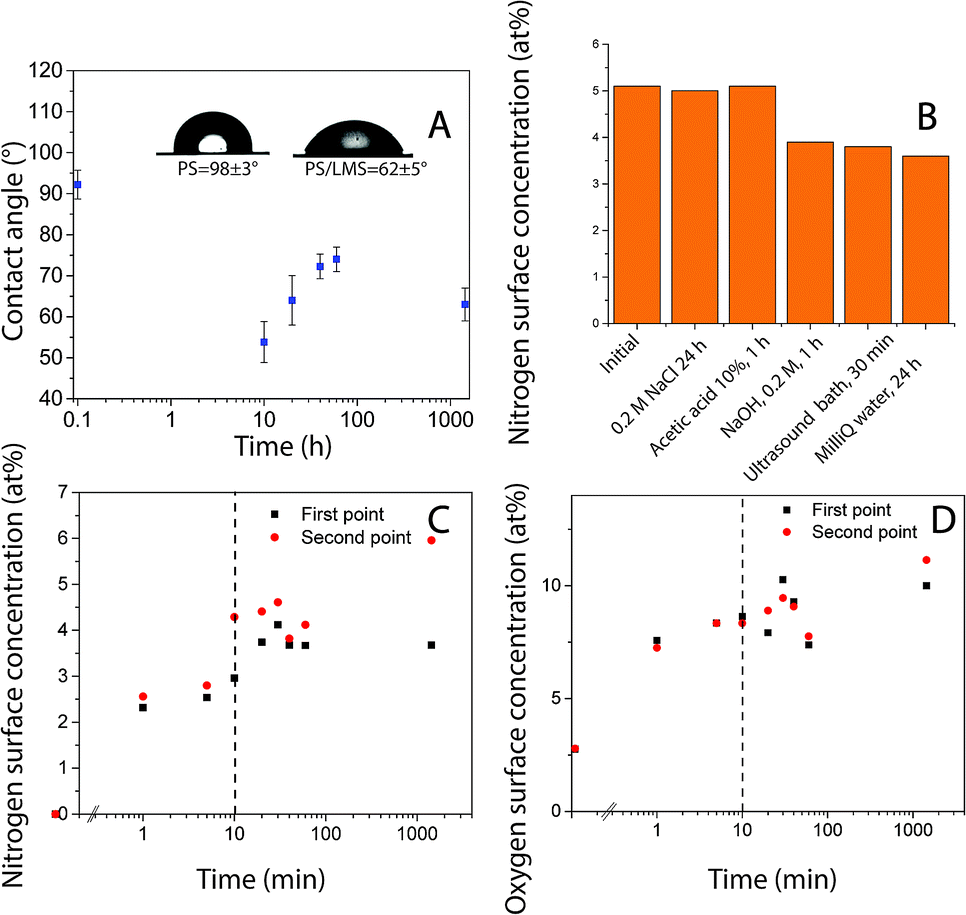
Polystyrene (PS) surface modification followed a similar protocol as used to functionalize HOPG surfaces.25 Robust layers suitable for functionalization were formed using laccase since the protein (enzyme) irreversibly adsorb quickly to hydrophobic surfaces. There was a change in surface hydrophobicity to more hydrophilic domains, directly correlated with the decrease in the surface water contact angle, Fig. 1A. This layer could withstand rinsing with solutions at various pH levels and ultrasound over long periods of time, Fig. 1B (Table S1†). Similar layer LM to the obtained on HOPG was formed.25 However, protein adsorption rate and coverage depends on time in contact with the surface, enzyme concentration, ionic strength, presence of other molecules in solution, surface affinity, among other factors.30 Surface elemental composition as a function of time in contact with laccase/maltodextrin suspension (LMS) was investigated using XPS for durations up to 24 hours. Both nitrogen and oxygen concentrations increase monotonically in time and saturated after ≈10 min (Table 2, Fig. 1C and D). Moreover, the enzyme was still active after being irreversible adsorbed on PS as confirmed by ABTS oxidation (Table S2†). Protein adsorption on PS surfaces could be driven by π–π interactions between protein and PS aromatic groups.31 The laccase is widely distributed in fungus that decomposes lignocellulosic materials and its sequence is composed of 49% of hydrophobic amino acids, suggesting that its function in nature is maybe set to have affinity for hydrophobic surfaces.32,33
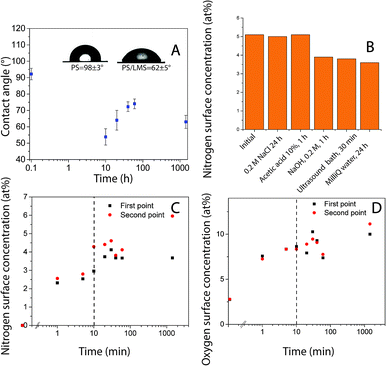 |
| | Fig. 1 (A) Water contact angle as a function of time for PS after exposure to LMS. Inset: sessile drop images for the unmodified PS control sample (left) and for the surface treated PS after 1 hour (right). XPS findings indicating (B) [N] after various rinsing and desorption procedures. Surface concentration as a function of time (C) nitrogen (D) oxygen; values of two independent samples are shown as first and second point. | |
Table 2 PS elemental surface concentration analyzed after contact with LMS, LMS and Tet-124-G-BrPh-DOPA-G. Average surface roughness (Ra) was measured using AFM topographic images
| Sample |
PS |
Tet124-G-BrPh-DOPA-G |
PS/LM |
LMS 10 min followed by the addition of the peptide solution for 50 minutes |
| O |
2.2 |
12.7 |
8.1 |
11.3 |
| N |
0.1 |
12.6 |
3.2 |
6 |
| C |
97.5 |
72.1 |
85.1 |
82.6 |
| S |
— |
— |
0.2 |
0.2 |
| Br |
— |
0.6 |
|
0.1 |
| Na |
— |
— |
0.1 |
|
| Si |
— |
— |
0.4 |
0.2 |
| Ra |
2.4 ± 0.9 nm |
— |
3.1 ± 0.7 nm |
3.8 ± 1.1 nm |
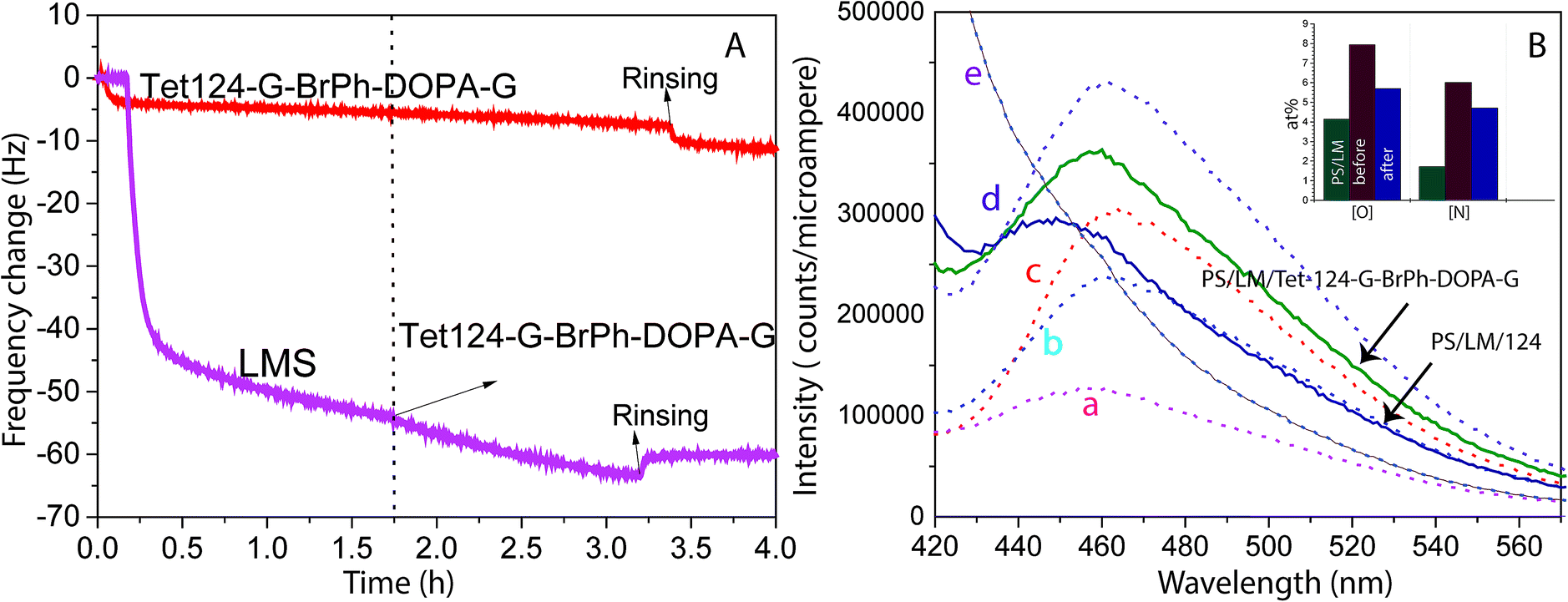
QCM-D was utilized to study the adsorption dynamics of peptide layer formation. Fig. 2A shows the resonant frequency shift arising from peptide adsorption on the PS surface and LM layer surface. Frequency shifts due to peptide adsorption on the LM layer (two-step functionalization process), and neat PS are detected and quantified. During the two-step functionalization process LM surface concentration increases until adsorption and desorption reach dynamic equilibrium. In the second step, the peptide solution was in contact with LM layers and a stable peptide layer was formed. Therefore, peptide layers on