
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAqueous colloidal systems of bovine serum albumin and functionalized surface active ionic liquids for material transport†
Gagandeep Singha,
Manvir Kaura,
Vinod Kumar Aswalb and
Tejwant Singh Kang *a
*a
aDepartment of Chemistry, UGC-Centre for Advance Studies – II, Guru Nanak Dev University, Amritsar, 143005, India. E-mail: tejwant.chem@gndu.ac.in; tejwantsinghkang@gmail.com; Tel: +91-183-2258802 ext. 3291
bSolid State Physics Division, Bhabha Atomic Research Centre, Mumbai 400085, India
First published on 17th February 2020
Abstract
Detailed physicochemical and computational investigation are made to explore different aspects of complexation between bovine serum albumin (BSA) and three structurally different surface active ionic liquids (SAILs), 1-dodecyl-3-methylimidazolium chloride, [C12mim][Cl]; 3-(2-(dodecylamino)-2-oxoethyl)-1-methyl-1H-imidazol-3-ium chloride, [C12Amim][Cl] and 3-methyl-1-dodecyloxy carbonyl methylimidazolium chloride, [C12Emim][Cl]. The interfacial and bulk complexation behavior has been monitored using tensiometry, conductivity, steady-state fluorescence and turbidity measurements. Thermodynamic insights about complexation have been obtained using isothermal titration calorimetry (ITC) measurements whereas molecular docking studies were used to predict the possible binding sites of SAILs on BSA. The information obtained from these studies helped in establishing the formed BSA–SAIL complex as a pH dependent colloidal transport system for controlled transport of a lipophilic dye, Rhodamine 6G (R6G), in aqueous phase, which is supported by confocal laser scanning microscopy (CLSM). In the present work, the effect of functionalization over the alkyl chain of SAILs, modulating the colloidal properties of SAIL–BSA systems, has been explored along with the utilization of these complexes as a pH dependent reversible carrier of lipophilic molecules. It is expected that besides providing basic understanding of colloidal complexes of BSA with SAILs, the present work is expected to be helpful in extending the applications of such colloidal systems for material transport.
1. Introduction
Serum albumins are the most utilized proteins due to their relatively large abundance, ease of purification and multi-faceted properties.1–3 Bovine serum albumin (BSA), is 77% identical and 87% homologous with human serum albumin (HSA),4,5 which makes it the most versatile model protein for different investigations. BSA is a globular protein having 583 amino acid residues with a molecular mass of 66.3 kDa, which functions as a chief transporter and distributor of various endogenous and exogenous ligand metabolites.4,6 The molecular structure of native BSA consists of three domains (domain I, II and III, Scheme 1), which are further divided into six sub-domains, two sub-domains for each domain. The globular structure of BSA possess 56% α-helical and 11% β-sheet content.7,8 BSA can reversibly adopt various spatial conformations at different pH values such as N-isoform at pH 7.4 (heart-like shape), F-isoform at pH 3.5 (cigar-like shape) and E-isoform at pH 2.7 (denatured form).9,10 The exceptional stability among other proteins such as long shelf-life (about 19 days), stability towards high temperature (at 60 °C up to 10 hours) and in a wide pH range (pH 4 to 9) along with reversible binding ability towards a variety of bioactive molecules and active pharmaceutical ingredients (APIs) make BSA a robust and imperative protein for physiological investigations.4,11–13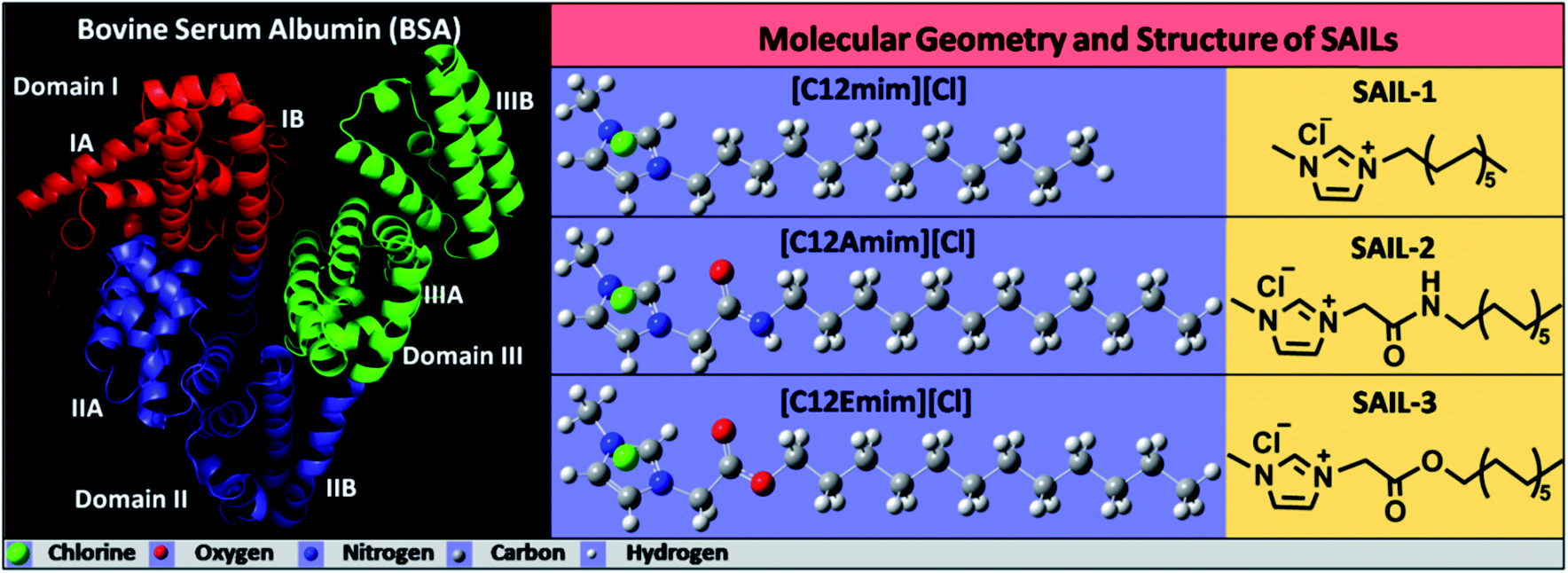 | ||
| Scheme 1 Molecular geometry and structure of BSA (left) and SAILs (right) used in present study. | ||
The use of surfactants in conjunction with proteins generally gives rise to improved physicochemical properties and functions of proteins.14,15 This renders the investigations on protein–surfactant interactions very important and therefore a large amount of work has been published in this regard.16–31 In last decade, a new class of surfactants called as surface active ionic liquids (SAILs) has gained great curiosity from the scientific community owing to their better colloidal and surface active properties as compared to conventional surfactants.32–45 The inherent amphiphilicity and tailorable properties of SAILs offers a precise control over the hydrophilicity/hydrophobicity of SAILs, required to control their colloidal behavior. The choice of cation and anion along with the possibility of functionalization over head group and alkyl chain adds to their applicability. Therefore, the properties of SAILs could offer more potential futuristic prospects for protein–surfactant applications as compared to conventional ionic surfactants. Till date, very limited reports on SAIL–protein colloidal systems are available.8,46–57 Different proteins such as BSA, Gelatin and β-lactoglobulin (β-LG) has been investigated for their complexation with SAILs, where the length of alkyl chain and nature of head group of SAIL has been found to exert significant influence on complexation behavior of BSA with SAILs.8,46–49,56,57 It is stressed that in most of the investigations, SAILs without any functionalization of ionic head group or alkyl chain have been employed with the exception of few studies.8,46,48 A contrasting complexation behavior of functionalized SAILs as compared to non-functionalized ones towards BSA,8 Gelatin48 and β-lactoglobulin (β-LG)46 has been observed. Different types of SAIL–protein complexes varying in morphology, internal structure and hydrophobicity etc., governed by the nature and extent of varying set of interactions has been observed. It has been found that both the nature of protein and functionalization of alkyl chain of SAIL affect the complexation phenomenon. This along with the formation of unique self-assembled structures of BSA and functionalized SAILs,8 previously reported by our group prompted us to further extent the study to explore physico-chemical aspects of such complexation, in detail. In past, no attempt has been made to identify the binding location of SAILs on BSA, which certainly would assist in understanding the nature of interactions at molecular level. Moreover, such BSA–SAIL complexes, owing to lack of structural functionality, has not been investigated for any potential application in past. Therefore, relying on the changes in pH dependent conformations of BSA and interactions between SAILs and BSA, we have also investigated the formed SAIL–BSA colloidal complexes as potential carriers of lipophilic molecules.
