
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new Ru(II) polypyridyl complex as an efficient photosensitizer for enhancing the visible-light-driven photocatalytic activity of a TiO2/reduced graphene oxide nanocomposite for the degradation of atrazine: DFT and mechanism insights†
Amir Hossein Cheshme Khavara,
Gholamreza Moussavi *b,
Kamyar Yaghmaeianc,
Ali Reza Mahjoub*d,
Neda Khedrid,
Michal Duseke,
Tereza Vaclavue and
Mahdieh Hosseinid
*b,
Kamyar Yaghmaeianc,
Ali Reza Mahjoub*d,
Neda Khedrid,
Michal Duseke,
Tereza Vaclavue and
Mahdieh Hosseinid
aDepartment of Basic Science, Farhangian University, Tehran, Iran
bDepartment of Environmental Health Engineering, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran. E-mail: moussavi@modares.ac.ir
cDepartment of Environmental Health Engineering, School of Public Health, Tehran University of Medical Sciences, Tehran, Iran
dDepartment of Chemistry, Faculty of Basic Sciences, Tarbiat Modares University, Tehran, Iran
eInstitute of Physics ASCR, Na Slovance 2, 182 21 Prague, Czech Republic
First published on 16th June 2020
Abstract
TiO2 is one of the most widely used semiconductors for photocatalytic reactions. However, its wide bandgap energy and fast charge recombination limit its catalytic activity. Thus, herein, a new Ru(II) polypyridyl complex, [RuII(tptz)(CH3CN)Cl2] (tptz = 2,4,6-tris(2-pyridyl)-1,3,5-triazine), was synthesized and used as a visible-light photosensitizer dye for improving the light harvesting and quantum efficiency of TiO2. Accordingly, a well-designed nanostructured photocatalyst was proposed using mesoporous TiO2 nanocrystals coupled with reduced graphene oxide (rGO) and the polypyridyl Ru(II) complex, which was tested for the photocatalytic degradation of atrazine (ATZ) as a model of emerging water contaminants. Specifically, the Ru complex (Ru-CMP) served as an electron donor, while rGO acted as an electron acceptor, and the synergistic effect between them promoted the separation of electron–hole pairs and suppressed the charge recombination in the hybridized species. Structural analysis indicated that the TiO2 nanoparticles with an anatase crystal structure had a mesoporous texture and were homogeneously coated on the rGO sheets. The detailed FT-IR, Raman, XPS and UV-vis absorption spectroscopic analyses combined with EDS mapping clearly confirmed the successful loading of the Ru complex onto the catalyst. The PL and EIS results revealed that the addition of the Ru-CMP photosensitizer enhanced the charge separation and transport. The gas-phase geometry and energies of the molecular orbitals of the Ru complex were evaluated via DFT calculations. The results from the DFT calculations were consistent with the experimental results. Compared to pure TiO2, the as-synthesized Ru-CMP-TiO2/rGO hybrid exhibited significantly enhanced photocatalytic activity for the degradation of ATZ. The rate of ATZ degradation in the developed photocatalytic process with the Ru-CMP-TiO2/rGO hybrid was 9 times that with commercial TiO2. The enhanced photocatalytic activity of the prepared catalyst can be attributed to its better light harvesting and efficient electron transportation due to its more suitable LUMO position than the conduction band of TiO2. Moreover, the excellent conductivity and adsorption capacity of graphene contributed to the increase in photocatalytic activity. Thus, these features make the Ru-CMP-TiO2/rGO hybrid nanomaterial an excellent candidate for the photocatalytic purification of contaminated water.
1. Introduction
Atrazine (ATZ) is a widely used pesticide for the control of broadleaf and grassy weeds in both agricultural and nonagricultural land.1 Its worldwide usage has made it a typical soil and water contaminant, which has harmful effects on human health including eye and skin irritation, endocrine disruption, cancer and reproductive defects.1–3 Previous reports have confirmed the presence of ATZ in surface and groundwater.2,3 Moreover, due to its low sorption affinity and resistance to biodegradation, it is considered an environmental concern.1 In recent decades, the heterogeneous photocatalytic oxidation (PCO) process has shown great potential for the decomposition of pesticides into less toxic products.4 In the conventional PCO process, reactive oxygen species (ROS) are produced through the irradiation of a semiconductor using UV light, which generate electron–hole pairs.4 The unique properties of TiO2 including high stability, high photoactivity, and non-toxicity make it an unrivaled semiconductor for PCO reactions.4–6 However, TiO2 suffers from the disadvantages of wide band gap energy, high electron–hole recombination rate and low quantum efficiency, which limit its catalytic activity.7 Therefore, modification of the electronic structure of TiO2 to enhance its light harvesting ability, quantum efficiency and charge carrier separation is necessary. To date, most modification efforts have focused on two strategies: doping TiO2 with metals and/or nonmetals8,9 and/or combining TiO2 with other semiconductors.10,11 However, these strategies mainly address one of the challenges, while the others remain unresolved. For instance, doping TiO2 with transition metals is an effective way to extend its absorption edge towards the visible region; however, this approach enhances the recombination rate of photoexcited electron–hole pairs.12 On the other hand, doping TiO2 with some metals and non-metals efficiently improves its charge carrier separation, but the shift in its band gap energy toward visible light is small.13 In addition, the solubility of coupled semiconductors in water and their stability are major disadvantages. Furthermore, sometimes the coupled semiconductors undergo photo-corrosion and thus, their photocatalytic activity is negatively affected. Accordingly, the use of metal-based complex photosensitizers may be an impressive approach to improve the catalytic activity of TiO2. In this strategy, the absorption range of TiO2 is extended to the visible region via the absorption of the narrow-band gap sensitizer material on the catalyst. Moreover, the photosensitizer can effectively reduce charge recombination. Under visible light illumination, the light absorption by metal complexes as photosensitizers excites electrons from the ground state (HOMO) to the excited state (LUMO). If the potential of the LUMO is more negative than the conduction band of the TiO2 semiconductor, the excited electrons can be injected from the excited state of the sensitizer to the conduction band (CB) of TiO2. Therefore, an efficient photosensitizer should be photo-stable, absorb a wide range of light, and possess suitable positioned excited- and ground-state potentials for effective electron transfer to the conduction band of TiO2.14 Transition metal complexes are promising candidates as photosensitizers because of their unique long term stability,15 attractive photophysical and photochemical properties,16 suitable excited state lifetime17 and wide absorption range.18 However, the recombination of injected electrons in TiO2 with oxidized sensitizers may still occur in these systems.19 Accordingly, an electron acceptor support may be a solution to this issue. Graphene oxide (GO) is known as a powerful electron sink material.4 The specific characteristics of GO such as easy chemical functionalization, mechanical flexibility and extreme charge carrier mobility make it an unrivaled electric shuttle.20–22 Furthermore, the reduction of GO (rGO) improves the mobility of electrons on the graphene-like layer.23 Therefore the combination of TiO2 with rGO has been considered an efficient way to improve its quantum efficiency, and thus catalytic activity.7 In previous studies, rGO was used as an effective agent for the separation of electron–hole pairs.4–6 Moreover, its excellent surface area could significantly increase the reaction sites of catalysts for photocatalytic reactions.7Accordingly, herein, the synthesis of a novel Ru(II) polypyridyl complex, [Ru(tptz)(ACN)Cl2] (tptz = 2,4,6-tris(2-pyridyl)-1,3,5-triazine, ACN = CH3CN), as a photosensitizer dye is reported for sensitizing TiO2 nanoparticles supported on rGO under visible light irradiation. The main goal of sensitizing TiO2 with an organic or inorganic dye is improving its light harvesting potential. Therefore, many efforts have been paid for enhancing the absorptivity and stability of dyes.24 Ru polypyridine complexes have shown significant performances owing to their strong absorption in the visible region and long-lived charge-separated excited states.24 Additionally, ruthenium(II) polypyridyl complexes display improved energy transfer properties due to their strong metal-to-ligand charge transfer (MLCT) absorption bands in the visible region.19 The synthesized complex exhibited excellent light harvesting ability, which resulted in a significant red shift in the band gap energy of the nanocomposite. A facile method was used for the preparation of the [Ru(tptz)(ACN)Cl2]–TiO2/rGO nanocomposite under ambient conditions. The synthesized complex was characterized via single crystal X-ray crystallography, and electrochemical and spectroscopic methods. In addition, the prepared nanocomposite was optimized and fully characterized, and its catalytic activity under visible light irradiation was investigated for the degradation of ATZ. The mechanism for the degradation of ATZ in the developed process was evaluated in the presence of various scavengers.
2. Materials and methods
2.1. Materials
All chemicals were of analytical grade, purchased from Merck Co. and used without further purification. Deionized water (DW) was used for the preparation of all solutions in the experiments.2.2. Catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 was used for the elution. The applied purification system achieved a violet fraction of the pure complex, which was dried at room temperature and recrystallized via slow evaporation. After 14 days, single crystals of [Ru(tptz)(ACN)Cl2] were formed, which were significantly stable and suitable for X-ray crystallography.:1, followed by sonication for 30 min to form a GO solution. 600 mg of TiO2 nanoparticles was homogeneously dispersed in 90 mL DW/ethanol solution under sonication and magnetic stirring for 2 h. Then, GO solution was added dropwise to the TiO2 dispersion and the suspension was stirred at 1000 rpm for 30 min, followed by sonication for 30 min. The stirring/sonication step was repeated twice. Finally, the prepared mixture was poured into a 120 mL Teflon-lined autoclave and heated at 130 °C for 3 h. The final product was centrifuged, washed 5 times with DW and finally dried at 60 °C overnight. The TiO2/rGO nanocomposite with x wt% of GO was labeled as TGx.