both PS and LM were stable against buffer rinsing. However, a thicker peptide layer was formed on LM layers. Peptide mass adsorbed on PS and PS/LM was calculated using Sauerbrey resulting in 92 and 167 ng cm−2, respectively, Fig. 2A. To enhance the antimicrobial properties, hydrophobic residues should face away from the surface in order to increase interactions with bacteria membrane. Differences in mass adsorption could be related to surface affinity as well as to differences in the orientation of molecules building up the film: e.g. molecular long-axes lying in the surface plane for neat PS or long-axes oriented perpendicular to the surface for LM layers. Laccase and peptides could interact not only by π–π stacking but also, by electrostatic interactions between the charged functional groups and formed hydrogen bonds.34 ITC titration curves of the Tet 124-G-BrPh-DOPA-G into laccase/maltodextrin suspension shows a characteristic curve for an enzymatic reaction on contrary to Tet-124-G-BrPh (Fig. S1†). Consequently, an intermolecular crosslinking could occur between laccase oxidized Tet-124-G-BrPh-DOPA-G peptide and the surface.35 The emission fluorescence spectra obtained from the Tet 124-G-BrPh-DOPA-G solution shows an increment of intensity as expected with peptide concentration. The peptide reacts with the benzylamine to form a benzoxazole derivate molecule that has a fluorescence emission at 467 nm. Moreover, the fluorescence emission peak of the solution removed from the PS/LM/Tet 124-G-BrPh-DOPA-G modified surface suggests the presence of catechol-free-moieties on the surface. In addition, the elemental concentration on the surface after the derivatization reaction indicates that the peptide could be partially desorbed after contact with this highly alkaline solution, [N] and [O] of the PS/LM/Tet-124-BrPh-DOPA-G after the derivatization reaction was lower than the surface before the derivatization reaction and higher than the PS/LM modified surface. The results suggest a peptide layer with good stability and a possible covalent attachment (Fig. 2B and S2†).
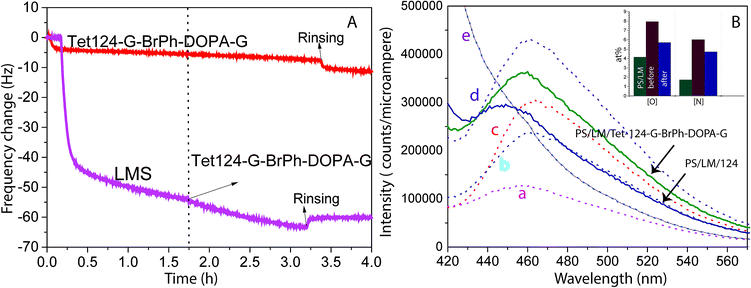 |
| | Fig. 2 (A) Resonance frequency shift Δf as a function of time for a PS coated crystal surface initially exposed to 0.1 mg mL−1 LMS and later to 0.05 mg mL−1 Tet-124-G-BrPh-DOPA-G (purple curve) and a PS coated crystal only exposed to 0.05 mg mL−1 Tet-124-G-BrPh-DOPA-G (red curve). (B) Fluorescence spectra of Tet-124-G-BrPh-DOPA-G solutions at concentration of: (a) 5 μg mL−1, (b) 10 μg mL−1, (c) 20 μg mL−1, (d) 50 μg mL−1 after reaction with 10-fold benzylamine; (e) only reagents (dotted line) and supernatant solution after contact with the peptide (Tet-124 or Tet-124-G-BrPh-DOPA-G) PS/LM modified surface (continuous line). Inset: comparison of [O] and [N] on PS/LM/before and after the surface derivatization reaction. XPS findings indicate peptide partial desorption from the surface. | |
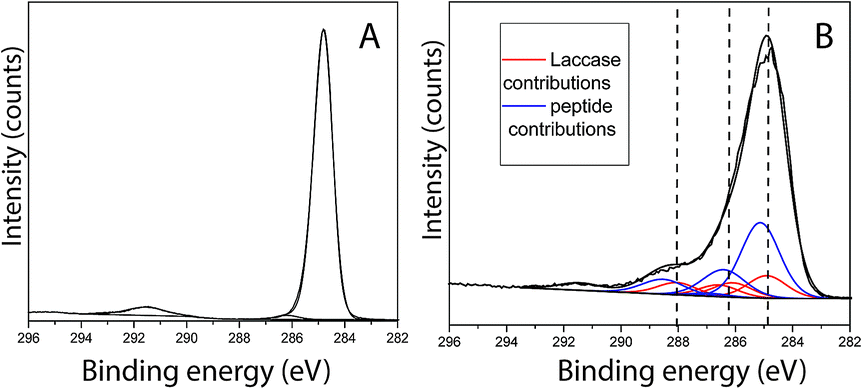