Herein, we have carried out detailed physicochemical studies on complexation behavior of BSA with three structurally different SAILs: 1-dodecyl-3-methylimidazolium chloride, [C12mim][Cl]; 3-(2-(dodecylamino)-2-oxoethyl)-1-methyl-1H-imidazol-3-ium chloride, [C12Amim][Cl] and 3-methyl-1-dodecyloxy carbonyl methylimidazolium chloride, [C12Emim][Cl]. A multi-technique approach is employed to explore the interfacial as well as bulk behavior of BSA–SAIL colloidal systems. Computed simulation (AutoDock Vina) has been used to further probe the binding site of SAILs on BSA. The knowledge gained from various physico-chemical studies has been used to establish the applicability of formed BSA–SAIL-2 colloidal complex (as a representative) as pH dependent transporter of a lipophilic dye, Rhodamine 6G (R6G), reversibly, which has been supported by confocal laser scanning microscopy (CLSM) studies. The interferences gained from different techniques has been corroborated and compared with the literature reports wherever possible. The present work highlight the dynamics of protein–SAILs interactions and is expected to help in establishing a new platform for encapsulating lipophilic drugs in protein–SAIL based colloidal complexes in aqueous medium.
2. Materials and methods
The interfacial and bulk complexation behavior of SAILs with BSA (Scheme 1) was investigated using titration method. Aqueous concentrated stock solutions of investigated SAILs were added to aqueous solution containing 0.1% BSA (w/v) in standard phosphate buffer solution (PBS, pH = 7.2, I = 5 mmol L−1) and the observations were made using various state of art techniques. The concentration of BSA employed (0.1%) is chosen, as one where maximum extent of interactions between BSA and SAILs has been observed without aggregation of BSA, based on fluorescence measurements performed at fixed concentrations (one above and one below to critical micelle concentration) of SAILs by varying the concentration of BSA (Fig. S1, ESI†). The detailed information about the experimental setup is, provided in Annexure S1, ESI.†3. Results and discussion
3.1. Interfacial behavior of SAIL–BSA colloidal systems
The comparative plots of tensiometric profiles of investigated SAILs in the presence and the absence of 0.1% BSA, in aqueous buffer solutions, are shown in Fig. 1A–C. The lower value of surface tension (γ ≈ 54 mN m−1) in aqueous solution containing BSA as compared to that without BSA (γ ≈ 71 mN m−1) is ascribed to surface active nature of BSA.56,57 The difference in nature of surface tension profiles of SAILs, with and without BSA, signifies the presence of interactions between SAILs and BSA. In case of SAIL-1, γ decreases continuously without showing any significant transition till C4(cmc) similar to that shown by SAIL-1 in the absence of BSA.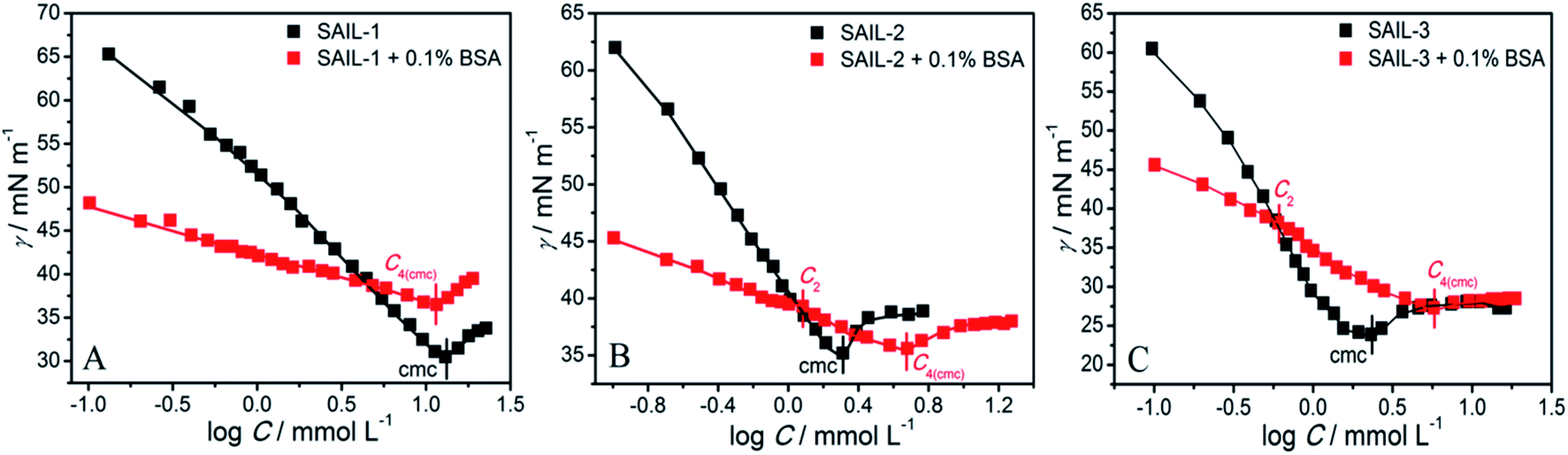 | ||
| Fig. 1 (A–C) Variation of surface tension in aqueous phosphate buffer solution with and without BSA as a function of concentration of different SAILs at 298.15 K. | ||
A higher value of γ around C4(cmc) suggests the presence of SAIL-1–BSA complex at air–solution interface, which hinders the adsorption of incoming SAIL ions. The monomeric complexation of investigated SAILs, in bulk, has been reported previously resulting in the formation of SAIL–BSA monomer complex (MC) till a concentration, C1, which even started to self-assemble in very dilute concentration range.8 Such self assembly of SAIL–BSA MCs becomes relatively more prominent near C2 which is marked as aggregate complex (AC) stage.8 However the absence of any break-point corresponding to change in surface behavior indicates that the complexation of SAILs with BSA is a continuous phenomenon. Once formed BSA–SAIL-1 MCs tends to grow in size via SAIL mediated self-assembly with increase in concentration of SAIL-1 at air–solution interface. SAIL-2 and SAIL-3 follow contrasting complexation behavior with BSA, as suggested by the presence of different transitions in respective tensiometric profiles, as compared to that observed in case of SAIL-1. There is a weak transition (C2) in tensiometric profile of SAIL-2 and SAIL-3 in the presence of BSA, where γ decreases with a marginally higher slope beyond C2 till C4(cmc). This is assigned to partial dissolution of SAIL–BSA complexes into the bulk after C2 and the occupancy of air–solution interface by respective SAIL ions, which is supported by rise in turbidity in similar concentration regime (discussed later). SAIL-1 interacts with negatively charged8 BSA (ζ-potential ≈ −18 mV at pH 7.4) via electrostatic and hydrophobic interactions whereas SAIL-2 and SAIL-3 offer additional synergistic H-bonding interactions, which affects the interactional process. It is inferred that SAIL-1–BSA complex (MCs and ACs) remains stable at air–solution interface in the whole concentration range whereas ACs formed by SAIL-2 and SAIL-3 undergo dissolution towards bulk at higher concentration of respective SAILs. This is also justified by the similarity in values of γ at or beyond C4(cmc) in the absence and presence of BSA owing to occupancy of air–solution interface by SAIL molecules.