:2 was used for the elution. The applied purification system achieved a violet fraction of the pure complex, which was dried at room temperature and recrystallized via slow evaporation. After 14 days, single crystals of [Ru(tptz)(ACN)Cl2] were formed, which were significantly stable and suitable for X-ray crystallography.:1, followed by sonication for 30 min to form a GO solution. 600 mg of TiO2 nanoparticles was homogeneously dispersed in 90 mL DW/ethanol solution under sonication and magnetic stirring for 2 h. Then, GO solution was added dropwise to the TiO2 dispersion and the suspension was stirred at 1000 rpm for 30 min, followed by sonication for 30 min. The stirring/sonication step was repeated twice. Finally, the prepared mixture was poured into a 120 mL Teflon-lined autoclave and heated at 130 °C for 3 h. The final product was centrifuged, washed 5 times with DW and finally dried at 60 °C overnight. The TiO2/rGO nanocomposite with x wt% of GO was labeled as TGx.2.3. Catalyst characterization
The prepared catalysts were characterized using specific analytical methods. X-ray diffraction (XRD) measurements were performed using a Philips X'pert diffractometer with monochromated Cu-Kα radiation (λ = 1.54056 Å). The surface morphology of the products was investigated using scanning electron microscopy (SEM, XL30 Philips) and high-resolution transmission electron microscopy (HRTEM) with a Philips CM30 instrument. Fourier transform infrared (FT-IR) spectra were recorded on a Shimadzu-8400S spectrometer in the wavenumber range of 400–4000 cm−1 using KBr pellets. An Almega Thermo Nicolet Dispersive Raman spectrometer (laser second harmonic @ 532 nm of an Nd:YLF laser) was employed to measure the Raman spectra of the samples. Diffuse reflectance spectroscopy (DRS) was performed using a Shimadzu-UV-2550-8030 spectrophotometer. The specific surface area, volume, and pore size of the prepared catalysts were determined from the N2 adsorption/desorption data obtained using a Micromeritic/Gemini-2372 surface area analyzer. Electrochemical impedance spectroscopy (EIS) measurements were conducted using Na2SO4 (0.3 M) in the frequency range of 0.1–106 Hz and amplitude of 0.01 V.2.4. X-ray crystallographic analysis
Crystallographic measurements were performed at 95 K with a four-circle CCD diffractometer SuperNova, Dual, Cu at home/near, AtlasS2, with graphite-monochromated Cu-Kα radiation (λ = 1.54184 Å). The crystal structures were solved by direct methods with the SIR 2002 program25 and refined with the Jana 2006 program package26 by the full-matrix least-squares technique on F2. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were added at the calculated positions and then refined using a riding model based on the parent atom. The isotropic atomic displacement parameters of the hydrogen atoms were evaluated as 1.2–1.5 Ueq of the parent atom. Structural artworks were drawn with MERCURY.27 The CIF files were deposited with the CCDC and given the deposition number 1923285.2.5. Computational
The Gaussian 09 program package55 was used to perform the calculations. The molecular geometry and frequency calculations were carried out using density functional theory (DFT) by employing the B3PW91 functional and the SDDall28,29 basis set for all atoms in the gas phase. The shapes and energies of the molecular orbitals, including the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), were determined using the calculated optimized structure of the complex at the same level of theory.2.6. Photocatalytic experiments
A batch-operated slurry glass circular photoreactor with an inner diameter of 10 cm and a total volume of 100 mL was used for the photocatalytic experiments. A quartz sheet was placed above the reactor at a distance of 1 cm from the top of the solution. The setup was placed in a box to prevent the influence of external light. The reactor was equipped with a water-cooling jacket, and illumination was provided by a 350 W (15 A) xenon lamp without any cut filter. The lamp was placed 20 cm from the surface of the reaction mixture. The photocatalytic activity of the prepared samples was evaluated via the photodegradation of ATZ under UV-vis light irradiation. In the photocatalysis test, 25 mg of catalyst was dispersed in 25 mL of ATZ aqueous solution having a known ATZ concentration and pH. The reaction solution was stirred for a predetermined time under UV-vis light irradiation. After the set reaction time, 3 mL of the suspension was taken and centrifuged (5000 rpm for 5 min) immediately and the filtrate was analyzed for residual ATZ and total organic carbon (TOC) (see ESI†). In addition, control experiments were also conducted in the dark (lamp was switched off) and in the absence of the catalyst. Commercial TiO2 (P25, Sigma Aldrich Co.) was also tested as a reference catalyst under the same conditions. The kinetics for the degradation and mineralization of ATZ was also determined. The main oxidizing species involved in the degradation of ATZ was investigated by performing the degradation process in the presence of various radical scavengers.3. Results and discussion
3.1. Catalyst characteristics
Single crystals of the [Ru(tptz)(ACN)Cl2] complex were obtained by slow evaporation at room temperature with good quality. The crystallographic data of this complex and its geometric parameters are summarized in Tables S1 and S2,† respectively. The X-ray crystallographic analysis revealed that this complex crystallizes in the triclinic crystal system and P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with Z = 2. Its molecular structure and crystal packing diagrams are shown in Fig. 1 and 2, respectively. In this structure, Ru is six-coordinated, in which the tptz ligand coordinates to the Ru atom in a tridentate-chelating mode. The coordination sphere around the ruthenium center can be described as octahedral with the nitrogen atoms of the tptz ligand [N1 (s-triazine), N6 (pyridine), and N7 (pyridine)] and acetonitrile [N(4)] forming the equatorial plane, while the two chloride atoms occupy the apical sites. The asymmetric unit of this complex consists of one Ru(II) metal center, one tptz ligand, two chloride anions, and one coordinated acetonitrile ligand. The Ru–N bond distances are in the range of 1.923(18)–2.062(18) Å and the Ru–Cl bond distances are 2.379(6) and 2.398(7) Å. The bond angles around the Ru center fall in the range of 78.3(7)–178.1(8)°. The relevant geometric parameters are listed in Table S2.†
space group with Z = 2. Its molecular structure and crystal packing diagrams are shown in Fig. 1 and 2, respectively. In this structure, Ru is six-coordinated, in which the tptz ligand coordinates to the Ru atom in a tridentate-chelating mode. The coordination sphere around the ruthenium center can be described as octahedral with the nitrogen atoms of the tptz ligand [N1 (s-triazine), N6 (pyridine), and N7 (pyridine)] and acetonitrile [N(4)] forming the equatorial plane, while the two chloride atoms occupy the apical sites. The asymmetric unit of this complex consists of one Ru(II) metal center, one tptz ligand, two chloride anions, and one coordinated acetonitrile ligand. The Ru–N bond distances are in the range of 1.923(18)–2.062(18) Å and the Ru–Cl bond distances are 2.379(6) and 2.398(7) Å. The bond angles around the Ru center fall in the range of 78.3(7)–178.1(8)°. The relevant geometric parameters are listed in Table S2.†
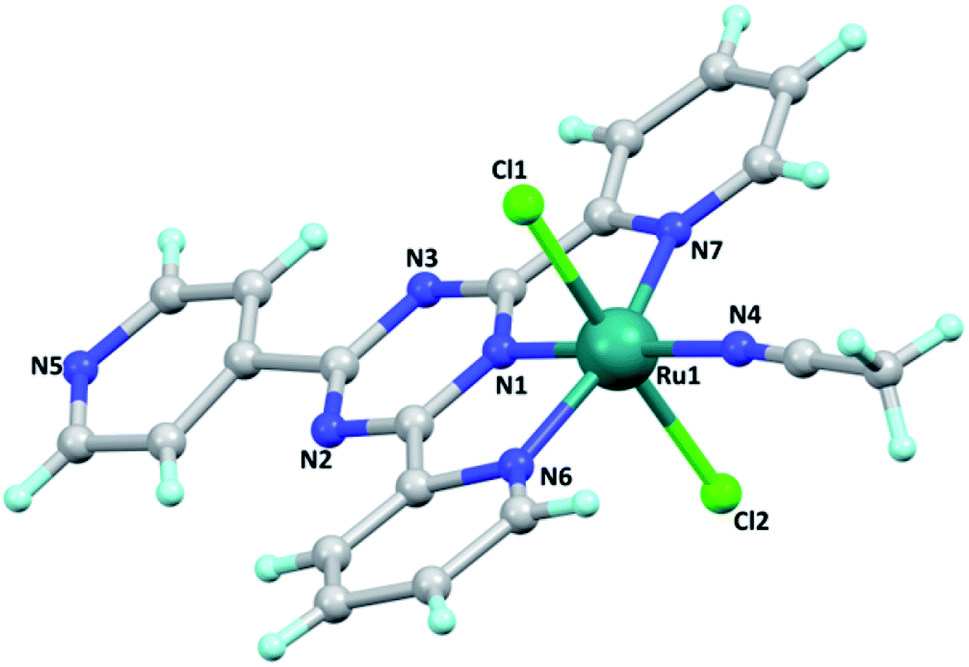 | ||
| Fig. 1 Ball and stick diagram of the molecular structure of [Ru(tptz)(ACN)Cl2]. | ||
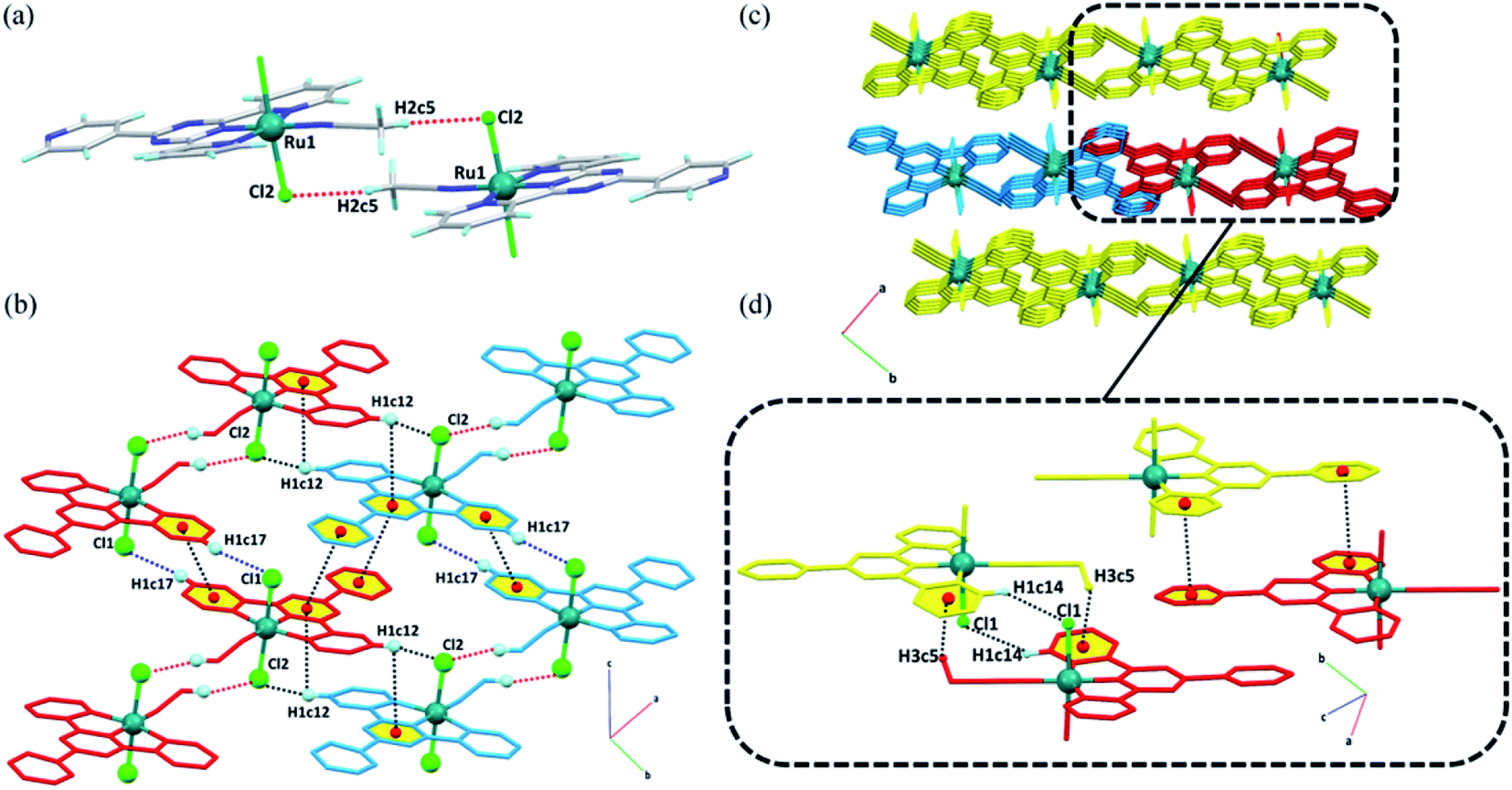 | ||