Peptide adsorption on the LM layers resulted in increased surface concentrations of [N] and [O] as detected by XPS. Moreover, the XPS results confirmed the presence of Br, as shown in Table 2. To determine the bulk composition of the peptide and to estimate the maximum expected Br concentration detected by XPS in a more than 10 nm thickness peptide film, a drop of aqueous peptide solution was dried, and the area probed using XPS. As shown in Table 2, the [Br] was determined to be 0.6 at%. From this characterization, it becomes clear that for the 0.1 at% Br concentration detected in the adsorbate, film obtained after 50 min of contact between PS/LM and the peptide solution, the peptide stoichiometry will predetermine a of [N] = 2.1 at% for the brominated peptide. On the other hand, any nitrogen species that contribute to a concentration [N] > 2.1 at% after such peptide adsorption will arise from the previous enzyme adsorption contributing to the formation of a hybrid enzyme-peptide layer. Fig. 3A and B show the fitting results for the C1s signal of neat PS and PS/LM/Tet-124-G-BrPh-DOPA-G surface. The C1s spectra for the PS and substrate modified with LM and the Tet-124-G-BrPh-DOPA-G is characterized by a major peak at 284.8 eV assigned to predominantly aromatic hydrocarbonaceous species in case of PS and predominantly aliphatic hydrocarbon species in case of the peptide and laccase adsorbates, with their prominent peaks situated at 285.1 ± 0.1 eV. Four additional peaks were allocated at higher binding energies for the PS/LM/Tet-124-G-BrPh-DOPA-G sample. The peak at 286.3 ± 0.1 eV is associated with amine-like C*–N; the signal related to C*–O from alcohol/alkoxy groups is characterized by a 0.5 eV higher binding energy; the peak centered at 288.2 ± 0.1 eV is characteristic of amide groups (–NH–C*![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); and, finally, the peak at 289.0 ± 0.1 eV is correlated with carboxyl groups.25 The layer thickness calculated following the protocol described by Corrales et al. is approximately 1 nm and 2.7 nm for LM and LM/Tet-124-G-BrPh-DOPA-G layers on PS, respectively.25
O); and, finally, the peak at 289.0 ± 0.1 eV is correlated with carboxyl groups.25 The layer thickness calculated following the protocol described by Corrales et al. is approximately 1 nm and 2.7 nm for LM and LM/Tet-124-G-BrPh-DOPA-G layers on PS, respectively.25
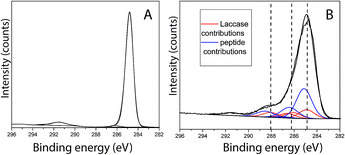 |
| | Fig. 3 C1s XPS findings for (A) PS control, (B) PS modified with LM/Tet-124-G-BrPh-DOPA-G when fitting the measured signal with contributions from the PS substrate, laccase and peptide bulk sample spectra. | |
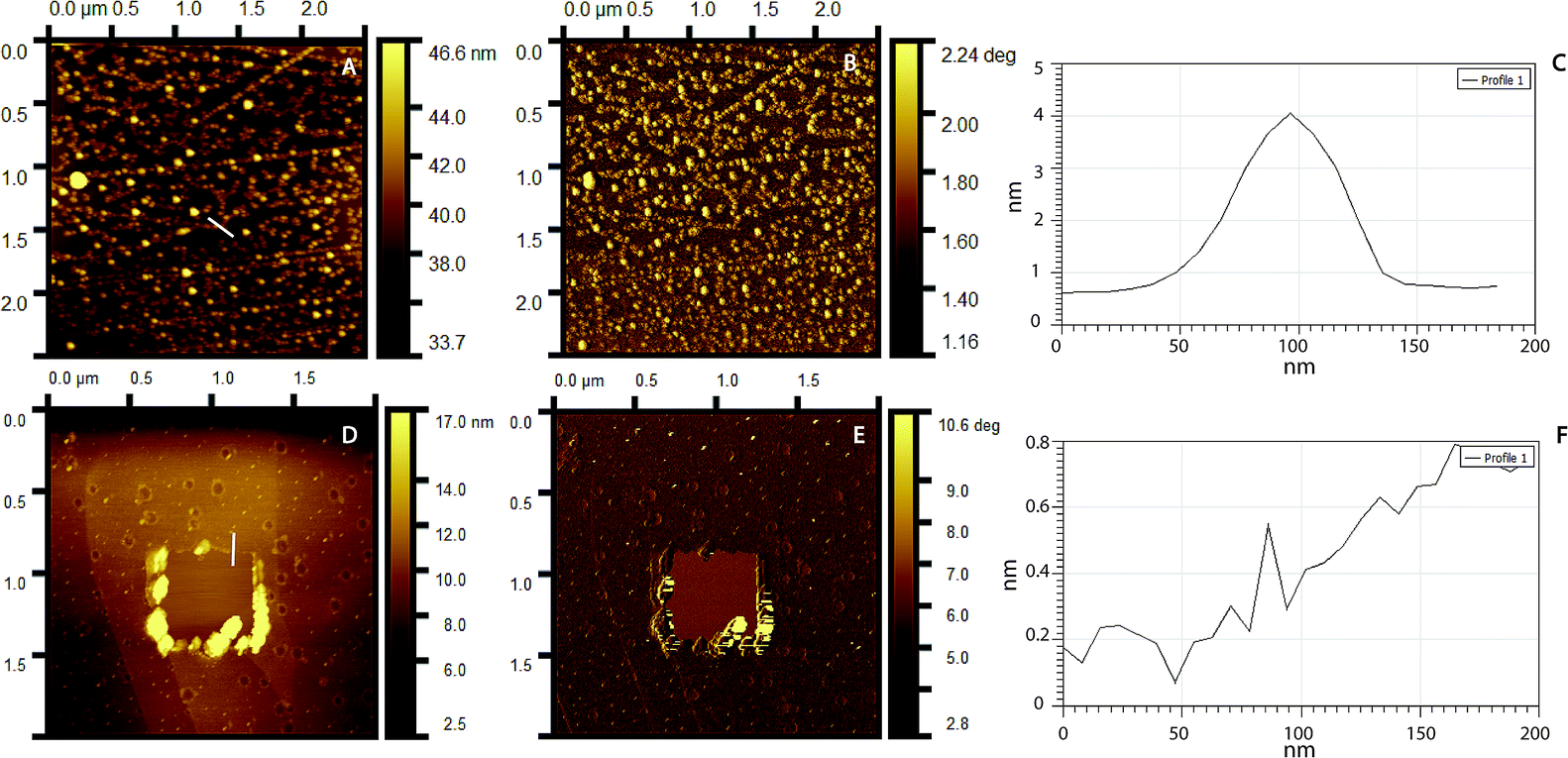
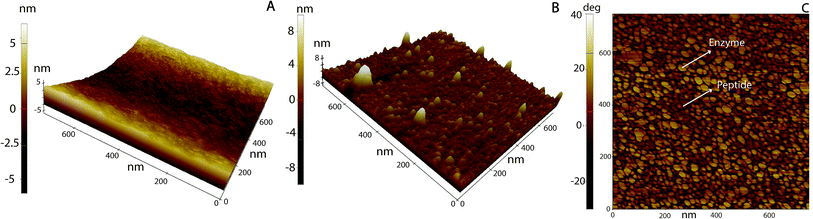
Surface topography and roughness of layers were studied using AFM. Initially, molecular dimensions of the laccase and peptide measured upon adsorption on HOPG as a model hydrophobic flat surface. Fig. 4A shows adsorbed laccase globular molecules having 3.5 ± 0.4 nm heights and larger 13 nm agglomerates appear to be randomly distributed. The peptide adsorbed on the surface forms a close thin monolayer of approximately 0.8 ± 0.2 nm on HOPG, Fig. 4D. The phase image shows contrast between the laccase molecules and substrate but not between the laccase molecules and the tip; suggesting similar adhesion forces, Fig. 4B. The peptide layer shows similar results, Fig. 4E. The layer thickness calculated between the bottom area a hole and upper LM/Tet-124-G-BrPh-DOPA-G adsorbed on HOPG is approximately 4.2 nm. The layer thickness correlates to one laccase and one peptide molecule (Fig. S3†).