As can be seen from Tables 1 and S2 (ESI†), critical micelle concentration (cmc) of SAIL-2 and SAIL-3 is found to be higher in presence of BSA as compared to their respective solutions without BSA, where as SAIL-1 exhibit relatively lower cmc in the presence of BSA. The presence of amide and ester-moiety in SAIL-2 and SAIL-3 enables these SAILs to interact synergistically through H-bonding along with other set of interactions with BSA and thus more number of SAIL ions of SAIL-2 and SAIL-3 interacts with BSA8 as compared to SAIL-1 leading to higher cmc in presence of BSA in case of SAIL-2 and SAIL-3. This is supported by experimental measurements where the number of SAIL ions associated per molecule of BSA is found to be 69, 111 and 210 for SAIL-1, SAIL-2 and SAIL-3, respectively (Table S1, Fig. S2 and S3, ESI†). Further, various interfacial parameters of interest i.e. interfacial tension at cmc (γcmc), the effectiveness of surface tension reduction (πcmc), minimum area per molecule at air–solution interface (Amin), Gibbs' surface excess (Γmax), and the standard free energy of adsorption 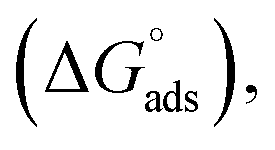 were calculated using surface tension data by employing standard procedures (Annexure S2, ESI†) and are provided in Table S3 (ESI†).
were calculated using surface tension data by employing standard procedures (Annexure S2, ESI†) and are provided in Table S3 (ESI†).
| SAIL-1 | SAIL-2 | SAIL-3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C1 | C2 | C3 | C4(cmc) | C1 | C2 | C3 | C4(cmc) | C1 | C2 | C3 | C4(cmc) | |
| ST | — | — | — | 11.62 | — | 1.21 | — | 4.76 | — | 0.59 | — | 5.69 |
| Cond. | — | — | — | 9.46 | 0.34 | — | — | 6.52 | 0.41 | — | — | 5.72 |
| I1/I3 | 0.84 | — | 2.85 | 3.34 | 0.35 | 1.24 | 2.47 | 2.95 | 0.25 | 0.65 | 2.46 | 2.89 |
| ITC | — | 3.14 | 9.14 | 14.56 | — | 1.74 | 3.42 | 5.03 | — | 1.74 | 2.90 | 4.46 |
| Turb. | 1.69 | 2.54 | — | 9.09 | 0.39 | 0.78 | — | 2.86 | 0.45 | 0.77 | — | 2.62 |
The higher values of γcmc for investigated systems in the presence of BSA as compared to that observed in the absence of BSA is indicative of decreased efficacy of SAILs to populate the air–solution interface in the presence of BSA. Amin of SAILs in the presence of BSA is found to be increased by 3.3, 3.7 and 3.3 times for SAIL-1, SAIL-2 and SAIL-3 respectively as compared to BSA free solution indicating relatively loose packing of investigated SAIL at air–solution interface in presence of BSA. However, these results are contrary to the previous reports, where Amin decreases for biamphiphilic SAILs (BAILs), [C4mim][C8OSO3] and [C12mim][C8OSO3].56,57 The additional electrostatic and hydrophobic interactions of amphiphilic anion of the investigated BAILs with BSA could have been resulted in such behavior. Relatively more negative values of 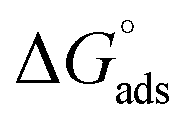 in presence of BSA suggest the greater adsorption efficacy of SAIL–BSA complexes over the micellization in bulk.
in presence of BSA suggest the greater adsorption efficacy of SAIL–BSA complexes over the micellization in bulk.
3.2. Bulk behavior of SAIL–BSA colloidal systems
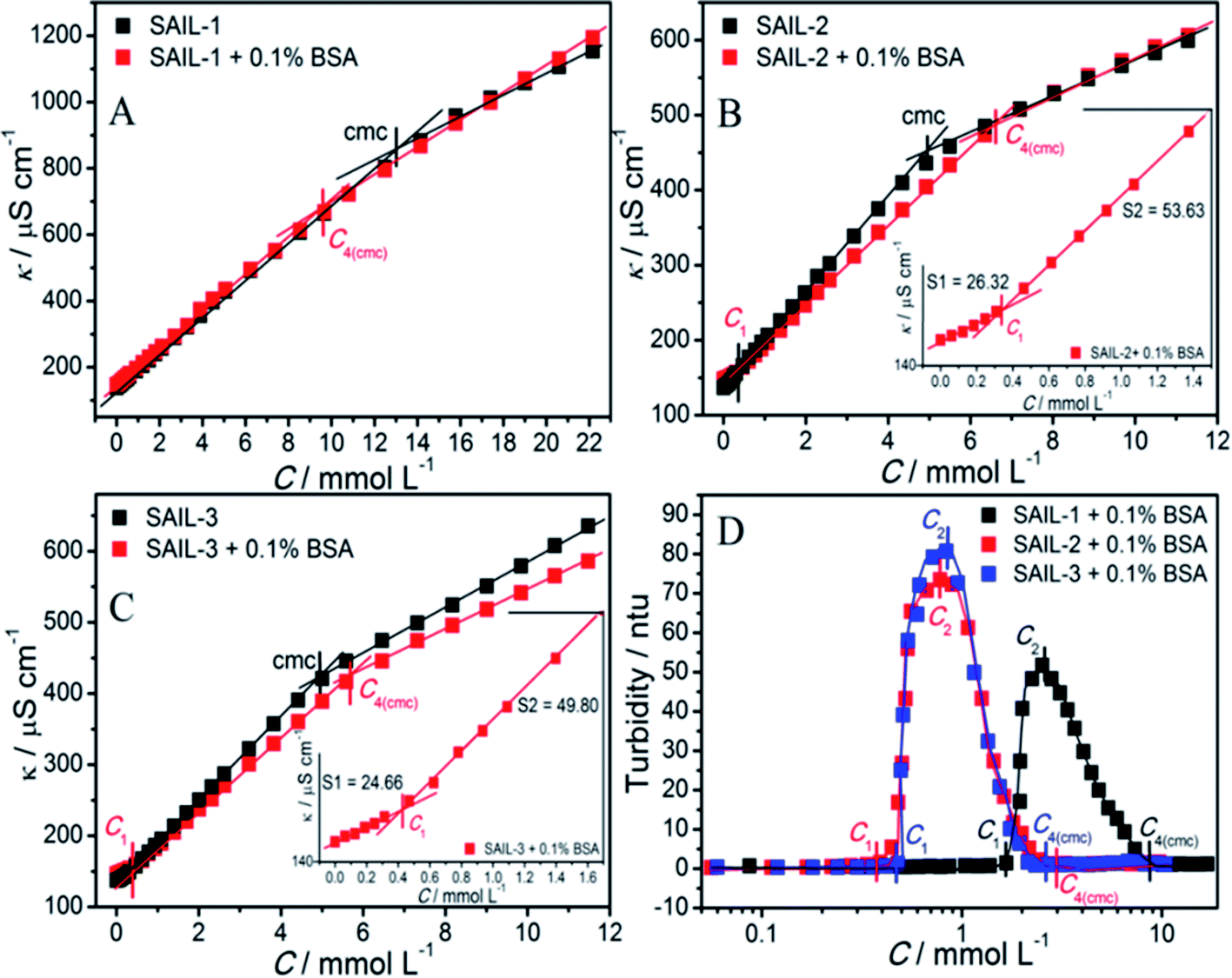 | ||
| Fig. 2 (A–C) Variation in specific conductivity with and without BSA and (D) turbidity of SAIL–BSA systems, in aqueous phosphate buffer solution as a function of concentration of different SAILs at 298.15 K. | ||
The concentrations corresponding to different transitions obtained from conductivity measurements are provided in Table 1. In case of SAIL-1, the profile of κ in the presence of BSA almost overlaps with that observed in the absence of BSA and only one transition corresponding to C4(cmc) is observed. This indicates weaker ionic interactions between SAIL-1 and BSA in the whole concentration range where the micellization of SAIL-1 seems not to be affected by the presence of BSA in terms of ionic environment. In case of SAIL-2 and SAIL-3, two transitions, namely C1 and C4(cmc) are observed. The presence of H-bond acceptor–donor and H-bond acceptor capability of amide and ester groups of SAIL-2 and SAIL-3, respectively, synergistically support already existing electrostatic interactions8 between these SAILs and BSA. This causes relatively stronger transient localization of ions of SAIL-2 and SAIL-3 near the polypeptide chains of BSA, which results in decreased contribution by these charge carriers per unit area leading to increase in κ with relatively lower slope till C1. In case of SAIL-2, below C4(cmc), κ increases with a lower slope in the presence of BSA, which further overlaps with κ observed in the absence of BSA beyond C4(cmc). The overlap of κ beyond C4(cmc) in the absence and presence of BSA, similar to that observed in case of SAIL-1, suggests the formation of BSA-free micelles. On the other hand, in case of SAIL-3, κ increases with relatively lower slope in whole concentration range in the presence of BSA as compared to BSA free systems. This is indicative of relatively stronger interactions SAIL-3 and BSA and is assigned to relatively flexible nature of ester-moiety, which enables SAIL ions to adopt different spatial orientations to maximize the extent of interactions with BSA. The transition at C2 is not observed from conductivity measurements for all the investigated systems despite the fact that SAIL ions interact with BSA leading to formation of MCs followed by ACs in different concentration regimes. This suggests that with increasing concentration of SAILs, after C1, the hydrophobic interactions begin to gain control over the complexation process, which triggers the self-assembly of SAIL–BSA MCs to ACs near C2. The substantial contribution of hydrophobic interactions in reorganization of MCs to form ACs could largely render the ionic environment of solution unaffected, which seems to be the reason behind the absence of C2 and C3 in the conductivity profile of SAILs. Such observations are quiet common in protein/polymer–SAIL systems.46,48 Further information about the degree of counter-ion binding (β) and standard free energy of micellization 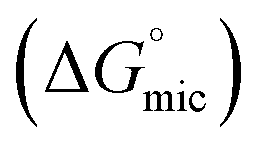 calculated using conductivity measurements (Annexure S2, ESI†) is provided in footnote of Table S3 (ESI†).
calculated using conductivity measurements (Annexure S2, ESI†) is provided in footnote of Table S3 (ESI†).