| Fig. 2 Crystal structure of [Ru(tptz)(ACN)Cl2]: (a) representation of its dimeric units, (b) association of dimers through hydrogen bonds and π–π interactions, creating a chain along the c-axis and joining the molecular chains in the bc-plane, (c) the three-dimensional supramolecular structure of the complex, and (d) intermolecular interactions linking the adjacent chains in the ac-plane (non-interacting hydrogen atoms have been omitted for clarity). Different colors represent different molecular chains. | ||
For the [Ru(tptz)(ACN)Cl2] complex in the solid state, each molecule is joined to an adjacent molecule by the head-to-tail non-classical hydrogen bond C5–H2c5⋯Cl2 to form dimeric molecular units (Fig. 2a). These dimers are further associated with each other through C17–H1c17⋯Cl1 hydrogen bonds and πpy–πpy interactions to generate molecular chains along the c-direction (Fig. 2b). The chains are laterally connected via several intermolecular interactions to form a 3D network (Fig. 2c). In the bc-plane, the chains are held together by hydrogen bonds (C12–H1c12⋯Cl2) and π stacking contacts (πpy–πs-triazine and C12–H1c12⋯πs-triazine), as shown in Fig. 2b. The molecular chains are firmly attached along the a-direction through a C14–H1c14⋯Cl1 hydrogen bond, C5–H3c5⋯πpy, and another πpy–πpy interaction (Fig. 2d). A summary of the parameters for the interactions mentioned above is presented in Tables S3 and S4.†
The crystal structure of the TiO2, GO, TiO2@rGO and Ru-CMP-TiO2/rGO samples were investigated using powder X-ray diffraction (PXRD), and the results are shown in Fig. 3. The as-synthesized GO shows the characteristic (002) peak at 2θ = 10.9°. For the other samples, all the diffraction peaks can be well indexed to the photocatalytically active anatase TiO2 (JCPDS No. 04-016-2837), which suggests that the synthesized Ru-CMP-TiO2/rGO hybrid still preserved the anatase crystal structure. The characteristic peaks at 29.6°, 44.2°, 56.4°, 63.7°, 64.8°, and 74.5° correspond to the crystal planes of (101), (004), (200), (105), (211) and (204) of anatase TiO2. There is no specific diffraction peak related to GO or the Ru-complex in the XRD pattern of Ru-CMP-TiO2/rGO, which is probably due to the low amount of these materials in the nanocomposite. Furthermore, no systematic peak position shift was observed upon sensitizing TiO2 with the [Ru(tptz)(ACN)Cl2] dye.
 | ||
| Fig. 3 XRD patterns of the prepared samples. (a) TiO2, (b) TiO2@rGO and (c) Ru-CMP-TiO2/rGO. | ||
The specific surface area of the synthesized TiO2 and Ru-CMP-TiO2/rGO samples was evaluated by measuring their nitrogen adsorption/desorption isotherms, as plotted in Fig. 4a. The isotherms of the samples can be classified as type IV, which is characteristic of mesoporous solid materials. The Ru-CMP-TiO2/rGO and TiO2 samples exhibit adsorption/desorption isotherms with H3-type and H4-type hysteresis loops at high P/P0 values, respectively. In the H3 hysteresis loop, the pores have a wedge or slit geometry, resulting from agglomerates of parallel plate-shaped particles.30 The H4-type hysteresis loop is related to pores with the same geometry as the H3 type, but with smaller diameters,30 which indicates the decorated TiO2 nanoparticles on the graphene sheets acted as a spacer to partially prevent the aggregation of the graphene sheets, resulting in a structure with specific macropores. The as-synthesized TiO2 had a BET surface area of 74.5 m2 g−1, which is related to its mesoporous structure. The BET surface area of the Ru-CMP-TiO2/rGO hybrid was 113.8 m2 g−1, which was predictable due to the high inherent surface area of GO (2600 m2 g−1).
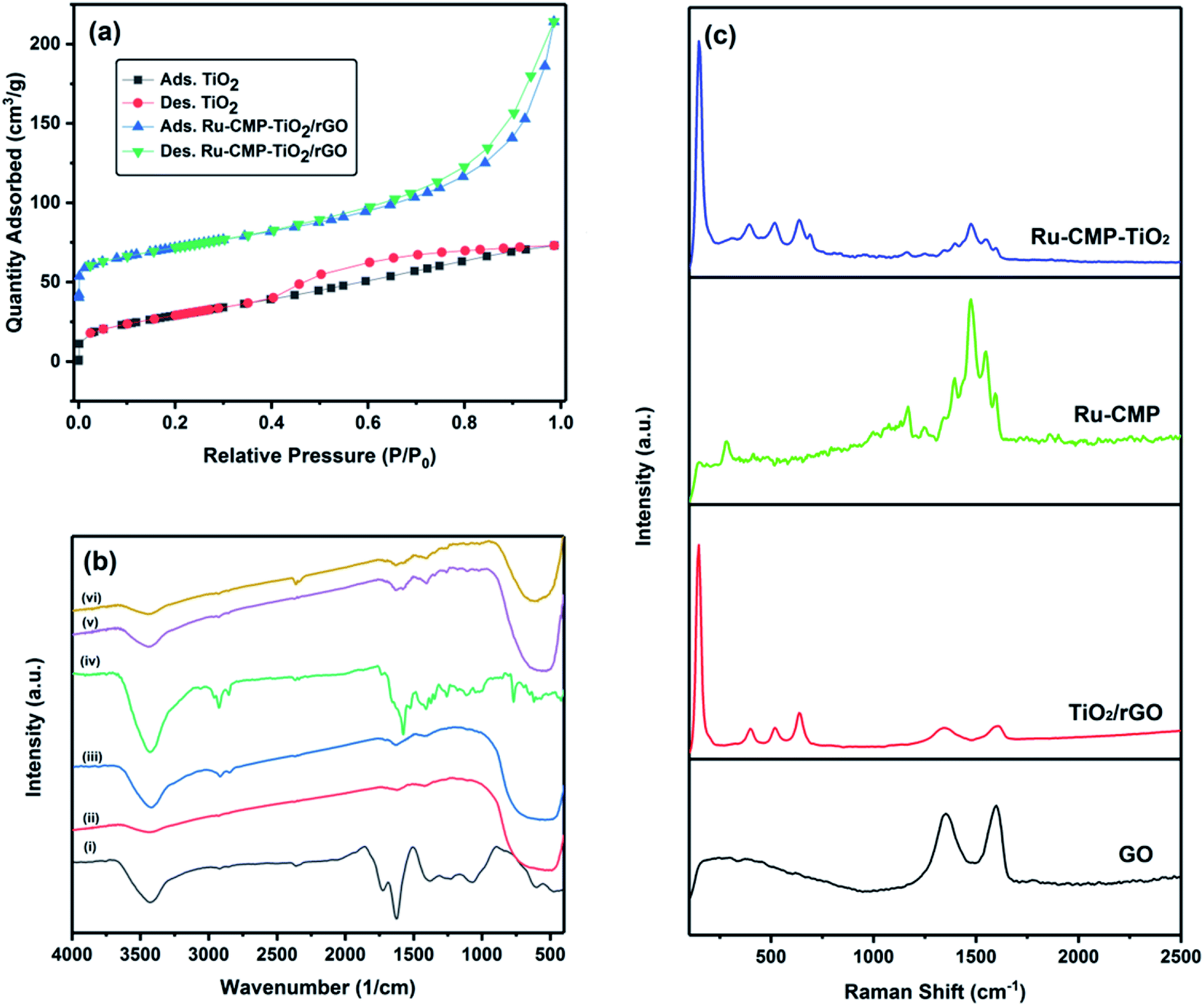 | ||
| Fig. 4 (a) Nitrogen adsorption and desorption isotherms of the TiO2 and Ru-CMP-TiO2/rGO samples. (b) FT-IR spectra of the synthesized (i) GO, (ii) TiO2, (iii) TiO2/rGO, (iv) Ru-CMP, (v) Ru-CMP-TiO2 and (vi) Ru-CMP-TiO2/rGO samples. (c) Raman spectra of GO, TiO2, Ru-CMP and Ru-CMP-TiO2. | ||
The FT-IR spectra of the GO, TiO2, TiO2/rGO, [Ru(tptz)(ACN)Cl2], Ru-CMP-TiO2 and Ru-CMP-TiO2/rGO samples are presented in Fig. 4b. For pure TiO2, three main peaks can be clearly observed. The broad absorption peak in the range of 3300–3500 cm−1 is related to the stretching vibration of the hydroxyl (–OH) group of physisorbed water on the surface of TiO2, the weak signal at 1636 cm−1 is attributed to the O–H bending modes of water molecules and the strong band below 800 cm−1 corresponds to the Ti–O and Ti–O–Ti bonds in the TiO2 lattice. Specifically the stretching vibration of the Ti–O bonds appear at higher wavenumbers and that of the Ti–O–Ti bonds at lower wavenumbers.7 The FT-IR spectrum of the as-prepared GO displayed absorption peaks at 3343 cm−1, 2925 and 2845 cm−1, 1730 cm−1, 1615 cm−1, 1370 cm−1, 1220 cm−1 and 1045 cm−1, corresponding to the stretching vibration of the hydroxyl (–OH) group, CH2 stretching vibration, stretching vibration of oxygen-containing functional groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), CC stretching vibration, C–O–C deformation vibration, epoxy (C–O) and (C–OH) of the residual COOH groups, respectively.31 These oxygen-containing functional groups are crucial for the immobilization of titania nanoparticles.32 As seen in the FT-IR spectrum of the TiO2/rGO nanocomposite, the absorption bands of most of the oxygen-containing functional groups disappeared. However, this sample showed a CO stretching band with a significantly lower intensity, implying that the initial GO was reduced to graphene in the presence of ethanol.33 The FT-IR spectrum of the [Ru(tptz)(ACN)Cl2] complex exhibits a strong signal at 1576 cm−1 with a shoulder at 1656 cm−1, which is related to the stretching vibration of CN and ν(CN) vibration of the triazine ring. Moreover, the absorption peaks at 1410 cm−1 and 1374 cm−1 are attributed to the ν(CC) vibrations of the pyridyl rings. The weak signals at 2344 cm−1 and 577 cm−1 correspond to the stretching vibration of C
O), CC stretching vibration, C–O–C deformation vibration, epoxy (C–O) and (C–OH) of the residual COOH groups, respectively.31 These oxygen-containing functional groups are crucial for the immobilization of titania nanoparticles.32 As seen in the FT-IR spectrum of the TiO2/rGO nanocomposite, the absorption bands of most of the oxygen-containing functional groups disappeared. However, this sample showed a CO stretching band with a significantly lower intensity, implying that the initial GO was reduced to graphene in the presence of ethanol.33 The FT-IR spectrum of the [Ru(tptz)(ACN)Cl2] complex exhibits a strong signal at 1576 cm−1 with a shoulder at 1656 cm−1, which is related to the stretching vibration of CN and ν(CN) vibration of the triazine ring. Moreover, the absorption peaks at 1410 cm−1 and 1374 cm−1 are attributed to the ν(CC) vibrations of the pyridyl rings. The weak signals at 2344 cm−1 and 577 cm−1 correspond to the stretching vibration of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N of acetonitrile and Ru–Cl bond, respectively.34 For the Ru-CMP-TiO2 sample, the absorption peaks located at around 1323–1628 cm−1 are attributed to the stretching vibration of CN and CN, and stretching vibrations of the pyridyl and triazine rings of the attached Ru-complex. Moreover, the absorption band related to the CN band of acetonitrile was also observed in this spectrum. The intense peak below 800 cm−1 is associated with the Ti–O band. FT-IR spectra of the Ru-CMP-TiO2/rGO hybrid showed a similar shape. Thus, the FT-IR results clearly confirm the reduction of GO into rGO and attachment of Ru-CMP on the TiO2/rGO nanocomposite.