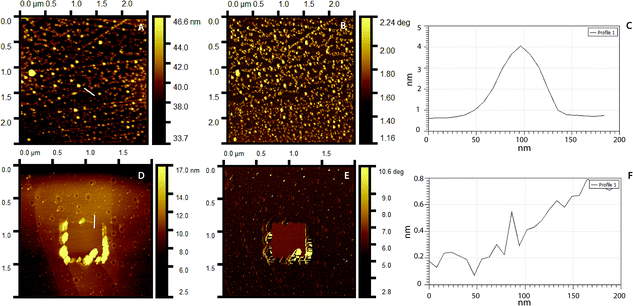 |
| | Fig. 4 AFM topography images of (A) laccase, (D) Tet-124-G-BrPh-DOPA-G. Phase image corresponding to (B) A, (E) D. Line height profile highlighted in (C) A, (F) D. | |
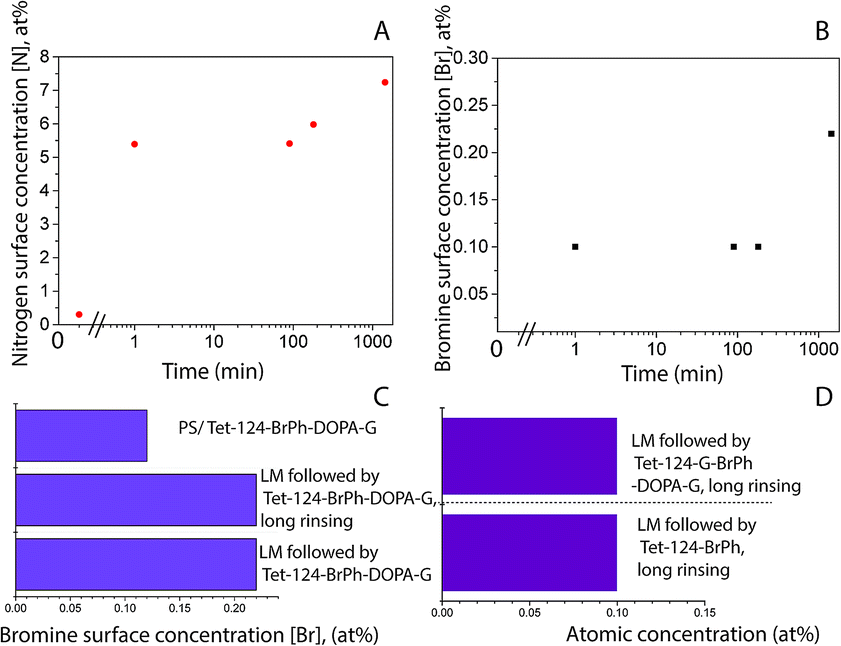
Fig. 5A shows a PS smooth, without globular nanostructures, surface. Fig. 5B shows a surface homogeneously covered with laccase enzyme molecules. Comparison between the phase images of the LM layers and LM/peptide allowed determining the peptide distribution. The darker areas could be associated to the peptide layer adsorption on top and surrounding of laccase molecules, Fig. 5C. The average roughness, a factor that influences bacterial adhesion,36,37 changed by less than 2 nm (see Table 2). The influence of the layers on a subsequent bacteria adhesion will be discussed further in this study. In order to maximize peptide concentration on LM interfactant layers, peptide adsorption time-dependence was investigated for time periods ranging from 1 min to 24 hours. Fig. 6A and B show an increase in surface concentrations of [N] and [Br] with time. After 24 hours, maximum concentrations of 7.2 at% and 0.22 at% were obtained for [N] and [Br], respectively. After comparing with the bulk composition of the peptide corresponding to a maximum surface concentration [Br] around 0.6 at%, we suggest that peptide layers characterized by [Br] = 0.1 at% are a few nanometers thin and may partially covering the surface. The [Br] concentration was higher for the peptide layers formed on LM layers than on PS even after 24 hours, Fig. 6C. Given that the isoelectric point pI value of the peptide Tet-124-G-BrPh-DOPA-G is ≈11.5, we expect that at a pH of 4.75, lysine and arginine side chains were positively charged and experience electrostatic repulsion that may prevent multilayer formation. Such supposition is in reasonable agreement with the presented AFM findings, and a more precise estimate of the peptide layer thickness may be provided following some XPS signal fitting that facilitates establishing the PS substrate signal attenuation. Another DOPA containing peptide that was previously published by the authors with charged +2 at pH 7, Ala-Lys-Pro-Ser-Tyr-Hyp-Hyp-Thr-DOPA-Lys,38 formed a thicker and stable layer on the laccase surface (Fig. S4†). Consequently, we suggest that the thin peptide layers formed could be result of electrostatic repulsion between peptide molecules; not favoring multilayer formation. The influence of the amino acid DOPA on the adsorption stability of antimicrobial peptides onto PS/LM was studied by comparing the surface concentrations [Br] after exposure to either peptide solution (Fig. 6D), in this way considering peptides with and without DOPA modification. Similar values suggest that catechol crosslinking at the surface does not play a significant role with respect to the achieved layer thickness and stability, even if the enzyme is active after immobilization on the PS surface and the laccase enzyme is able to oxidize and to polymerize the DOPA containing peptide in solution (Fig. S1†).