Turbidity measurements have been performed to understand the colloidal stability of SAIL–BSA complexes. Till C1, almost no change in turbidity (Fig. 2D) and DLS8 profiles of SAILs in BSA solution indicates the formation of very small BSA–SAIL MCs, which don't scatter appreciably and remains well dispersed in solution. This observation is supported by small angle neutron scattering (SANS) measurements (Fig. 3 and Table S4, ESI†). No appreciable change in shape and size of BSA has been observed in the form of BSA–SAIL MCs for all the investigated SAILs. The shape of BSA–SAIL complexes at low SAIL concentration has been found to be oblate-ellipsoid as suggested by SANS measurements, which is similar to that formed by the BSA protein in aqueous medium. Further, the observed size of BSA–SAIL complexes (Table S4A, ESI†) is marginally larger than that observed for BSA (oblate ellipsoid; ε < 1; having semi-major axes 39.7 Å and semi-minor axes 14.6 Å), which is in good agreement with that reported earlier.58
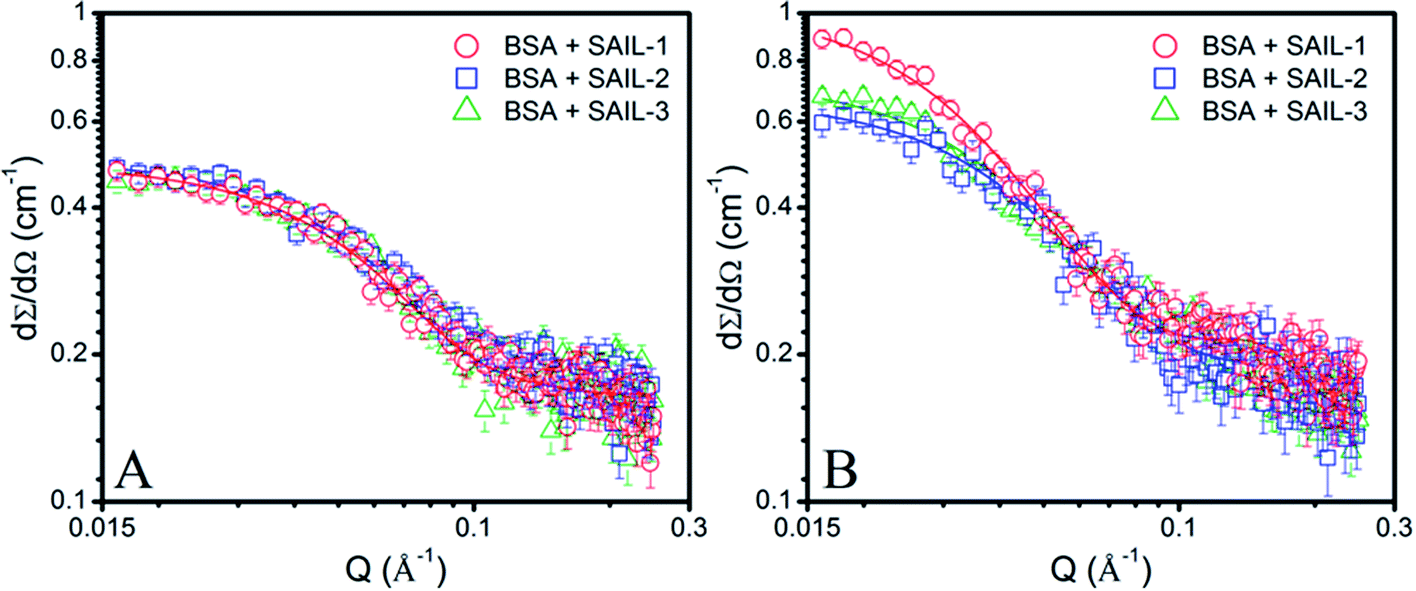 | ||
| Fig. 3 (A and B) SANS profiles of 1% BSA in presence of different SAIL systems (A) near C1 and (B) near C4(cmc), along with model fits on obtained data points shown in solid lines. | ||
However, between C1 to C2, turbidity increases abruptly indicating the appearance of large colloidal particles, which gets microscopically phase separated from the solution, in the form of BSA–SAIL ACs formed via SAIL mediated self-assembly of BSA–SAIL MCs. Such microscopic phase separation is also supported by the charge neutralization of formed BSA–SAIL MCs.8 Beyond C2, the addition of respective SAILs leads to dissolution of formed SAIL–BSA ACs as indicated by sharp decrease in turbidity, which reaches a plateau beyond C4(cmc). Near C4(cmc), BSA gets denatured as also suggested by CD measurements and the unfolding of BSA exposes the hydrophobic regions towards aqueous environment, which is an entropically unfavorable condition. To counter this change, the BSA gets wrapped around the thus forming micelles of surfactants or SAILs by entrapping water molecules. This leads to the formation of micelle-clusters of BSA–SAIL complexes in the form of necklace-bead like structure.59,60 The corresponding data obtained by fitting the SANS data using random flight model31 for oblate-ellipsoidal micelles is provided in Table S4.† It is natural to assume that the electrostatic and hydrophobic interactions between amino acids residues of BSA and SAIL ions at higher SAIL concentration would affect the curvature of forming micelles and this could result in formation of oblate-ellipsoid micelles in comparison to spherical micelles observed in aqueous medium (Fig. S4†). In our previous study, necklace-bead like structures of BSA–SAIL complexes have not been observed8 from TEM measurements, which is assigned to the fact that such protein–SAIL colloidal systems when dried don't retain their solution phase structure. It is important to mention that no transition corresponding to C3 has been observed from turbidity measurements contrary to that reported earlier by using DLS measurements.8 This is due to the fact that different techniques different aspects of complexation.
 | ||
| Fig. 4 (A–C) Variation in I1/I3 of pyrene in aqueous buffer solution with and without BSA as function of concentration of different SAILs at 298.15 K. | ||
A lower value of I1/I3 (1.23) of pyrene in BSA solution as compared to that observed in phosphate buffer (1.71) indicates the adsorption of pyrene in the hydrophobic pockets located on the surface of BSA.46,56 I1/I3 decreases with a relatively lower slope in all of the investigated systems till C1 indicating a marginal increase in hydrophobicity of forming MCs due to the adsorption of SAIL ions. At C1, I1/I3 value of respective SAIL–BSA MCs follows the order: SAIL-3 (1.32) < SAIL-2 (1.46) < SAIL-1 (1.56), which indicates the higher hydrophobicity of SAIL-3–MCs complexes. This is in expectation with stronger interactions of H-bonding prone SAIL-2 and SAIL-3 with BSA resulting in the formation of relatively more hydrophobic MCs. After C1, I1/I3 decreases abruptly in all cases till C3 following different paths. This sharp decrease in I1/I3 indicates the lower polarity of ACs formed through self-assembly of MCs mediated via hydrophobic interactions offered by SAIL ions8 in this concentration regime. There is no change in hydrophobicity index of ACs between C3 and C4(cmc) for all the investigated systems, although in this concentration region, ACs tends to dissolve to form smaller complexes.8 This suggest negligible change in internal structure of BSA–SAIL complexes in this concentration regime. After C4(cmc), I1/I3 begin to rise in all the cases indicating a relative increase in the polarity of SAIL–BSA aggregates. It is quite probable that beyond C4(cmc), the forming micelles containing high surface charge8,46,48 disintegrate the respective SAIL–BSA ACs into smaller micelle bound/unbound ACs. A similarity in values of I1/I3 in the absence and presence of BSA beyond C4(cmc) supports the partition of pyrene into the micelle bound to ACs.