N of acetonitrile and Ru–Cl bond, respectively.34 For the Ru-CMP-TiO2 sample, the absorption peaks located at around 1323–1628 cm−1 are attributed to the stretching vibration of CN and CN, and stretching vibrations of the pyridyl and triazine rings of the attached Ru-complex. Moreover, the absorption band related to the CN band of acetonitrile was also observed in this spectrum. The intense peak below 800 cm−1 is associated with the Ti–O band. FT-IR spectra of the Ru-CMP-TiO2/rGO hybrid showed a similar shape. Thus, the FT-IR results clearly confirm the reduction of GO into rGO and attachment of Ru-CMP on the TiO2/rGO nanocomposite.
To further evaluate the chemical structure of the as-prepared catalyst, Raman spectroscopy was employed (Fig. 4c), which is used as a powerful nondestructive tool to elucidate the carbon structure in composite networks. As observed in Fig. 2c, the two Raman shifts at 1353 and 1599 cm−1 correspond to the D (for vacancies, disorders and edge defects) and G (for sp2 carbon atoms) bands, respectively.35,36 Compared to GO, the D band and G band for the TiO2/rGO nanocomposite shifted to 1342 cm−1 and 1604 cm−1, respectively, indicating the successful reduction of GO. In addition, the D/G intensity (ID/IG) ratio increased from 0.86 for GO to 0.97 for the TiO2/rGO nanocomposite, which further confirmed that the reduction of GO occurred during the annealing process. The broader D bands for the TiO2/rGO nanocomposite indicate a reduction in the average size of the sp2 hybridized carbon and oxygen-functional groups on GO and the generation of more defect sites during the annealing process.7 The TiO2/rGO nanocomposite presented characteristic anatase features at 182 cm−1, 395 cm−1, 512 cm−1 and 633 cm−1, corresponding to the Eg, B1g, and A1g and Eg modes, respectively, with no distinctive features for the rutile phase, which is in accordance with the XRD results. To further characterize the attachment of the Ru-complex on TiO2, the Raman spectra of Ru-complex and Ru-CMP-TiO2 were measured, and the changes before and after complex loading were monitored. The backbone peaks in the range of 1250–1600 cm−1 can be assigned to modes of the C–C and CN stretching vibration in the polypyridine skeleton, while the peaks at 1163 and 1047 cm−1 are related to the C–H bending vibration of the ligand.37,38 The weak band at 290 cm−1 can be attributed to the Ru–N stretching mode of the polypyridine complex.37 Moreover, the signal at 1620 cm−1 is related to the N–Ru–N in-plane bending with respect to the connected pyridine group.45 Compared with the Raman spectra of the pure [Ru(tptz)(ACN)Cl2] and TiO2/rGO samples, all the vibration modes of TiO2 and [Ru(tptz)(ACN)Cl2] are observed for the [Ru(tptz)(ACN)Cl2]–TiO2 hybrid, which further confirms that [Ru(tptz)(ACN)Cl2] was successfully loaded on TiO2.
To obtain further information on the chemical composition and valence state of the elements, energy dispersive X-ray (EDX) mapping and XPS of Ru-CMP-TiO2/rGO were carried out and the results are shown in Fig. S1† and 5, respectively. The elemental mapping for Ru-CMP-TiO2/rGO for titanium, ruthenium, carbon, and nitrogen can be observed in Fig. S1,† which confirms the presence of these elements with a uniform distribution in the prepared catalyst. The XPS and EDX results are in good agreement, as confirmed by the presence of peaks related to Ti, O, C, N, and Ru in the high-resolution XPS spectra. The peak located at 284.6 is related to the C 1s core-level spectrum of GO. This peak could be devolved into three contributions, corresponding to C–C, CC/C–H of the sp2 carbon of rGO, C–O and CO.6 The weak signal for the oxygen-containing functional groups suggests the reduction of GO into rGO during the solvothermal process.39 The binding energies of O 1s centered at 529.8 eV could be deconvoluted into two main bands at 529.1 and 530.1, which are ascribed to O2− in the transition metal oxides and O–O, respectively.40 The signal located at 532.4 eV is attributed to the C–O bond.41 The Ti core-level XPS spectrum of TiO2 exhibits two signals at 458.6 and 464.1, which are related to the Ti 2p3/2 and Ti 2p1/2 spin–orbital splitting photoelectrons in the Ti4+ state, respectively.42 The adsorption of the Ru-complex was evidenced by the presence of N 1s and Ru 3d signals in the XPS spectrum. As shown in Fig. 5d, the binding energies of N 1s show 2 types of N bonding at 398.6 and 400.1, which are assigned to the sp2 hybridized nitrogen (CN–C) involved in the triazine rings and the nitrogen-center atom in the tri-s-triazine units, respectively.43 The peak centered at 278.5 eV is associated with Ru 3d5/2, confirming that ruthenium is in the Ru2+ oxidation state in the sample.44 The peak related to Ru 3d3/2 was not observed for this sample because the signal of C 1s core-level overlaps with this peak. The signal of Ru 3d5/2 appeared at a lower binding energy compared to that reported for Ru2+ (281.3 eV) in previous works.45 This is due to the electron transfer from the ligands to the central metal in the complex. In this process, the lone pairs provided by the ligands can transfer to Ru, resulting in an enhancement in the electron density on ruthenium. The increase in electron density in ruthenium leads to a decrease in the attraction for an extra nuclear electron, and therefore, a decrease in the binding energy of ruthenium compared to the normal binding energy of Ru2+.45
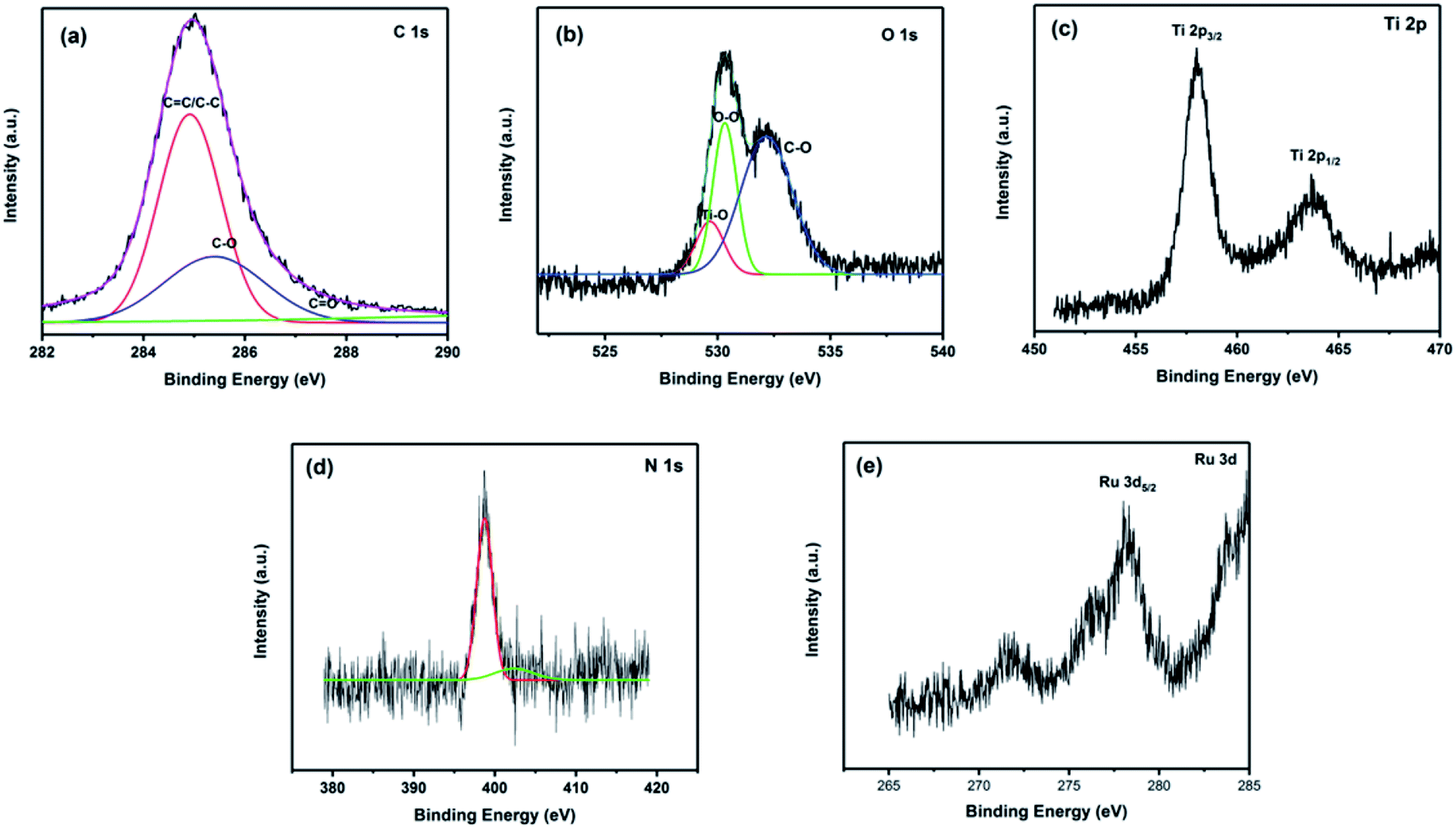 | ||
| Fig. 5 High-resolution XPS spectra of the Ru-CMP-TiO2/rGO sample: (a) C 1s, (b) O 1s, (c) Ti 2p, (d) N 1s and (e) Ru 3d. | ||
The detailed microstructures and morphologies of the prepared samples were evaluated by FESEM and TEM, as shown in Fig. 6. Fig. 6a illustrates the structure of the two-dimensional sheet for the as-prepared GO with silk veil waves, which is the typical morphology for this material. The length and width of the GO sheets are in the micrometer range. As seen in Fig. 6b, the TiO2 nanoparticles were found to be almost spherical in shape and uniform in size. As driven by the oxygen-containing functional groups on the surface of GO, the TiO2 nanoparticles were uniformly fixed on the GO shells. Fig. 6c and d show the SEM images of the TiO2/rGO nanocomposite under low and high magnification, respectively. Based on the low magnification image (Fig. 6c), the TiO2 nanoparticles were homogeneously dispersed on the surface of rGO. As can be seen in Fig. 6c, the layered structure of GO and the shape and size of the TiO2 nanoparticles remained unchanged after the solvothermal reaction. According to the high magnification SEM image (Fig. 6d), it can be clearly seen that the TiO2/rGO product was composed of three distinct parts, including a small part of bare rGO sheet, a trace amount of unconnected TiO2 and a major portion of TiO2 particles deposited on the rGO sheets. This rGO-connected TiO2 network plays a critical role in efficient electron collection during the photocatalytic process.4 It can be found in Fig. 6e that the loading of the Ru-complex had no remarkable effect on the microstructure of the TiO2@rGO nanocomposite. To obtain more detailed structural information, TEM imaging was carried out for the Ru-CMP-TiO2/rGO sample. The TEM image totally supports the SEM results, displaying that the TiO2 nanoparticles are well-dispersed and occupy most of the available surface of rGO and directly anchored to its surface.