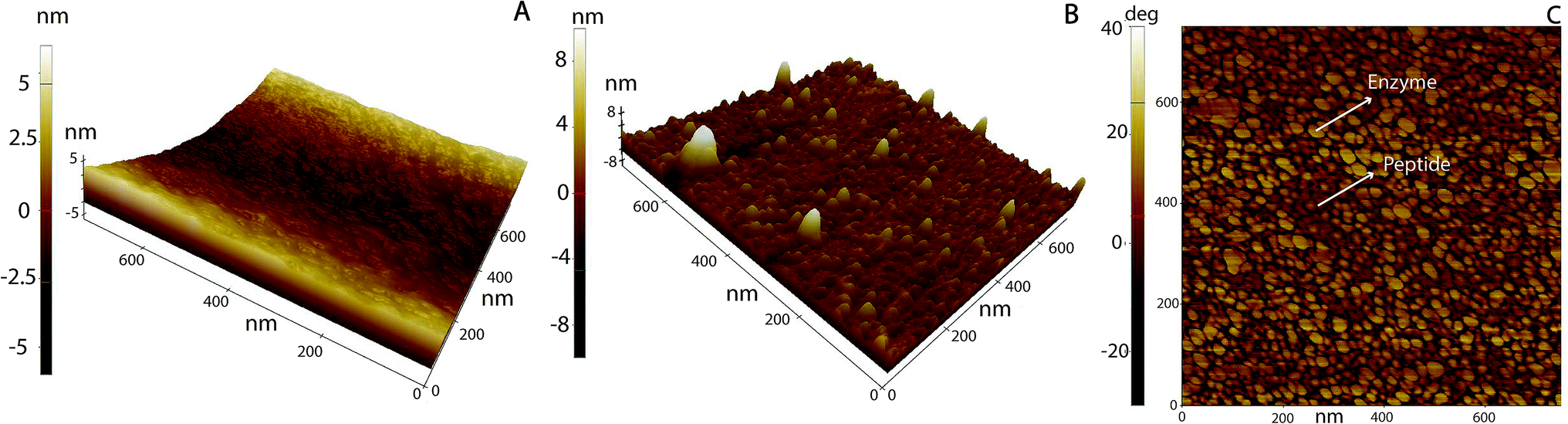
 |
| | Fig. 5 AFM 3D topography images of (A) neat PS, (B) PS/LM (10 min) and then, exposed to a Tet-124-G-BrPh-DOPA-G (50 min). (C) B phase image. | |
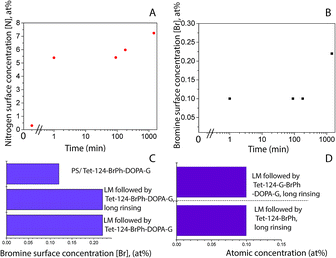 |
| | Fig. 6 XPS-based elemental surface concentration as a function of time (A) [N], (B) [Br]; LMS contact time 10 min and then, Tet-124-G-BrPh-DOPA-G solution up to 24 h. Comparison of adsorption of (C) Tet-124-G-BrPh-DOPA-G on PS and PS/LM surface, assessing the stability after long rinsing time (0.1 M aqueous NaCl, 24 hours exposure under shaking). (D) Comparison of [N] and [Br] between Tet-124-G-BrPh-DOPA-G and Tet-124-G-BrPh layers formed on PS/LM. | |
We suggest that the adhesion promoting interfactant properties of the laccase layer are dominant and that the electrostatic interactions and hydrogen bonding between the functional groups facing the laccase layer surface drive the adsorption and stabilization of the peptide layer and consequently, the transport of the respective catechol moieties inwards or outwards to the catalytically active sites of the laccase. We infer that the molecular environment of such lock-and-key system may affect the reaction probabilities between laccase and catechol moieties. We attribute such hindrance to the limited diffusive mobility and the restricted conformational freedom of the adsorbed peptide on top of the bio-interfactant layer, which is due to non-covalent interactions. Further studies are necessary to determine differences in the adsorption coverage density and stability of a peptide with the DOPA modification at the N terminus modification or a comparison of hydrophilic peptides with and without DOPA.
Antimicrobial properties and biocompatibility
Given that it is a common bacterium colonizing medical device surfaces and potentially may cause infections, E. coli was used for antimicrobial studies.39 MIC in solution was calculated to determine whether the changes on the C-terminus caused antibacterial activity depletion.
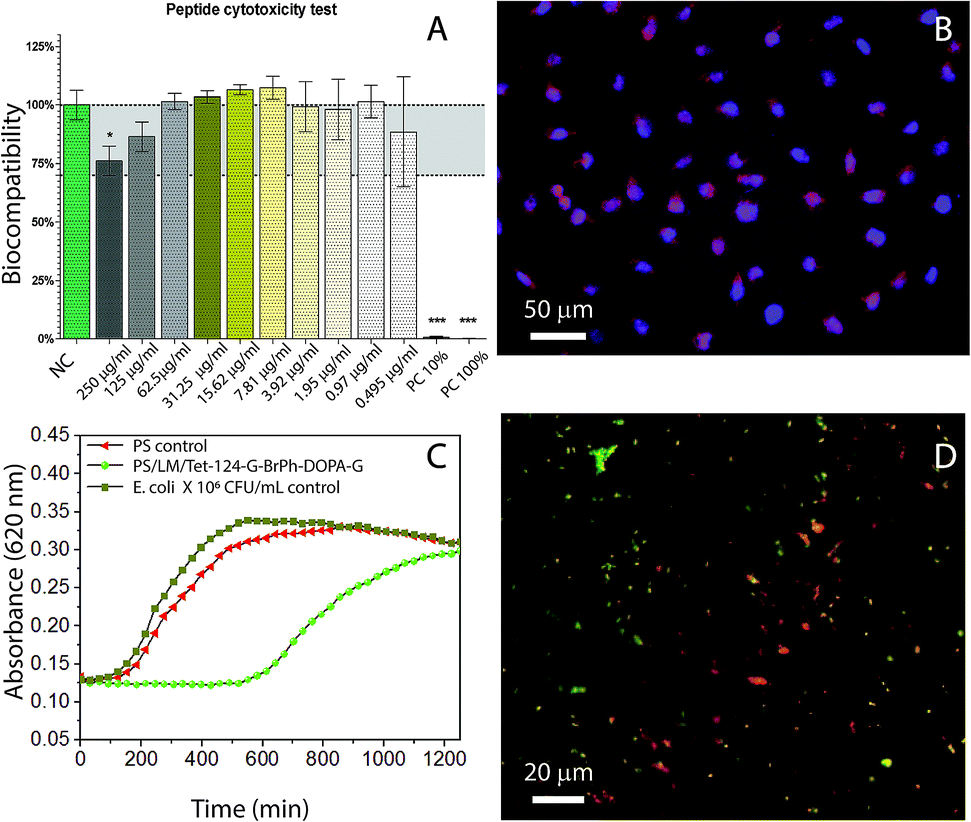
As it can be seen in the data shown in Table 3, the antimicrobial activity was decreased by G-BrPh and G-BrPh-DOPA-G C-terminus sequence modification. For instance, significant changes in antimicrobial activity has been reported due to the exchange of an L-amino acid to a D-amino acid in other peptides sequences.40 Circular dichroism spectra shown in ESI Fig. S5† indicates that both peptides have α-helical conformation in solution.41 Further studies are necessary to determine if changes in the factors such as hydrophobicity and steric hindrance influenced the antimicrobial activity.42–46 The antimicrobial activity of the Tet-124-G-BrPh-DOPA-G and Tet-124-G-BrPh were lower than the Tet-124; however, the peptide is still active. They show higher antimicrobial activity than the Tet-000 (peptide without reported antimicrobial activity), Table 3. The peptide Tet-124-G-BrPh-DOPA-G was chosen for further antimicrobial tests due to the lower MIC in comparison with the Tet-124-G-BrPh. Fibroblast viability in solution was not affected at MIC of the peptides under studied conditions, Fig. 7A. There are no significant differences between cells growth negative control and cell in contact with the different peptides tested in solution at concentration of 250 μg mL−1 for all the peptides according to one-way ANOVA statistical analysis. Fibroblast cell morphology was compared between the cell's growth on PS, PS/LM and PS/LM/Tet-124-G-BrPh-DOPA-G substrates, Fig. 7B and S6.† For both PS surface modifications, cells adhered and survived in similar way that the control PS sample. Image processing (Image J) was used to measure the average cell surface density. Cell densities were determined to be 95 ± 5, 100 ± 10 and 130 ± 20 fibroblast cells/mm2 for the PS, PS/LM and PS/LM/Tet-124-G-BrPh-DOPA-G, respectively. We did not find a significant difference applying ANOVA test (p < 0.05) between the samples (Fig. S6†). To determine the influence of roughness and chemical functional groups, bacterial adhesion was tested. There was no increase in E. coli adhesion in between the neat control surface (Fig. S7†) and PS/LM/Tet-124-G-BrPh-DOPA-G surface. E. coli grown on LM/Tet-124-G-BrPh-DOPA-G modified surfaces