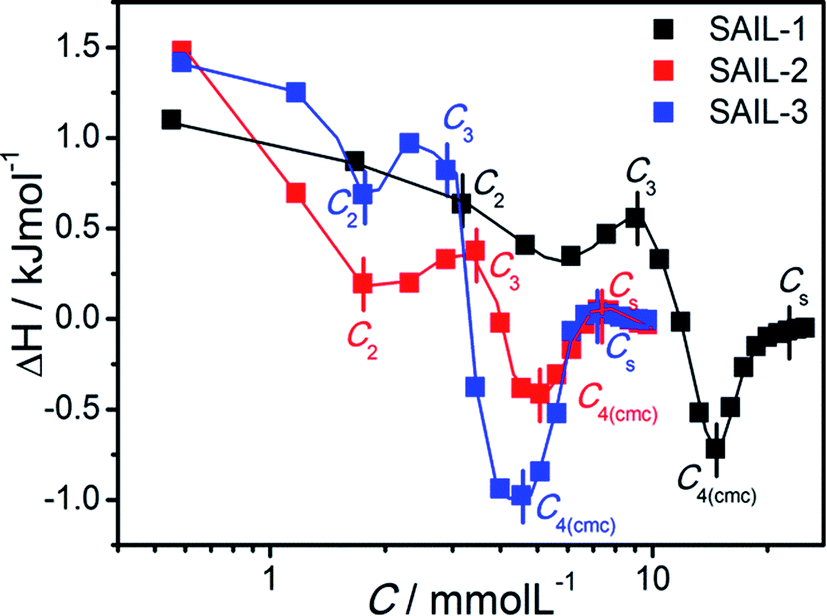 | ||
| Fig. 5 Difference plot of enthalpograms of SAIL–BSA systems in buffer solution of BSA as the function of concentration of different SAIL at 298.15 K. | ||
It is problematic to describe and quantify precisely the types of processes40,62–65 occurring during complexation, therefore qualitative information has been derived from the obtained thermodynamic data. For all the investigated SAIL–BSA systems, in dilute solution, ΔH° is endothermic as compared to corresponding aqueous SAILs (Fig. S6A–C, ESI†), where the enthalpic changes are more extensive in case of SAIL-2 and SAIL-3 as compared to SAIL-1. This indicates the significant role of entropic factors associated with the dehydration of SAILs and BSA upon interaction. The breaking of intramolecular H-bonds, specifically in case of SAIL-2 and SAIL-3, results in relatively large endothermic changes. Thereafter, the enthalpic processes favored by attractive H-bonding, electrostatic and hydrophobic interactions between SAIL and BSA compensate the initial entropy factor in the succeeding part of complexation process till C4(cmc). The concentrations corresponding to different transitions obtained from the difference plots (Fig. 5) of enthalpograms corroborate well with other techniques (Table 1). The difference plot of SAILs indicates the dominance of entropic control after C4(cmc) till Cs (Cs, saturation of ITC curve).
For the sake of simplicity, the enthalpy change for interactional process is divided into four parts as C1–C2 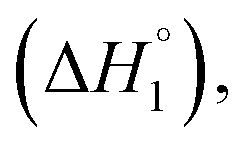 C2–C3
C2–C3 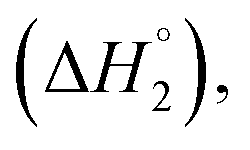 C3–C4(cmc)
C3–C4(cmc) 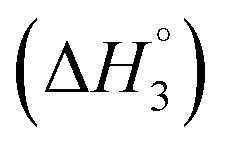 and C4–Cs
and C4–Cs 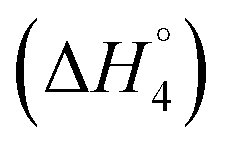 and the corresponding values of ΔH° are provided in Table S5 (ESI†). Between C1 and C2, exothermic enthalpy change is observed for all of the investigated systems, which is assigned to dominance of electrostatic and hydrophobic interactions in case of SAIL-1, assisted by H-bonding interactions in case of SAIL-2 and SAIL-3. This is justified by the magnitude of
and the corresponding values of ΔH° are provided in Table S5 (ESI†). Between C1 and C2, exothermic enthalpy change is observed for all of the investigated systems, which is assigned to dominance of electrostatic and hydrophobic interactions in case of SAIL-1, assisted by H-bonding interactions in case of SAIL-2 and SAIL-3. This is justified by the magnitude of 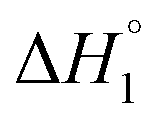 which follows the order: SAIL-2 (−1.30 kJ mol−1) > SAIL-3 (−0.73 kJ mol−1) > SAIL-1 (−0.46 kJ mol−1). For all the investigated systems, between C2 to C3, a slight endothermic rise (entropic process) is observed. Near C2, the ζ-potential value of BSA–SAIL systems becomes almost zero8 and remain constant till C3, indicating the electrostatic saturation of accessible binding sites of BSA. Therefore, such endothermic enthalpy change could be attributed to hydrophobic interactions between the formed complexes and incoming SAIL ions.8 The values of
which follows the order: SAIL-2 (−1.30 kJ mol−1) > SAIL-3 (−0.73 kJ mol−1) > SAIL-1 (−0.46 kJ mol−1). For all the investigated systems, between C2 to C3, a slight endothermic rise (entropic process) is observed. Near C2, the ζ-potential value of BSA–SAIL systems becomes almost zero8 and remain constant till C3, indicating the electrostatic saturation of accessible binding sites of BSA. Therefore, such endothermic enthalpy change could be attributed to hydrophobic interactions between the formed complexes and incoming SAIL ions.8 The values of 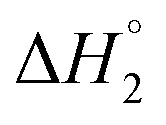 follows the similar trend as observed for
follows the similar trend as observed for 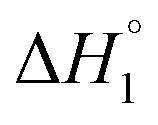 : SAIL-2 (+0.19 kJ mol−1) > SAIL-3 (+0.14 kJ mol−1) > SAIL-1 (−0.08 kJ mol−1). With further rise in concentration of SAILs, between C3 and C4(cmc), relatively larger exothermic drop in values of enthalpy change
: SAIL-2 (+0.19 kJ mol−1) > SAIL-3 (+0.14 kJ mol−1) > SAIL-1 (−0.08 kJ mol−1). With further rise in concentration of SAILs, between C3 and C4(cmc), relatively larger exothermic drop in values of enthalpy change 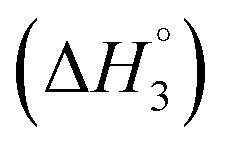 is observed, which for different SAILs, follows the order: SAIL-3 (−1.79 kJ mol−1) > SAIL-1 (−1.28 kJ mol−1) > SAIL-2 (−0.79 kJ mol−1). The order indicates the greater extent of interactions between SAIL-3 and BSA–SAIL-3 ACs between C3 and C4(cmc), which resulted in the formation of highly ordered self-assembled molecular architecture of SAIL-3–BSA into helical fibers in this concentration regime (at 1.5 mmol L−1 of SAIL-3 in 0.1% BSA).8 Further, the observed endothermic change, after C4(cmc) in all cases, establishes the dominance of entropically favored processes, which is assigned to the dissolution of SAIL–BSA complexes in micellar solution of SAILs.
is observed, which for different SAILs, follows the order: SAIL-3 (−1.79 kJ mol−1) > SAIL-1 (−1.28 kJ mol−1) > SAIL-2 (−0.79 kJ mol−1). The order indicates the greater extent of interactions between SAIL-3 and BSA–SAIL-3 ACs between C3 and C4(cmc), which resulted in the formation of highly ordered self-assembled molecular architecture of SAIL-3–BSA into helical fibers in this concentration regime (at 1.5 mmol L−1 of SAIL-3 in 0.1% BSA).8 Further, the observed endothermic change, after C4(cmc) in all cases, establishes the dominance of entropically favored processes, which is assigned to the dissolution of SAIL–BSA complexes in micellar solution of SAILs.