 | ||
| Fig. 6 (a–e) SEM images of prepared samples: (a) GO, (b) TiO2, (c and d) TiO2/rGO, (e) Ru-CMP-TiO2/rGO and (f) TEM image of Ru-CMP-TiO2/rGO hybrid. | ||
The optical properties of the prepared samples were evaluated by UV-vis (Fig. 7a) and PL (Fig. 7b) spectroscopy at room temperature. The optical band gaps were estimated from the absorption data by plotting (αhν)1/2 against the photon energy, hν, (Fig. S2†). Based on Fig. 7a, pure TiO2 exhibited an absorption edge at around 389 nm, having a band gap energy of 3.23 eV. As can be seen in Fig. 7a, the TiO2/rGO sample possessed a similar absorption edge with a small shift toward longer wavelengths, which is related to the interaction between TiO2 and GO.31 The value of the conduction and valence band edge potential of a semiconductor can be predicted using the following formula:
| EVB = χ − Ee + 0.5Eg | (1) |
| ECB = EVB − Eg | (2) |
 | ||
| Fig. 7 (a) UV-vis spectra of the prepared photocatalysts. The inset shows the UV-vis spectrum of the [Ru(tptz)(ACN)Cl2] complex, (b) PL spectra of the synthesized materials, (c) Nyquist plots of the TiO2 and Ru-CMP-TiO2/rGO samples and (d) cyclic voltammograms of the [Ru(tptz)(ACN)Cl2] complex and Ru-CMP-TiO2/rGO hybrid. | ||
Therefore, the EVB and ECB values for TiO2 were calculated to be about +2.93 eV and −0.31 eV and that for the TiO2/rGO nanocomposite about +2.91 eV and −0.29 eV, respectively. According to the UV-vis absorption results, loading the Ru-complex into TiO2/rGO led to a significant shift in its absorption edge toward the visible region and this Ru-CMP-TiO2/rGO sample exhibited strong visible light absorption. The UV-vis spectrum of the complex was recorded in ethanol solution, as shown in the inset of Fig. 7a. This spectrum featured the most intense absorption bands in the UV region at around 219, 259, and 287 nm, arising from the intraligand n → π* and/or π → π* transitions for the tptz ligand. The complex also exhibits a broad absorption peak in the visible region at 450 nm due to the metal-to-ligand charge transfer from dπ (RuII) to the empty π* orbitals of the tptz ligand.46,47 As seen in Fig. 7a, the heterojunction between rGO and TiO2 did not have the capacity to change the electronic structure, while inserting the [Ru(tptz)(ACN)Cl2] sensitizer led to the absorption edge being extended toward the visible light region, which is ascribed to the photosensitizing effect of the Ru-complex. Indeed, the Ru-CMP-TiO2/rGO sample exhibited a broad absorption band at 450 nm, a fingerprint of the electron transfer from the d orbitals of the ruthenium atom to the empty π* orbitals of the tptz ligand, which further confirms the successful loading of the dye on the catalyst.
The separation and recombination of charge carriers play a critical role in the photocatalytic activity of semiconductors, and their photocatalytic performance is improved when charge recombination is prevented.48 Thus, to study the charge recombination process, the photoluminescence (PL) spectra of prepared samples were recorded, as shown in Fig. 7b. As observed in Fig. 7b, all the samples exhibit similar PL spectra, while, the intensity of their emission bands is different. It is assumed that quenching of the PL emission indicates a weaker electron–hole recombination rate and higher charge carrier lifetime.49,50 As shown in Fig. 7b, the insertion of GO in the composite reduced the emission intensity of the product in comparison with pure TiO2. GO with a two-dimensional π-conjugated structure is known as a competitive electron-accepting material, and thus it could inhibit the recombination of photogenerated electron–holes.51 Moreover, the results reveal that loading Ru-complex into the sample significantly reduced the PL peak intensity, which confirms that the ruthenium complex greatly improved the effective separation of photogenerated electron–hole pairs. Considering the CB band position of TiO2 and the LUMO of the complex, the efficient electron transportation at the heterojunction interface can induce a higher efficiency of electron–hole pair transport, more efficient charge separation and rapid interfacial charge transfer. Moreover, it is noted that the PL emission exhibits a red shift from 386 nm for TiO2 to 410 nm for the Ru-CMP-TiO2 hybrid due to the lower π* orbital of the tptz ligand.52
Electrochemical impedance spectroscopy was carried out for the TiO2 and Ru-CMP-TiO2/rGO samples at the charged state, and the corresponding Nyquist plots are shown in Fig. 7c. Both curves show similar characteristics. The results indicate the reduced charge transfer resistance at the solid-state interface after the introduction of rGO and addition of the Ru-complex. The electron accepting and transporting features of rGO and efficient charge transport due to Ru-complex can contribute to the suppression of charge recombination, thereby resulting in a high photocatalytic performance.
The redox behaviors of the Ru-complex and Ru-CMP-TiO2@rGO samples were investigated using cyclic voltammetry. The voltammograms were recorded in acetonitrile solution with 0.1 M TBAH as the supporting electrolyte and a glassy carbon working electrode under an Ar atmosphere at room temperature, as shown in Fig. 7d. Three distinction reduction peaks were observed in the voltammogram of the Ru-complex at −0.87, −1.12, and −1.50 V vs. Ag/AgCl. The irreversible peak at −0.87 V can be assigned to the Ru(II/I) couple. The couples at potentials below −1.00 V are attributed to two sequential reduction couples of the tptz ligand. The reduction of Ru(II) to Ru(I) led to a change in electron configuration from a d6 low spin with a very strong crystal field stabilization energy, CFSE, to d7 with a weaker CFSE. Therefore, it is expected that the reduction of Ru(II) to Ru(I) occurred at a negative potential and the oxidation of Ru(I) to Ru(II) occurred at a positive potential. Similar redox behavior was achieved for the Ru-complex after it was loaded in the TiO2/rGO nanocomposite, with a small shift toward less positive potentials.
DFT calculations were performed to optimize the configurations and electronic structure of the complex. Fig. 8 indicates the optimized structure of the Ru-complex, which is consistent with its crystal (solid-state) structures. The distribution and energy gap of the HOMO and LUMO are also shown in Fig. 8. As seen in Fig. 8, the HOMO of the complex, [Ru(tptz)(ACN)Cl2], possesses a mixed ligand/metal character of chlorine as the coordinated ligand and ruthenium as the central metal. In addition, the LUMO is localized on the π* orbitals of the tptz ligand and d-orbitals of Ru(II). The energies of HOMO and LUMO were calculated to be −5.12 eV and −3.21 eV vs. vacuum, which give the band gap energy of 1.91 eV. Considering the conduction band (CB) edge of TiO2 (−4.5 eV vs. vacuum) and the band gap energy of 3.2 eV, the prepared complex can act as an efficient visible light photosensitizer for TiO2. The prepared Ru-CMP with a narrow band gap energy can simply be photoexcited under visible light, and since the CB of TiO2 stays at a more negative potential than the LUMO of the Ru-complex, the photo-excited electrons from the LUMO of the complex can be injected with high efficiency into the conduction band of TiO2, which results in improved charge separation.
 | ||
| Fig. 8 Optimized structures of the complex and schematic representation of the calculated HOMO and LUMO. | ||
3.2. Photocatalytic activity of the prepared TGA catalysts
TiO2 has been widely studied as a photocatalyst for the degradation of various pollutants53,54 due to its inherent properties and high photocatalytic activity. However, some limitations such as its fast charge recombination and wide band gap (3.2 eV) make it inactive under visible light, hindering the widespread use of TiO2 for industrial applications.55,56 Hence, in this study, two general approaches were used to enhance the photocatalytic efficiency of TiO2, loading a trace amount of Ru-complex and decorating the catalyst on rGO sheets. The photocatalytic activity of the prepared TiO2/rGO nanocomposites with different TiO2 to GO ratios was investigated for the degradation of 20 mg L−1 ATZ in aqueous solution (pH = 5.4) in the PCO reaction with 1 g L−1 of catalyst under visible light irradiation for 60 min. Control experiments were also conducted in the absence of the catalyst (direct photolysis) and in the dark (adsorption) under similar conditions.Before the photo-induced degradation reaction, the ATZ solution containing the photocatalyst was stirred for 1 h in the dark to reach adsorption equilibrium, and the results are shown in Fig. 9a. It was found that ATZ adsorption increased with an increase in GO to TiO2 weight ratio due to the inherent specific surface area of GO (2600 m2 g−1) and the π–π conjugation accumulation effect between the rGO sheets and ATZ molecules. The photocatalytic performance of the nanocomposites was also evaluated under UV-vis irradiation. According to the control experiments, no ATZ degradation was observed in the absence of the catalyst (direct photolysis condition). A similar result was obtained when GO was used as a catalyst (not shown). Thus, GO does not function as a photocatalyst independently. It was found that all the TiO2/rGO nanocomposites possessed greater photocatalytic capacity than pure TiO2. The improved photocatalytic performance of the TiO2/rGO nanocomposites compared to that of pure TiO2 can be explained based on the various benefits provided by compositing TiO2 with GO, including better photogenerated electron–hole separation, increase in light harvesting and promoted reaction potential due to the enhanced reactant adsorbability.31 GO with a two-dimensional π-conjugated structure is a competitive acceptor material and acts as an electron sink for photogenerated electrons from the conductive band of TiO2. Therefore, decorating titania nanoparticles on rGO sheets provides the channels for electron transportation, leading to a decrease in the charge recombination rate.4 Reducing the recombination rate of photogenerated electron–hole pairs leads to more electrons and holes remaining available to interact with water molecules and the production of more active radical species, and thereby higher photocatalytic activity. It can be clearly seen in Fig. 9a that an increase in GO content to 3.0 wt% in the nanocomposite resulted in an enhancement of the removal efficiency (adsorption + photodegradation) to 41%. However, a further increase in the GO percentage caused a slight reduction in the photocatalytic performance of the catalyst. Although a higher amount of GO may adsorb a greater amount of ATZ, it can disadvantageously occupy some active sites of the titania nanoparticles, resulting in partial shielding of the photons arriving at TiO2, and thus reducing the photocatalytic activity and performance.7 Thus, based on the above discussion, the TiO2/rGO nanocomposites with 3.0 wt% of GO was used for evaluating the effect of sensitizing with the [Ru (tptz)(ACN)Cl2] complex.