displayed a log phase delay of 10 hours indicating enhanced antimicrobial surface activity. Adsorbed peptides exhibit low mobility, and therefore, antimicrobial effects can be attributed to cell membrane disruption.47 Bacterial cell membrane disruption must arise from electrostatic destabilization rather than peptide penetration since the membrane thickness of E. coli (45–55 nm) is approximately 10 times larger than peptide length (≈5.9 nm). The membrane of Gram-negative-bacteria consist of a peptidoglycan layer between a lipid bilayer in which a high density of negatively charged lipopolysaccharides acids are in the outer lipid membrane. Cationic peptides interact with the outer cell membrane allowing for the entry of exterior molecules to diffuse into the cell.47,48 This disrupts the electrochemical gradient across the membranes, increasing water and ion diffusion through the membrane which leads to cell swelling and osmolysis.49,50 The sample surface modified with the peptide showed dead and alive bacteria after 18 hours of contact with E. coli. This result suggests that this surface modification caused a partial depletion of the bacteria, which may be related to a non-homogeneous peptide surface distribution as can be seen in Fig. 7D. Overall, E. coli grown on PS/LM/Tet-124-G-BrPh-DOPA-G surfaces displayed a log phase delay of 10 hours indicating enhanced antimicrobial activity. Our results demonstrated that the immobilization process was successful and preserved (at least partially) antimicrobial peptide activity. For better understanding the effect of AMP immobilization technique on the antimicrobial activity further analysis is necessary because the antimicrobial mode of action could change according to the immobilization strategy.51 Therefore, this methodology could be used to immobilize mixtures of peptides that could act in synergy against biofilm forming bacteria.
Table 3 MIC of peptides against E. coli, 18 h incubation in minimal medium
| Peptide |
Sequence |
MIC peptide, μg mL−1 |
| Tet000 |
GATPEDLNQKLS |
500< |
| Tet-124 |
KLWWMIRRW |
14 |
| Tet-124-G-BrPh-G |
KLWWMIRRWG-BrPh-G |
225 |
| Tet-124-G-BrPh-DOPA-G |
KLWWMIRRWG-BrPh-DOPA-G |
100 |
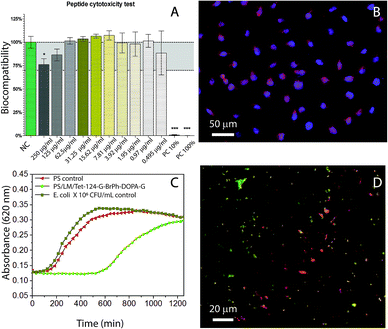 |
| | Fig. 7 (A) WST-1 fibroblast viability test (NC: negative control, PC: positive control, Tet-124-G-BrPh-DOPA-G at different concentrations). (B) Fibroblast L-929 cell growth on PS/LM/Tet-124-G-BrPh-DOPA-G modified surface. Cells were stained using rhodamine phalloidin (actin filaments) and DAPI (nucleus). (C) E. coli growth as a function of time following the detachment protocol after 18 h in contact with the surface. (D) Fluorescence image of live (green) and dead (red) bacteria (LIVE/DEAD staining) on a PS/LM/Tet-124-G-BrPh-DOPA-G modified surfaces, surface rinsed with buffer. | |
Conclusions
We reported a new bio-functionalization method to increase surface energy and add functional groups that provide binding sites and potential active sites for hydrophobic polymer surfaces. Layers exemplarily formed on polystyrene from aqueous laccase/maltodextrin formulations serve as a bio-interfactant layers. They allow for immobilization of antimicrobial peptides based on Tet-124 resp. KLWWMIRRW with and without DOPA C-terminus modification and for formation of layers that are stable and bioactive at physiological conditions. The antibacterial properties against Escherichia coli and the fibroblast L-929 growth viability were tested. Fibroblasts survived when in contact with both, the PS and PS/bio-functionalized surface. Antibacterial properties of the PS modified with laccase/maltodextrin/Tet-124-G-BrPh-DOPA-G surface were tested against E. coli. The surface functionalization does not increase bacteria adhesion and it caused partial depletion of bacteria growth.
Conflicts of interest
The authors declare that they do not have a conflict of interest.
Acknowledgements
This work benefited from support from the Consejo Nacional para Investigaciones Científicas y Tecnológicas CONICIT of Costa Rica and the Swiss National Science Foundation through the National Center of Competence in Research Bio-Inspired Materials. We thank Dr Miguel Spuch from the Bionanomaterials group at Adolphe Merkle Institute for designing the graphical abstract.