In docking studies, nine modes of binding conformations of SAILs are obtained (Table S6, ESI†). The first three poses with high free energy of binding affinities of SAIL-1, SAIL-2 and SAIL-3 towards BSA are shown in Fig. S8 (ESI†), 6A–F and S9 (ESI†), respectively. The investigated SAILs, due to their structural resemblance with hydrophobic moieties like fatty acid and octanoate, bind in domain III as first preference followed by domain II. SAIL-1 being non-functionalized analogue among the investigated SAILs interacts through electrostatic as well as hydrophobic interactions and binds in domain IIIB (Fig. S8A–C†) along with other locations on BSA similar to SAIL-2 and SAIL-3. SAIL-2 (Fig. 6A–F) and SAIL-3 (Fig. S9A–E†), owing to the presence of amide and ester functionality, respectively, forms H-bond acceptor–donor pairs and exhibit various other polar interactions with amino acid residues present at their binding cavity. This corroborates well with the previously made claims that SAIL-2 and SAIL-3 interacts more strongly with BSA as compared to SAIL-1.
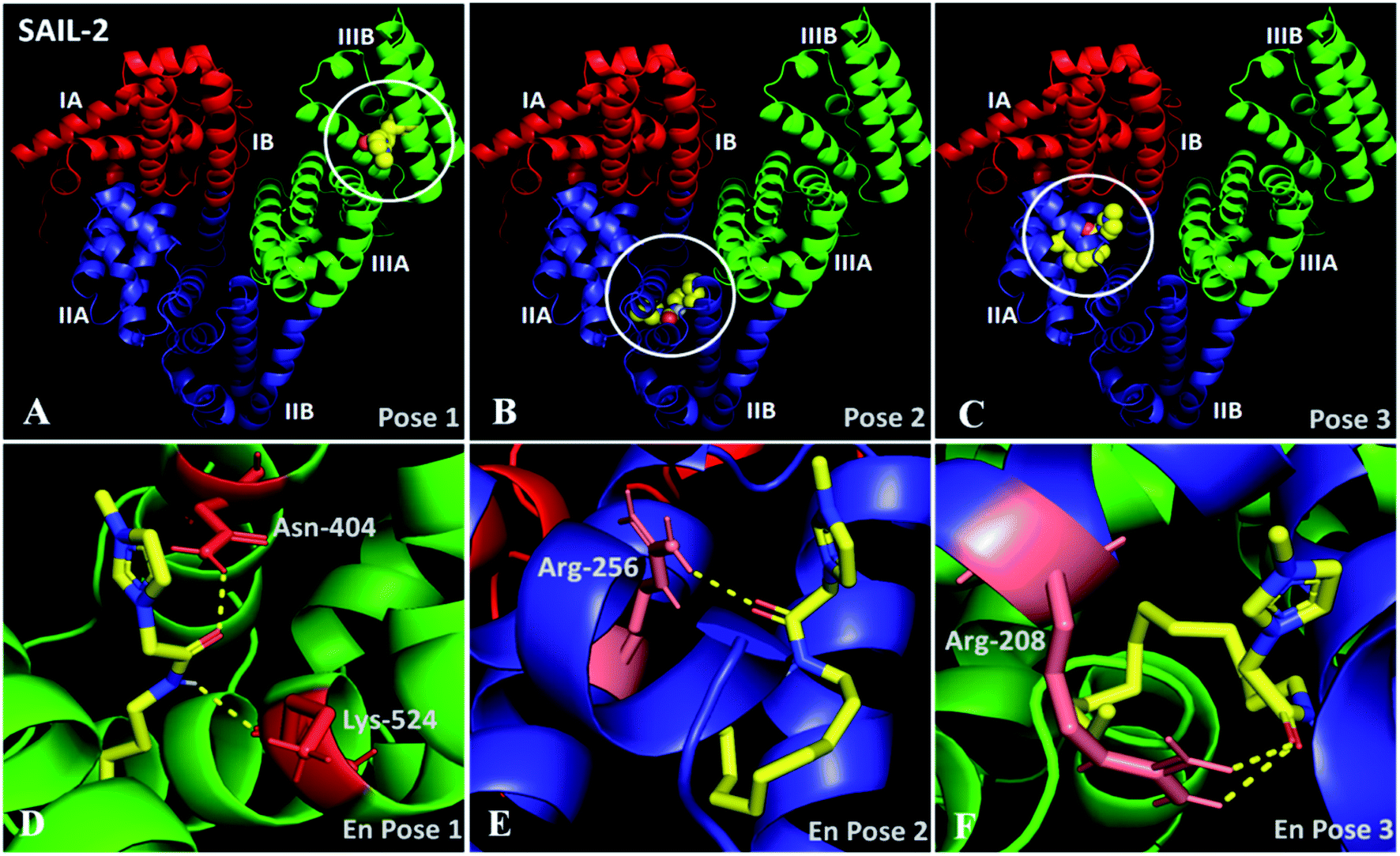 | ||
| Fig. 6 (A–C) Docked conformation of SAIL-2–BSA system showing the high affinity binding sites for SAIL-2 on BSA in different poses; (D–F) enlarged view of respective poses. | ||
In pose 1 (Fig. 6A and D), SAIL-2 is shown to bind in hydrophobic cavity formed between the helices of domain IIB, where it undergoes H-bonding with –NH2 group of Asn-404 and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of Lys-524 through –CO and –NH moiety, respectively, of amide group of SAIL-2. In pose 2 (Fig. 6B and E), SAIL-2 is shown to bind between the helices of sub-domain IIA and IIB where it forms H-bonding with –NH2 group of Arg-256 through –CO of amide group. In pose 3 (Fig. 6C and F), SAIL-2 binds in sub-domain IIA, where it makes two polar interactions with –NH2 groups of Arg-208 through its carbonyl group. Similar binding locations have been obtained for SAIL-3, which however differ in their mode of interactions (Fig. S9A–E, ESI†). In pose 1 (Fig. S9A, ESI†), no such polar interactions are observed which is attributed to flexible nature of ester moiety of SAIL-3. The absence of favorable space orientations required for making such polar interactions cannot be ruled out at this stage. In pose 2 (Fig. S9B and D, ESI†), SAIL-3 makes three polar interactions in its binding cavity, one with –NH2 group of Arg-208 via polar oxygen atom of ester linkage and another two between two –NH2 groups of Arg-208 and oxygen atom of carbonyl group. In pose 3 (Fig. S9C and E, ESI†), SAIL-3 makes two polar interactions of oxygen atom of ester linkage with –NH2 group of Arg-198. It is inferred that the investigated SAILs which differ in their molecular structure prefer different binding sites, however the H-bonding prone SAIL-2 and SAIL-3 exhibit similar tendency towards binding in sub-domain IIA with Arg-208.
O of Lys-524 through –CO and –NH moiety, respectively, of amide group of SAIL-2. In pose 2 (Fig. 6B and E), SAIL-2 is shown to bind between the helices of sub-domain IIA and IIB where it forms H-bonding with –NH2 group of Arg-256 through –CO of amide group. In pose 3 (Fig. 6C and F), SAIL-2 binds in sub-domain IIA, where it makes two polar interactions with –NH2 groups of Arg-208 through its carbonyl group. Similar binding locations have been obtained for SAIL-3, which however differ in their mode of interactions (Fig. S9A–E, ESI†). In pose 1 (Fig. S9A, ESI†), no such polar interactions are observed which is attributed to flexible nature of ester moiety of SAIL-3. The absence of favorable space orientations required for making such polar interactions cannot be ruled out at this stage. In pose 2 (Fig. S9B and D, ESI†), SAIL-3 makes three polar interactions in its binding cavity, one with –NH2 group of Arg-208 via polar oxygen atom of ester linkage and another two between two –NH2 groups of Arg-208 and oxygen atom of carbonyl group. In pose 3 (Fig. S9C and E, ESI†), SAIL-3 makes two polar interactions of oxygen atom of ester linkage with –NH2 group of Arg-198. It is inferred that the investigated SAILs which differ in their molecular structure prefer different binding sites, however the H-bonding prone SAIL-2 and SAIL-3 exhibit similar tendency towards binding in sub-domain IIA with Arg-208.