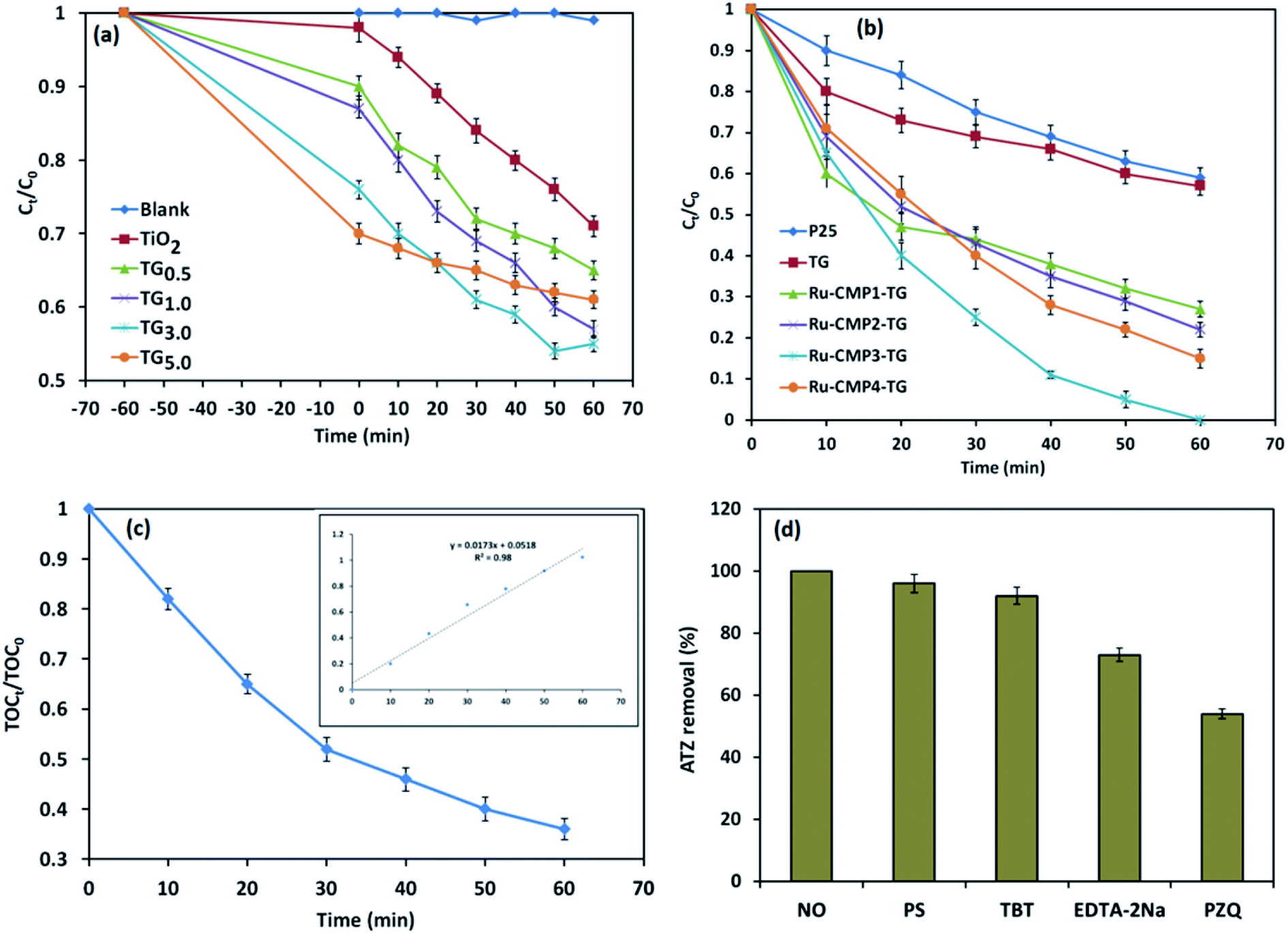 | ||
| Fig. 9 (a) Photodegradation of ATZ under UV-vis irradiation over the TiO2/rGO nanocomposite with various rGO contents. (b) Photocatalytic activity of the different synthesized photocatalysts under visible light irradiation. (c) Mineralization of ATZ in the PCO process as a function of contact time. (d) ATZ degradation efficiency under visible light irradiation in the presence of selected radical scavengers (reaction conditions: pH: 5.4; ATZ concentration: 20 mg L−1; reaction time: 60 min. The concentration of radical scavengers/inhibitors was 200 mg L−1). | ||
Fig. 9b presents the photocatalytic activity of the TiO2/rGO nanocomposites sensitized with different concentrations of [Ru(tptz)(ACN)Cl2] as a function reaction time up to 60 min. For comparison, the photocatalytic activity of commercial TiO2 (Sigma Aldrich P25) was also investigated under the same conditions. When Ru-CMP0.0-TG was used as the photocatalyst, the ATZ removal efficiency was only 41%. Significantly, the removal of ATZ increased for the [Ru(tptz)(ACN)Cl2]-treated samples to 68%, 88%, 100% and 86% over Ru-CMP1.0-TG, Ru-CMP2.0-TG, Ru-CMP3.0-TG and Ru-CMP5.0-TG as the photocatalyst, respectively. Hence, the Ru-complex was used as a visible light sensitizer, and electron transfer from the excited dye to TiO2 occurred efficiently. The PFO kinetics of the degradation of ATZ in the PCO process with the prepared hybrids was also calculated and the results are presented in Table 1. As observed in Table 1, the loaded Ru-complex as a visible light sensitizer exhibited a highly enhanced PFO rate of 0.42 mg L−1 min−1. Moreover, inserting the Ru-complex at an optimal concentration (0.003 mol L−1) resulted in a more than 9 times higher rate constant for the degradation of ATZ than that with the commercial TiO2.
| Reaction | Catalyst | R2 | kobs (min−1) | robs (mg L−1 min−1) |
|---|---|---|---|---|
| Degradation | P25 | 0.996 | 0.009 | 0.14 |
| TG | 0.942 | 0.008 | 0.2 | |
| Ru-CMP1-TG | 0.951 | 0.019 | 0.54 | |
| Ru-CMP2-TG | 0.987 | 0.024 | 0.82 | |
| Ru-CMP3-TG | 0.979 | 0.060 | 1.26 | |
| Ru-CMP4-TG | 0.998 | 0.031 | 0.42 | |
| Mineralization | Ru-CMP3-TG | 0.980 | 0.017 | 0.36 |
Based on Table 1, loading the Ru-complex as a visible light sensitizer dye promoted the photocatalytic performance. The improvement in the photocatalytic activity of the samples after loading the Ru-complex photosensitizer can be attributed to its better light harvesting and efficient charge separation. The photocatalytic performance of a photocatalyst directly depends on its light absorption behavior, and light harvesting ability is a key factor to improve its photocatalytic efficiency. The wide band gap of TiO2 makes it inactive under visible light irradiation. However, after loading the Ru-complex, the absorption edge significantly extended toward the visible region. These results suggest the Ru-complex with extended visible light absorption could act as an efficient photosensitizer for TiO2 and improve the light harvesting in the final product. This increase in absorption in the visible light led to the generation of more electron–hole pairs, which resulted in higher photocatalytic activities. Furthermore, based on the PL and EIS results, sensitizing the catalyst using the Ru-complex inhibited the recombination of photogenerated electrons–holes and improved the photocatalytic performance (Fig. 7). When the prepared hybrid was irradiated by visible light photons, the photogenerated electrons in the LUMO of the Ru-complex could be directly injected to TiO2 rather than recombine with the holes by itself. Therefore, the formed junction between TiO2 and Ru-complex could serve as an electron sink to efficiently inhibit the recombination of photo-generated e−/h+ pairs, and thus significantly improve the photocatalytic activity. As the concentration of the complex solution increased, the complex loading capacity and ATZ removal efficiency increased. This result was predictable because the higher complex level in the catalyst led to the absorption of more visible light photons, resulting in the generation of more electron–hole pairs, and thus a better photocatalytic performance. However, above a complex solution concentration of 3 mM, the ATZ removal efficiency decreased because the dye precipitated on the TiO2 nanoparticles.57 This phenomenon led to an enhancement in inactive dye molecules on the surface of TiO2 and prevented the photogenerated electron injection processes.57 Moreover, as mentioned before, the source of illumination was a lamp without any cut filter, which emitted both UV and visible irradiation. Therefore, some of the contaminant could be directly degraded using TiO2 under UV irradiation. As seen in Fig. 9, 29% of ATZ was degraded over the TiO2/rGO photocatalyst without the Ru-complex. Therefore, there was a synergistic effect between TiO2 and Ru-complex, but a high content of Ru-complex resulted in the agglomeration of this dark material on the surface of the TiO2 nanostructure, which could block out the light, resulting in fewer photogenerated charges and weaker photocatalytic performances.58,59 Furthermore, based on electrochemical impedance spectroscopy (EIS) analysis, Tang and co-workers60 indicated that with an increase in Ru-complex concentration in dye-sensitized solar cells above the optimum concentration, the lifetime of photogenerated electrons increased, and thus the charge recombination was reduced. A similar trend was observed in this study. Electron–hole recombination and light harvesting are the most important factors limiting the photocatalytic efficiency of materials. Therefore, adjusting the complex concentration is a crucial factor for the efficient performance of a photocatalyst.