References
- L. W. McKeen, Plastics used in medical devices, in Handbook of Polymer Applications in Medicine and Medical Devices, 2014, pp. 21–53 Search PubMed.
- A. Muñoz-Bonilla and M. Fernández-García, Polymeric materials with antimicrobial activity, Prog. Polym. Sci., 2012, 37(2), 281–339 CrossRef.
- V. Sambhy, M. MacBride, B. Peterson and A. Sen, Silver bromide nanoparticle/polymer composites: dual action tunable antimicrobial materials, J. Am. Chem. Soc., 2006, 128(30), 9798–9808 CrossRef CAS PubMed.
- J. A. Lichter, K. J. Van Vliet and M. F. Rubner, Design of antibacterial surfaces and interfaces: polyelectrolyte multilayers as a multifunctional platform, Macromolecules, 2009, 42(22), 8573–8586 CrossRef CAS.
- Q. Wei, T. Becherer, P. L. M. Noeske, I. Grunwald and R. Haag, A universal approach to cross-linked hierarchical polymer multilayers as stable and highly effective antifouling coatings, Adv. Mater., 2014, 26(17), 2688–2693 CrossRef CAS PubMed.
- M. Yoshinari, T. Kato, K. Matsuzaka, T. Hayakawa and K. Shiba, Prevention of biofilm formation on titanium surfaces modified with conjugated molecules comprised of antimicrobial and titanium-binding peptides, Biofouling, 2010, 26(1), 103–110 CrossRef CAS PubMed.
- R. B. H. W. Huang, Free energies of molecular bound states in lipid bilayers: lethal concentrations of antimicrobial peptides, Biophys. J., 2009, 96(8), 3263–3272 CrossRef PubMed.
- D. Easton, A. Nijnik, M. Mayer and R. Hancock, Potential of immunomodulatory host defense peptides as novel anti-infectives, Trends Biotechnol., 2009, 27(10), 582–590 CrossRef CAS PubMed.
- A. A. Slavokhotova, A. A. Shelenkov, Y. A. Andreev and T. I. Odintsova, Hevein-like antimicrobial peptides of plants, Biochemistry, 2017, 82(13), 1659–1674 CAS.
- B. Bechinger and K. Lohner, Detergent-like actions of linear amphipathic cationic antimicrobial peptides, Biochim. Biophys. Acta, 2006, 1758(9), 1529–1539 CrossRef CAS PubMed.
- R. Hancock and H. Sahl, Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies, Nat. Biotechnol., 2006, 24(12), 1551–1557 CrossRef CAS PubMed.
- S. Maria-Neto, C. de Almeida, M. Rodrigues and M. Luiz-Franco, Understanding bacterial resistance to antimicrobial peptides: from the surface to deep inside, Biochim. Biophys. Acta, 2015, 1848(11), 3078–3088 CrossRef CAS PubMed.
- L. T. Nguyen, E. F. Haney and H. J. Vogel, The expanding scope of antimicrobial peptide structures and their modes of action, Trends Biotechnol., 2011, 29(9), 464–472 CrossRef CAS PubMed.
- W. C. Wimley, Describing the mechanism of antimicrobial peptide action with the interfacial activity model, ACS Chem. Biol., 2010, 15(10), 905–917 CrossRef PubMed.
- K. Hilpert, M. R. Elliott, R. Volkmer-Engert, P. Henklein, O. Donini, Q. Zhou, D. F. Winkler and R. E. Hancock, Sequence requirements and an optimization strategy for short antimicrobial peptides, Chem. Biol., 2006, 13(10), 1101–1107 CrossRef CAS PubMed.
- A. Shukla, K. Fleming, H. Chuang, T. Chau, C. Loose, S. Stephanopoulos and P. Hammond, Controlling the release of peptide antimicrobial agents from surfaces, Biomaterials, 2010, 31(8), 2348–2357 CrossRef CAS PubMed.
- F. Costa, I. Carvalho, R. C. Montelaro, P. Gomes and M. C. Martins, Covalent immobilization of antimicrobial peptides (AMPs) onto biomaterial surfaces, Acta Biomater., 2011, 7(4), 1431–1440 CrossRef CAS PubMed.
- H. Yazici, G. Haib, K. Boone, M. Urgen, F. Sermin and C. Tamerler, Self-assembling antimicrobial peptides on nanotubular titanium surfaces coated with calcium phosphate for local therapy, Mater. Sci. Eng., C, 2019, 94, 333–343 CrossRef CAS PubMed.
- H. Yazici, G. Haib, K. Boone, M. Urgen, F. Sermin and C. Tamerler, Engineered chimeric peptides as antimicrobial surface coating agents toward infection-free implants, ACS Appl. Mater. Interfaces, 2016, 8(8), 5070–5081 CrossRef CAS PubMed.
- Y. R. Corrales Ureña, L. Wittig, M. Vieira Nascimento, J. Faccioni and P. Noronha, Influences of the pH on the adsorption properties of an antimicrobial peptide on titanium surfaces, Appl. Adhes. Sci., 2015, 3(7), 1–17 Search PubMed.
- M. Xiao, J. Jasensky, J. Gerszberg, J. Chen, J. Tian, T. Lin, T. Lu, J. Lahann and Z. Chen, Chemically immobilized antimicrobial peptide on polymer and self-assembled monolayer substrates, Langmuir, 2018, 34(43), 12889–12896 CrossRef CAS PubMed.
- K. Hilpert, M. Elliott, H. Jenssen, J. Kindrachuk, C. Fjell, J. Körner, D. Winkler, L. Weaver, P. Henklein, A. Ulrich, S. H. Chiang, S. Farmer, N. Pante, R. Volkmer and R. Hancock, Screening and characterization of surface-tethered cationic peptides for antimicrobial activity, Chem. Biol., 2009, 16(1), 58–69 CrossRef CAS PubMed.
- K. Hilpert, et al., High-throughput generation of small antibacterial peptides with improved activity, Nat. Biotechnol., 2005, 23(8), 1008–1012 CrossRef CAS PubMed.
- D. G. Moussa, A. Fok and C. Aparicio, Hydrophobic and antimicrobial dentin: a peptide-based 2-tier protective system for dental resin composite restorations, Acta Biomater., 2019, 88, 251–265 CrossRef CAS PubMed.
- Y. R. Corrales-Ureña, P. N. Lisboa-Filho, M. Szardenings, L. Gätjen, P. L. M. Noeske and K. Rischka, Formation and composition of adsorbates on hydrophobic carbon surfaces from aqueous laccase-maltodextrin mixture suspension, Appl. Surf. Sci., 2016, 385, 216–224 CrossRef.
- F. Macul-Perez, Y. R. Corrales-Ureña, K. Rischka, W. Leite-Cavalcanti, P. L. M. Noeske, A. Safari, G. Wei and L. Colombi-Ciacchi, Bio-interfactants as double-sided tapes for graphene oxide, Nanoscale, 2019, 11(10), 4236–4247 RSC.