3.3. Efficacy of BSA–SAIL complexes as colloidal scaffolds for transport
It is inferred that the ACs formed by SAIL-2 and SAIL-3 are relatively more hydrophobic and could show pH dependent structural organization owing to presence of electrostatic interactions between SAILs and BSA. Going with this background, we have investigated pH dependent loading and unloading of a lipophilic dye, Rhodamine 6G (R6G, 25 μM) as a model compound, employing BSA–SAIL-2 colloidal complexes (0.1%/1 mM, near C2 of SAIL-2) system, as a representative. The entire process has been monitored using CSLM (Fig. 7A–D) equipped with the facility of UV-visible absorption measurements (Fig. 7E and F). The change in absorption intensity corresponding to absorption maxima of R6G (λmax ≈ 575 nm) was monitored both in the colloidal complexes (red-cross, Fig. 7A–D) as well as in bulk (green-cross, Fig. 7A–D) as a function of pH. A decrease in absorption corresponding to λmax of R6G (Fig. 7E) present in BSA–SAIL-2 colloidal complex (red-cross, Fig. 7A–D) with simultaneous increase in absorption of R6G (Fig. 7F) present in bulk solution (green-cross, Fig. 7A–D) with decrease in pH from 7 to 3 indicates the unloading of R6B from BSA–SAIL-2 colloidal complex in solvent rich phase.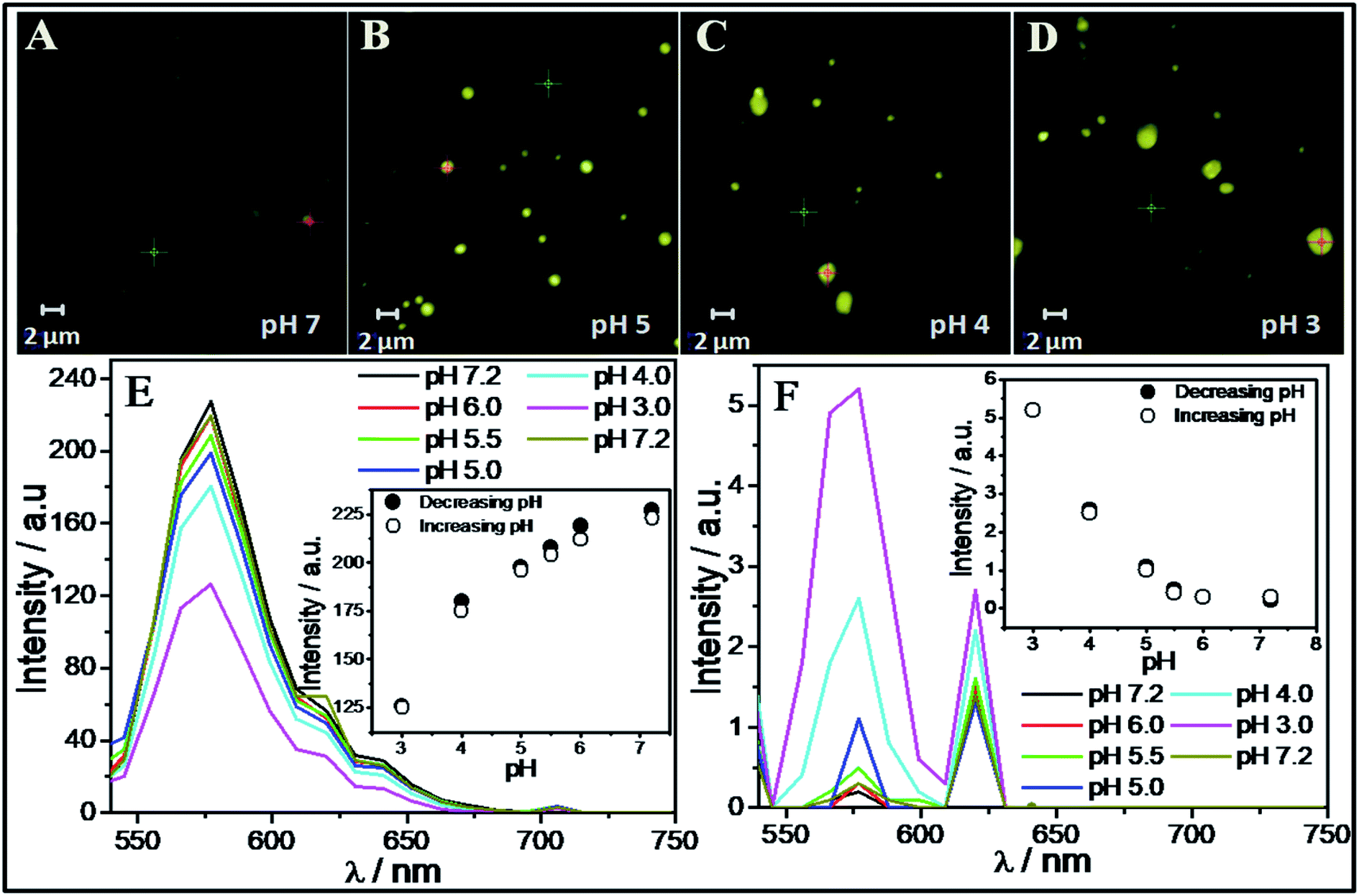 | ||
| Fig. 7 (A–D) CLSM images of R6G loaded in SAIL-2–BSA aggregates complexes at different pH; (E and F) UV spectra of R6G taken at the colored spots and from background solution as marked in CLSM images. | ||
Further with increase in pH, the system show reversible uptake of R6G. The pH induced structural changes in investigated BSA–SAIL-2 colloidal complex are supposed to be the reason behind the reversible loading and unloading of R6G. Such changes have been probed by ζ-potential measurements as the alteration in surface charge and size of the ACs was thought to govern the dye loading and delivery. An increase in the values of ζ-potential from −4 mV to 12 mV has been observed while going from pH 7 to 2 (Fig. S10, ESI†). The protonation of negatively charged amino acid residues of BSA, namely Asp, Glu and His at pH 3 and below results in highly positive ζ-potential. Such high extent of protonation could weaken electrostatic interactions between BSA and SAIL-2 in BSA–SAIL-2 colloidal complexes leading to structural reorganization of complexes. The reversible structural transition from N (pH 7.4) to F-isoform (pH 3.5) with opening up of the inherent globular structure of BSA could diminish the binding site present in the cavity formed by three domains and thus facilitates the release of R6G from its binding site (Fig. S11 and Table S7, ESI†).
The role of increasing hydrophobicity of colloidal complex owing to structural transition from N (pH 7.4) to F isoform (pH 3.5)10 in release of R6G into the solution along with SAIL ions can't be ruled out as solubility of R6G decreases with decrease in polarity of the solvent.71 The studies conducted on BSA–SAIL-2 colloidal complex and R6G establishes a new platform for developing new colloidal systems comprising proteins and SAILs (inherently non-toxic) for transport of biologically important lyophobic/lipophilic molecules along with their targeted and controlled release. Further the role of SAILs used in conjunction with proteins for such applications can be easily modified by the choice of anion or cation or by functionalization of SAILs, if needed. Therefore, easily tunable structural and chemical nature of SAIL–protein based scaffold makes such systems as potential candidates for different biomedical applications.
4. Conclusion
The nature of SAILs is found to affect their complexation behavior with BSA. The H-bond acceptor/donor and H-bond acceptor abilities of SAIL-2 and SAIL-3, respectively, produce distinct alternations in the structure of BSA, which resulted in the formation of SAIL–BSA complexes of different shape and morphology at different stages of complexation. The highest surface activity of SAIL-3–BSA complexes at air–solution interface reflects relatively stronger interactions of SAIL-3 with BSA, which modifies the hydrophobic–hydrophilic index of the formed colloidal aggregates at air–solution interface. On the other hand the amide group of SAIL-2 resembles the peptide linkages of protein and stabilizes the polypeptide network at low concentrations. The molecular docking profile of SAIL-2 and SAIL-3 clearly showed the presence of H-bonding and other polar interactions of these SAILs at their binding locations. The studies conducted to understand the physiochemical behavior of SAIL–BSA system inspired us to develop pH dependent reversible carrier of lipophilic molecule such as R6G dye (as a model hydrophobic drug) in SAIL-2–BSA colloidal system, which has been established successfully.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to DST, Govt. of India wide project number (SB/FT/CS-057/2013) for providing the research grant for this work. We are thankful to UGC, India, for their UGC-CAS program and DST, India, for the FIST program awarded to the Department of Chemistry, Guru Nanak Dev University, Amritsar. M. K. is thankful to CSIR, Govt. of India, for SRF.References
- G. J. Quinlan, G. S. Martin and T. W. Evans, Hepatology, 2005, 41, 1211–1219 CrossRef CAS PubMed
.