Fig. 9c displays the ATZ mineralization in the developed PCO process with the most efficient catalyst (Ru-CMP3.0-TG). It can be observed in Fig. 9c that the mineralization efficiency reached 71.8% with an increase in the irradiation time to 60 min. The PFO reaction rate (R2 = 0.99) constant for ATZ mineralization in the selected process was determined to be 0.017 min−1. The difference between the degradation and mineralization efficiencies implies that a small amount of the intermediates from the oxidation of ATZ remained in the solution, which was further mineralized with an increase in the reaction time.
To further explore the possible photocatalytic degradation mechanism of ATZ in the developed PCO process, the removal of ATZ was compared in the absence and the presence of various sacrificial agents. Four different quenchers, including tert-butanol (TBT), p-benzoquinone (PZQ), persulfate (PS) and EDTA-2Na, were used as ˙OH, O2−˙, electron and hole (h+) scavengers, respectively, in the trapping experiments, and the results are shown in Fig. 9d. Fig. 9d indicates that the addition of persulfate and TBT to the photocatalytic reaction did not have a considerable effect on the photocatalytic performance, while upon the introduction of EDTA-2Na, the photodegradation efficiency of ATZ decreased. In comparison, the presence of BZQ significantly decreased the degradation of ATZ, suggesting that ˙O2− was the main active reactive oxidation species (ROS) contributing to the ATZ degradation reaction. Besides, holes can play a relatively important role and participate in the oxidation reactions to a minor extent.
Thus, based on the above discussion, a possible mechanism for the photoactivity of Ru-CMP-TiO2/rGO under visible light can be proposed, as shown in Fig. 10. TiO2 as a wide band gap semiconductor absorbs only a small portion of solar irradiation (UV region). By sensitizing titania with the Ru-complex, two fundamental processes can occur, an improvement in visible light harvesting and the separation of charge carriers from each other.61 At the heart of the photocatalyst, the [Ru(tptpz)(ACN)Cl2] charge-transfer dye is illuminated and excited by visible light to an excited state. Polypyridyl Ru(II)-type complexes are known as the most efficient photosensitizers due to their broad range of visible light absorption owning to their metal-to-ligand charge transfer (MLCT) in this region, high photochemical stability, high light-to-electricity conversion, long excited state lifetime and tunable redox properties.15,62,63 The calculations suggest that the synthesized Ru-complex possesses a suitable ground state (HOMO) and excited state (LUMO) for efficient electron injection to TiO2. The HOMO and LUMO edge potentials are located at −5.12 eV and −3.21 eV for the Ru-complex, and −4.05 eV and −0.85 eV for TiO2, respectively. Therefore, since the CB of TiO2 remains at a less negative potential than the LUMO of the Ru-complex, the photo-excited electrons from the LUMO of the complex can be injected with high efficiency into the conduction band of TiO2. Owning to the band alignment of the Ru-complex and TiO2, this electron transfer process is thermodynamically more favorable and efficiently reduces the electron–hole recombination and promotes the photocatalytic efficiency. Besides this favorable pathway for the light-induced electron transfer process, the direct recombination of excited electrons with holes in the complex can occur as a competing reaction.64 Previous studies indicated that the recombination between photoinjected electrons with oxidized Ru-complex molecules occurs within microseconds, while electron injection from the excited dye to TiO2 occurs on the femtosecond scale (10−15 s).65 Therefore, because the electron transfer rate from the photo-excited complex is more than twice the photogenerated electron–hole recombination rate of the dye molecules, the contribution of the recombination mechanism can usually be neglected.65 The electrons in the CB of TiO2 can react with the oxygen molecules dissolved in the solution to produce superoxide anion radicals (O2−˙). The produced reactive oxygen species can directly oxidize organic pollutant molecules finally into CO2 and H2O and mineral acids. The holes (h+) on the HOMO of the Ru-complex cannot react with OH−/H2O to form ˙OH because its potential is more negative (0.995 eV vs. NHE) than that of ˙OH/OH− (2.72 eV vs. NHE, 1.99 C2).66 Hence, the holes migrate from the HOMO of the Ru-complex to the surface of the catalyst and directly oxidize the contaminant molecules, and therefore improve the photocatalytic activity. Therefore, as the aggregation of photogenerated electrons and holes increases, the generation of reactive oxidizing species is enhanced, resulting in the more efficient degradation of ATZ. In addition, due to the interaction between the oxygen-containing functional groups of GO and Ru-CMP-TiO2, GO as an excellent electron sink, provides a conduction path to move excited electrons from TiO2 to rGO. These trapped electrons on GO can also be captured directly by oxygen molecules and OH− dissolved in the solution and generate more O2−˙ and ˙OH.4 The barrier formed at the network, which is termed the Schottky barrier, separates the electron–hole pairs, and thus overcomes charge recombination.64 Therefore, the synergistic effect between the broad spectral response in the visible region and appropriate band alignment of the Ru-complex, and the close contact between rGO and TiO2 lead to effective light harvesting and adequate charge anti-recombination, and thus the greater generation of effective radical species and much greater photocatalytic activity compared to that of the bare TiO2 catalyst.
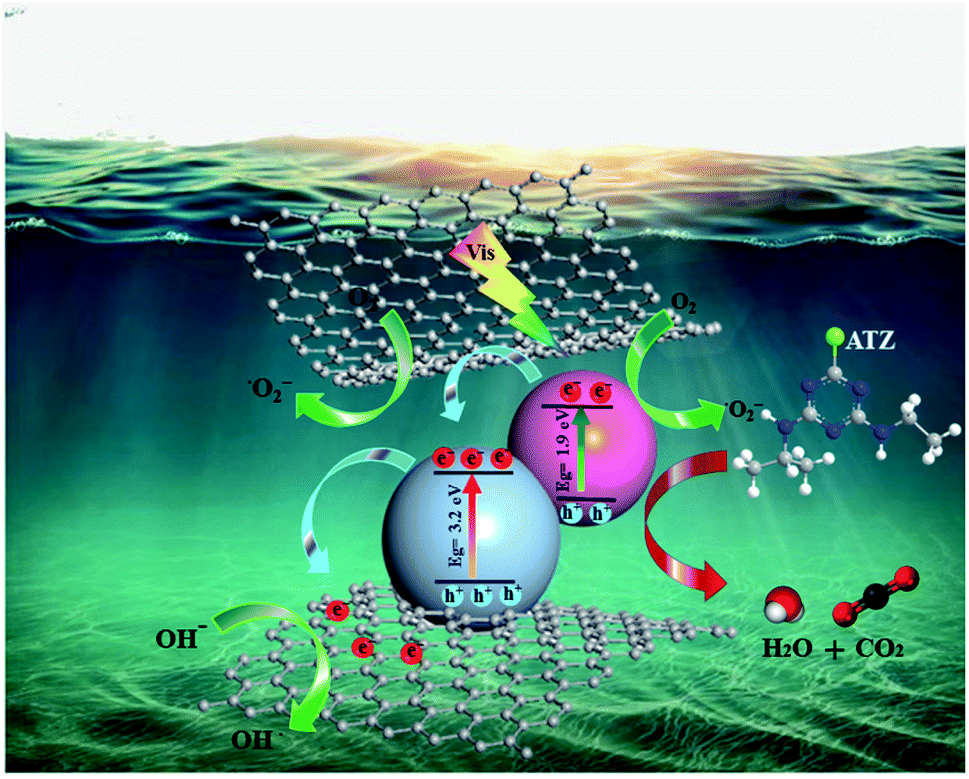 | ||
| Fig. 10 Mechanism of the visible-light activity of the Ru-CMP-TiO2/rGO photocatalyst. | ||
4. Conclusions
To enhance the visible light photoactivity of TiO2, an effective protocol was proposed based on the use of a novel Ru(II) polypyridyl complex, [Ru(tptz)(ACN)Cl2], as photosensitizer dye for sensitizing TiO2 nanoparticles supported on rGO. The molecular structure of the complex indicated that the coordination sphere around the ruthenium center was octahedral with the nitrogen atoms of the tptz ligand and acetonitrile forming the equatorial plane, while the two chloride atoms occupied the apical sites. By using a one-step approach, a hybrid Ru-CMP-TiO2/rGO nanomaterial was synthesized, where the loading of the Ru-complex molecules into catalyst was confirmed based on spectroscopic analyses. The XRD, BET, SEM and TEM results revealed that the prepared hybrids with a hexagonal anatase crystal structure had a mesoporous texture and TiO2 nanoparticles were attached to the surface of rGO. The reduction of GO during solvothermal treatment was proven trough FT-IR and Raman spectroscopy. The ruthenium(II) polypyridyl complex exhibited improved energy transfer properties due to the strong MLCT absorption bands in the visible region. The PL results confirmed that supporting the catalyst on rGO sheets as an inherent electron acceptor material improved the separation efficiency of the photogenerated electrons and holes pairs. Further studies revealed that loading the synthesized ruthenium(II) polypyridyl complex as a photosensitizer not only efficiently promoted charge separation but also usefully extended the light absorption of the photocatalyst. The gas-phase geometry and energies of the molecular orbitals of the Ru-complex was evaluated using DFT calculations. The DFT calculations were consistent with the experimental results. The prepared nanomaterial hybrid was examined for is activity in a lab-scale photocatalytic oxidation process for the degradation of ATZ solution. Compared to the pure TiO2, the prepared hybrid exhibited significantly enhanced photocatalytic activity. The rate of ATZ photodegradation was 9 times higher with the best catalyst compared to that of pure TiO2. Based on the comprehensive investigations, it was concluded that presence of rGO and loading the Ru-complex photosensitizer remarkably increased the photocatalytic activity of the catalyst by synergistically enhancing the quantity of photon absorption, adsorption capacity and charge transport through the reduction of charge recombination. The prepared polypyridyl Ru(II) complex with the band gap energy of 1.9 eV could simply be photoexcited under visible light, and due to its more suitable LUMO position than the CB of TiO2, the excited electrons in the complex could efficiently be injected to TiO2, which resulted in improved charge separation. Moreover, the excellent conductivity and adsorption capacity of graphene also contributed to the increase in photocatalytic activity. Therefore, the synergistic effect between TiO2, rGO and the Ru-complex resulted in the advantages of chemical stability, extended solar spectral utilization, reduction in electron–hole recombination and facilitation of charge transport. According to the results from the trapping experiments, photogenerated holes and ˙O2− radicals were the main active species in the ATZ photodegradation process. Thus, this study provides new insights into a new strategy to prepare visible light-active TiO2 for the degradation and detoxification of emerging contaminants and can be extended to other applications such as dye-sensitized solar cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Tarbiat Modares University, Tehran, Iran for the technical and financial support under the research group grant no. IG-39801. The authors are also grateful to the Ministry of Health, Iran, for providing financial support.References
- G. Granados-Oliveros, E. A. Páez-Mozo, F. M. Ortega, C. Ferronato and J. M. Chovelon, Appl. Catal., B, 2009, 89, 448–454 CrossRef CAS.