- Q. Wei, T. Becherer, R. C. Mutihac, P. L. M. Noeske, F. Paulus, R. Haag and I. Grunwald, Multivalent anchoring and cross-linking of mussel-inspired antifouling surface coatings, Biomacromolecules, 2014, 15(8), 3061–3071 CrossRef CAS.
- R. V. Ulijn and A. M. Smith, Designing peptide based nanomaterials, Chem. Soc. Rev., 2008, 37(4), 664–675 RSC.
- V. S. Sharov, E. S. Dremina, J. Pennington, J. Killmer, C. Asmus, M. Thorson, S. J. Hong, X. Li, J. F. Stobaugh and C. Schöneich, Selective fluorogenic derivatization of 3-nitrotyrosine and 3,4-dihydroxyphenylalanine in peptides: a method designed for quantitative proteomic analysis, Methods Enzymol., 2008, 441, 19–32 CAS.
- M. Rabe, D. Verdes, M. Rankl, G. R. Artus and S. Seeger, A comprehensive study of concepts and phenomena of the nonspecific adsorption of beta-lactoglobulin, Chemphyschem, 2007, 8(6), 862–872 CrossRef CAS PubMed.
- A. Baty, P. Suci, B. Tyler and G. Geesey, Investigation of mussel adhesive protein adsorption on polystyrene and poly(octadecyl methacrylate) using angle dependent XPS, ATR-FTIR, and AFM, J. Colloid Interface Sci., 1996, 177(2), 307–315 CrossRef CAS.
- M. Karim, M. G. Daryaei, J. Torkaman, R. Oladi, M. A. T. Ghanbary, E. Bari and N. Yilgor, Natural decomposition of hornbeam wood decayed by the white rot fungus Trametes versicolor, An. Acad. Bras. Cienc., 2017, 89(4), 2647–2655 CrossRef CAS PubMed.
- T. Saarinen, H. Orelma, S. Gronqvist, M. Andberg, S. Holappa and J. Laine, Adsorption of different laccases on cellulose and lignin surfaces, Bioresources, 2009, 4(1), 94–110 CAS.
- N. Artemova, V. Stein-Margolina, E. Smirnova and B. Gurvits, Formation of supramolecular structures of a native-like protein in the presence of amphiphilic peptides: Variations in aggregate morphology, FEBS Lett., 2012, 586(2), 186–190 CrossRef CAS.
- M. L. Mattinen, K. Kruus, J. Buchert, J. H. Nielsen, H. J. Andersen and C. L. Steffensen, Laccase-catalyzed polymerization of tyrosine-containing peptides, FEBS J., 2005, 272(14), 3640–3650 CrossRef CAS PubMed.
- C. Sousa, P. Teixeira and R. Oliveira, Influence of surface properties on the adhesion of Staphylococcus epidermidis to acrylic and silicone, Int. J. Biomater., 2009, 2009, 718017 Search PubMed.
- E. P. Ivanova, V. K. Truong, J. Y. Wang, C. C. Berndt, R. T. Jones, I. I. Yusuf, I. Peake, H. W. Schmidt, C. Fluke, D. Barnes and R. J. Crawford, Impact of nanoscale roughness of titanium thin film surfaces on bacterial retention, Langmuir, 2010, 26(3), 1973–1982 CrossRef CAS PubMed.
- K. Richter, G. Diaconu, K. Rischka, M. Amkreutz, F. Muller and A. Hartwig, Adsorption studies of mussel-inspired peptides, Bioinspired, Biomimetic Nanobiomater., 2013, 2(1), 45–53 CrossRef CAS.
- C. Beloin, A. Roux and J. Guigo, Escherichia coli biofilms, Curr. Top. Microbiol. Immunol., 2008, 322, 249–289 CAS.
- Z. Ye, X. Zhu, S. Acosta, D. Kumar, T. Sang and C. Aparicio, Self-assembly dynamics and antimicrobial activity of all L- and D-amino acid enantiomers of a designer peptide, Nanoscale, 2018, 11(1), 266–275 RSC.
- N. Greenfield, Using circular dichroism spectra to estimate protein secondary structure, Nat. Protoc., 2006, 1(6), 2876–2890 CrossRef CAS PubMed.
- L. Lombardi, A. Falanga, V. Del Genio and S. Galdiero, A new hope: self-assembling peptides with antimicrobial activity, Pharmaceutics, 2019, 11(4), 1–17 CrossRef PubMed.
- X. Han, J. Uzarski, C. Mello and Z. Chen, Different interfacial behaviors of N- and C-terminus cysteine-modified cecropin P1 chemically immobilized onto polymer surface, Langmuir, 2013, 29(37), 11705–11712 CrossRef CAS PubMed.
- X. Han, Y. Liu, F. Wu, J. Jansensky, T. Kim, z. Wang, c. Brooks, j. Wu, c. Xi, c. Mello and Z. Chen, Different interfacial behaviors of peptides chemically immobilized on surfaces with different linker lengths and via different termini, J. Phys. Chem. B, 2014, 118(11), 2904–2912 CrossRef CAS PubMed.
- Y. Li, S. Wei, J. Wu, J. Jasensky, L. Honglin, Y. Xu, Q. Wang, N. Marsh, C. Brooks and Z. Chen, Effects of peptide immobilization sites on the structure and activity of surface-tethered antimicrobial peptides, J. Phys. Chem. C, 2015, 119(13), 7146–7155 CrossRef CAS.
- X. T. Mai, J. Huang, J. Tan, Y. Huang and Y. Chen, Effects and mechanisms of the secondary structure on the antimicrobial activity and specificity of antimicrobial peptides, J. Pept. Sci., 2015, 21(7), 561–568 CrossRef CAS PubMed.
- J. Li, J. J. Koh, S. Liu, R. Lakshminarayanan, C. S. Verma and R. W. Beuerman, Membrane Active antimicrobial peptides: translating mechanistic insights to design, Front. Neurosci., 2017, 11, 73 Search PubMed.
- Y. Shai, Mode of action of membrane active antimicrobial peptide, Biopolym. Pept. Sci., 2002, 66, 236–248 CrossRef CAS.
- R. B. M. Zasloff, Antimicrobial peptides of multicellular, Nature, 2002, 24(415), 389–395 CrossRef PubMed.
- A. Strömstedt, L. Ringstad, A. Schmidtchen and M. Malmsten, Interaction between amphiphilic peptides and phospholipid membranes, Curr. Opin. Colloid Interface Sci., 2010, 10, 467–478 CrossRef.
- K. Rapsch, F. Bier, M. Tadros and M. von Nickisch-Rosenegk, Identification of antimicrobial peptides and immobilization strategy suitable for a covalent surface coating with biocompatible properties, Bioconjugate Chem., 2014, 25(2), 308–319 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07380a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Ziani Souza-Schiaberc,
Paulo Noronha Lisboa-Filho
*ab,
Ziani Souza-Schiaberc,
Paulo Noronha Lisboa-Filho