- X. Zhao, C. Zhang, S. Wang, C. Songc and X. Li, RSC Adv., 2017, 7, 1581–1587 RSC.
- J. A. Santacruz-Chávez, S. Oros-Ruiz, B. Prado and R. Zanella, J. Environ. Chem. Eng., 2015, 3, 3055–3061 CrossRef.
- A. H. Cheshme Khavar, G. Moussavi and A. R. Mahjoub, Appl. Surf. Sci., 2018, 440, 963–973 CrossRef CAS.
- W.-K. Jo, T. Adinaveen, J. J. Vijaya and N. C. S. Selvam, RSC Adv., 2016, 6, 10487–10497 RSC.
- M. S. A. Sher Shah, A. R. Park, K. Zhang, J. H. Park and P. J. Yoo, ACS Appl. Mater. Interfaces, 2012, 4, 3893–3901 CrossRef CAS PubMed.
- A. H. C. Khavar, G. Moussavi, A. R. Mahjoub, M. Satari and P. Abdolmaleki, Chem. Eng. J., 2018, 345, 300–311 CrossRef CAS.
- X. w. Liang, L. Wang, F. Ma, H. Lou, X. Jiang and Z. Lia, RSC Adv., 2016, 6, 89994–90001 RSC.
- N. Sharotri and D. Sud, Sep. Purif. Technol., 2017, 183, 382–391 CrossRef CAS.
- C. Xue, T. Wang, G. Yang, B. Yanga and S. Ding, J. Mater. Chem. A, 2014, 2, 7674–7679 RSC.
- H. Khan, M. G. Rigamonti, G. S. Patience and D. C. Boffito, Appl. Catal., B, 2018, 226, 311–323 CrossRef CAS.
- M. Zukalova, M. Bousa, Z. Bastl, I. Jirka and L. Kavan, J. Phys. Chem. C, 2014, 118, 25970–25977 CrossRef CAS.
- M. Khan, J. Xu, N. Chen, W. Cao, S. Asadullah, Z. Usman and D. F. Khan, Res. Chem. Intermed., 2013, 39, 1633–1644 CrossRef CAS.
- S. Sinn, B. Schulze, C. Friebe, D. G. Brown, M. Jäger, J. Kübel, B. Dietzek, C. P. Berlinguette and U. S. Schubert, Inorg. Chem., 2014, 53, 1637–1645 CrossRef CAS PubMed.
- Ç. Şahin, A. Apostolopoulou and E. Stathatos, Inorg. Chim. Acta, 2018, 478, 130–138 CrossRef.
- R. K. Chitumalla, K. S. V. Gupta, C. Malapaka, R. Fallahpour, A. Islam, L. Han, B. Kotamarthi and S. P. Singh, Phys. Chem. Chem. Phys., 2014, 16, 2630–2640 RSC.
- K. Katsumata, H. Matsui, T. Yamaguchi and N. Tanabe, Inorg. Chim. Acta, 2017, 463, 118–125 CrossRef CAS.
- A. Mahmood, Sol. Energy, 2016, 123, 127–144 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- H. M. Yadav and J. S. Kim, J. Alloys Compd., 2016, 688, 123–129 CrossRef CAS.
- M. Baghayeri, Sens. Actuators, B, 2017, 240, 255–263 CrossRef CAS.
- M. Khan, M. N. Tahir, S. F. Adil, H. U. Khan, M. R. H. Siddiqui, A. Al-warthan and W. Tremel, J. Mater. Chem. A, 2015, 3, 18753–18808 RSC.
- B. Saravanakumar, S. Vadivel, S. S. Dhar, D. Maruthamani, M. Kumaravel, G. Ramadoss, A. Manikandan, B. Paul and A. Habibi-Yangjeh, J. Colloid Interface Sci., 2017, 498, 449–459 CrossRef PubMed.
- G. Li, P. G. Bomben, K. C. D. Robson, S. I. Gorelsky, C. P. Berlinguette and M. Shatruk, Chem. Commun., 2012, 48, 8790–8792 RSC.
- M. C. Burla, M. Camalli and G. L. Cascarano, J. Appl. Crystallogr., 2003, 36, 1103 CrossRef CAS.
- V. Petrícek, M. Dušek and L. Palatinus, Z. Kristallogr., 2014, 229, 345–352 Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- D. Farhat, J. Educ. Work, 2014, 27, 1–42 CrossRef.
- D. M. Oliveira and A. S. Andrada, Cerâmica, 2019, 65, 170–179 CrossRef CAS.
- A. H. Cheshme Khavar, G. Moussavi, A. R. Mahjoub and M. Satari, Sol. Energy, 2018, 173, 848–860 CrossRef CAS.
- Z. Q. Li, H. L. Wang, L. Y. Zi, J. J. Zhang and Y. S. Zhang, Ceram. Int., 2015, 41, 10634–10643 CrossRef CAS.
- L. Pan, S. Liu, O. Oderinde, K. Li, F. Yao and G. Fu, Appl. Surf. Sci., 2018, 427, 779–786 CrossRef CAS.
- Y. Q. Zheng, W. Xu, F. Lin and G. S. Fang, J. Coord. Chem., 2006, 59, 1825–1834 CrossRef CAS.
- J. Wu, M. Lin, X. Cong, H. Liua and P. Tan, Chem. Soc. Rev., 2018, 47, 1822–1873 RSC.
- H. Zhu, J. Wu, M. Fang, L. Tan, C. Chen, N. S. Alharbi, T. Hayat and X. Tan, RSC Adv., 2017, 7, 36231–36241 RSC.
- J. Liu, H. Shi, Q. Shen, C. Guo and G. Zhao, Appl. Catal., B, 2017, 210, 368–378 CrossRef CAS.
- C. G. Coates, T. E. Keyes, H. P. Hughes, P. M. Jayaweera, J. J. McGarvey and J. G. Vos, J. Phys. Chem. A, 1998, 102, 5013–5018 CrossRef CAS.
- L. Gu, J. Wang, H. Cheng, Y. Zhao, L. Liu and X. Han, ACS Appl. Mater. Interfaces, 2013, 5, 3085–3093 CrossRef CAS PubMed.
- B. Li, S. Xiong, Y. Liao, X. Xiao, N. Huang, Y. Geng, S. Zou and S. Yang, J. Phys. Chem. C, 2016, 120, 23511–23522 CrossRef CAS.
- X. Y. Chen, Q. J. Pan and Y. R. Guo, Mater. Res. Bull., 2018, 107, 164–170 CrossRef CAS.
- M. S. A. Sher Shah, A. R. Park, K. Zhang, J. H. Park and P. J. Yoo, ACS Appl. Mater. Interfaces, 2012, 4, 3893–3901 CrossRef CAS PubMed.
- G. Jiang, K. Geng, Y. Wu, Y. Han and X. Shen, Appl. Catal., B, 2018, 227, 366–375 CrossRef CAS.
- S. Sayan, S. Suzer and D. O. Uner, J. Mol. Struct., 1997, 410–411, 111–114 CAS.
- X. Lin, K. Yang, R. Si, X. Chen, W. Dai and X. Fu, Appl. Catal., B, 2014, 147, 585–591 CrossRef CAS.
- P. Kumar, A. K. Singh, J. K. Saxena and D. S. Pandey, J. Organomet. Chem., 2009, 694, 3570–3579 CrossRef CAS.
- K. Abdi, H. Hadadzadeh, M. Salimi, J. Simpson and A. D. Khalaji, Polyhedron, 2012, 44, 101–112 CrossRef CAS.
- N. T. T. Truc, D. Sy Duc, D. Van Thuan, T. Al Tahtamouni, T. Dong Pham, N. Thi Hanhg, D. Trinh Tran, M. Viet Nguyen, N. Min Dang, N. T. P. Le Chib and V. Noi Nguyen, Appl. Surf. Sci., 2019, 489, 875–882 CrossRef CAS.
- R. M. Mohamed and F. M. Ibrahim, J. Ind. Eng. Chem., 2015, 22, 28–33 CrossRef CAS.
- J. Qin and H. Zeng, Appl. Catal., B, 2017, 209, 161–173 CrossRef CAS.
- H. Sun, S. Zeng, Q. He, P. She, K. Xu and Z. Liu, Dalton Trans., 2017, 46, 3887–3894 RSC.
- M. K. Nazeeruddin, S. M. Zakeeruddin, R. Humphry-Baker, M. Jirousek, P. Liska, N. Vlachopoulos, V. Shklover, C.-H. Fischer and M. Grätzel, Inorg. Chem., 2002, 38, 6298–6305 CrossRef PubMed.
- G. Wang, W. Feng, X. Zeng, Z. Wang, C. Feng, D. T. McCarthy, A. Deletic and X. Zhang, Water Res., 2016, 94, 363–370 CrossRef CAS PubMed.
- Z. Hua, J. Zhang, X. Bai, Z. Ye, Z. Tang, L. Liang and Y. Liu, Sci. Total Environ., 2016, 539, 196–205 CrossRef CAS PubMed.
- L. Zhang, J. Zhang, H. Jiu, C. Ni, X. Zhang and M. Xu, J. Phys. Chem. Solids, 2015, 86, 82–89 CrossRef CAS.
- L. C. Sim, K. H. Leong, S. Ibrahim and P. Saravanan, J. Mater. Chem. A, 2014, 2, 5315–5322 RSC.
- Z. Arifin, S. Soeparman, D. Widhiyanuriyawan and S. Suyitno, Int. J. Photoenergy, 2017, 2017, 1–5 CrossRef.
- P. Ma, H. Yu, Y. Yu, W. Wang, H. Wang, J. Zhang and Z. Fu, Phys. Chem. Chem. Phys., 2016, 18, 3638–3643 RSC.
- X. Liu, D. Li, X. Zhang, S. Liu and L. Zhang, J. Alloys Compd., 2015, 655, 38–43 Search PubMed.
- X. Tang, Y. Wang and G. Cao, J. Electroanal. Chem., 2013, 694, 6–11 CrossRef CAS.
- T. V. Arjunan and T. S. Senthil, Mater. Technol., 2012, 28, 9–14 CrossRef.
- B. Athanas Anish, S. Thangaraj and S. Kalaiyar, Chem. Phys. Lett., 2018, 699, 32–39 CrossRef CAS.
- A. S. Polo, M. K. Itokazu and N. Y. Murakami Iha, Coord. Chem. Rev., 2004, 248, 1343–1361 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- J. Gong, J. Liang and K. Sumathy, Renew. Sustain. Energy Rev., 2012, 16, 5848–5860 CrossRef CAS.
- E. D. Sherly, J. J. Vijaya and L. J. Kennedy, J. Mol. Struct., 2015, 1099, 114–125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06704c |
| This journal is © The Royal Society of Chemistry 2020 |