- J. P. Doweiko and D. J. Nompleggi, JPEN, J. Parenter. Enteral Nutr., 1991, 15, 207–211 CrossRef CAS PubMed
- M. Roche, P. Rondeau, N. R. Singh, E. Tarnus and E. Bourdon, FEBS Lett., 2008, 582, 1783–1787 CrossRef CAS PubMed
- S. Moghaddassi, Modification of the Bovine Genome for the Large-Scale Production of Human Serum Albumin, PhD dissertation, Wake Forest University Graduate School of Arts and Sciences, Winston-Salem, North Carolina, December 2013 Search PubMed
- E. L. Gelamo, C. H. T. P. Silva, H. Imasato and M. Tabak, Biochim. Biophys. Acta, 2002, 1594, 84–99 CrossRef CAS
- A. M. Merlot, D. S. Kalinowski and D. R. Richardson, Front. Physiol., 2014, 5, 299 Search PubMed
- A. Bujacz, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 1278–1289 CrossRef CAS PubMed
- G. Singh and T. S. Kang, J. Phys. Chem. B, 2015, 119, 10573–10585 CrossRef CAS PubMed
- M. Y. Khan, Biochem. J., 1986, 236, 307–310 CrossRef CAS PubMed
- K. Baler, O. A. Martin, M. A. Carignano, G. A. Ameer and J. A. Vila, J. Phys. Chem. B, 2014, 118, 921–930 CrossRef CAS PubMed
- S. Fujiwara and T. Amisaki, Biochim. Biophys. Acta, 2013, 1830, 5427–5434 CrossRef CAS PubMed
- U. Kragh-Hansen, Pharmacol. Rev., 1981, 33, 17–53 CAS
- J. J. Vallner, J. Pharm. Sci., 1977, 66, 447–465 CrossRef CAS PubMed
- C. Pinholt, R. A. Hartvig, N. J. Medlicott and L. Jorgensen, Expert Opin. Drug Delivery, 2011, 8, 949–964 CrossRef CAS PubMed
- M. C. Manning, J. Liu, T. Li and R. E. Holcomb, Adv. Protein Chem. Struct. Biol., 2018, 112, 1–59 CAS
- D. Otzen, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 562–591 CrossRef CAS PubMed
- J. Maldonado-Valderrama and J. M. R. Patino, Curr. Opin. Colloid Interface Sci., 2010, 15, 271–282 CrossRef CAS
- T. A. Khan, H.-C. Mahlera and R. S. K. Kishore, Eur. J. Pharm. Biopharm., 2015, 97, 60–67 CrossRef CAS PubMed
- A. Chakraborty, D. Seth, P. Setuna and N. Sarkar, J. Phys. Chem. B, 2006, 110, 16607–16617 CrossRef CAS PubMed
- S. F. Santos, D. Zanetti, H. Fischer and R. Itri, J. Colloid Interface Sci., 2013, 662, 400–408 Search PubMed
- S. H. Chen and J. Teixeiria, Phys. Rev. Lett., 1986, 57, 2583 CrossRef CAS PubMed
- D. Kelley and D. J. McClements, Food Hydrocolloids, 2003, 17, 73–85 CrossRef CAS
- E. L. Gelamo, R. Itri, A. Alonso, J. V. da Silva and M. Tabak, J. Colloid Interface Sci., 2004, 277, 471–482 CrossRef CAS PubMed
- T. Chakraborty, I. Chakraborty, S. P. Moulik and S. Ghosh, Langmuir, 2009, 25, 3062–3074 CrossRef CAS PubMed
- Y. Moriyama, Y. Kawasaka and K. Takeda, J. Colloid Interface Sci., 2003, 257, 41–46 CrossRef CAS PubMed
- Y. Li, X. Wang and Y. Wang, J. Phys. Chem. B, 2006, 110, 8499–8505 CrossRef CAS PubMed
- N. J. Turro, X. G. Lei, K. P. Ananthapadmanabhan and M. Aronson, Langmuir, 1995, 11, 2525–2533 CrossRef CAS
- D. Wu, G. Xu, Y. Sun, H. Zhang, H. Mao and Y. Feng, Biomacromolecules, 2007, 8, 708–712 CrossRef CAS PubMed
- K. P. Ananthapadmanabhan, in Interactions of Surfactants with Polymers and Proteins, ed. E. D. Goddard and K. P. Ananthapadmanabhan, CRC Press, Inc, London, UK, 1993, ch. 8 Search PubMed
- M. N. Jones, Chem. Soc. Rev., 1992, 21, 127–136 RSC
- S. Hirlekar, D. Ray, V. K. Aswal, A. Prabhune, A. Nisal and S. Ravindranathan, Langmuir, 2019, 35, 14870–14878 CrossRef CAS PubMed
- T. Singh, M. Drechsler, A. H. E. Müller, I. Mukhopadhyay and A. Kumar, Phys. Chem. Chem. Phys., 2010, 12, 11728–11735 RSC
- Y. Zhao, S. J. Gao, J. J. Wang and J. M. Tang, J. Phys. Chem. B, 2008, 112, 2031–2039 CrossRef CAS PubMed
- B. Dong, N. Li, L. Zheng, L. Yu and T. Inoue, Langmuir, 2007, 23, 4178–4182 CrossRef CAS PubMed
- O. A. El Seoud, P. A. R. Pires, T. Abdel-Moghny and E. L. Bastos, J. Colloid Interface Sci., 2007, 313, 296–304 CrossRef CAS PubMed
- P. Brown, C. P. Butts, J. Eastoe, D. Fermin, I. Grillo, H.-C. Lee, D. Parker, D. Plana and R. M. Richardson, Langmuir, 2012, 28, 2502–2509 CrossRef CAS PubMed
- H. Wang, J. Wang, S. Zhang and X. Xuan, J. Phys. Chem. B, 2008, 112, 16682–16689 CrossRef CAS PubMed
- C. Jungnickel, J. Łuczak, J. Ranke, J. F. Fernandez, A. Muller and J. Thöming, Colloids Surf., A, 2008, 316, 278–284 CrossRef CAS
- R. Dutta, S. Ghosh, P. Banerjee, S. Kundu and N. Sarkar, J. Colloid Interface Sci., 2017, 490, 762–773 CrossRef CAS PubMed
- J. Bowers, P. Butts, J. Martin, C. Vergara-Gutierrez and K. Heenan, Langmuir, 2004, 20, 2191–2198 CrossRef CAS PubMed
- P. Brown, C. Butts, R. Dyer, J. Eastoe, I. Grillo, F. Guittard, S. Rogers and R. Heenan, Langmuir, 2011, 27, 4563–4571 CrossRef CAS PubMed
- P. Brown, C. P. Butts, J. Eastoe, D. Fermin, I. Grillo, H.-C. Lee, D. Parker, D. Plana and R. M. Richardson, Langmuir, 2012, 28, 2502–2509 CrossRef CAS PubMed
- N. V. Sastry, N. M. Vaghela and V. K. Aswal, Fluid Phase Equilib., 2012, 327, 22–29 CrossRef CAS
- I. Goodchild, L. Collier, S. L. Millar, I. Prokeš, J. C. D. Lord, C. P. B. Butts, J. Bowers, J. R. P. Webster and R. K. Heenan, J. Colloid Interface Sci., 2007, 307, 445–468 CrossRef PubMed
- N. V. Sastry, N. M. Vaghela, P. M. Macwan, S. S. Soni, V. K. Aswal and A. Gibaud, J. Colloid Interface Sci., 2012, 371, 52–61 CrossRef CAS PubMed
- G. Singh, G. Singh, S. Kancharla and T. S. Kang, J. Phys. Chem. B, 2019, 123, 2169–2181 CrossRef CAS PubMed
- S. Chabba, R. Vashishat and R. K. Mahajan, J. Mol. Liq., 2018, 259, 134–143 CrossRef CAS
- G. Singh, G. Singh and T. S. Kang, Phys. Chem. Chem. Phys., 2016, 18, 25993–26009 RSC
- T. Singh, S. Boral, H. B. Bohidar and A. Kumar, J. Phys. Chem. B, 2010, 114, 8441–8448 CrossRef CAS PubMed
- P. Bharmoria, M. J. Mehta, I. Pancha and A. Kumar, J. Phys. Chem. B, 2014, 118, 9890–9899 CrossRef CAS PubMed
- P. Bharmoria, T. J. Trivedi, A. Pabbathi, A. Samanta and A. Kumar, Phys. Chem. Chem. Phys., 2015, 17, 10189–10199 RSC
- H. Yan, J. Wu, G. Dai, A. Zhong, H. Chen, J. Yang and D. Han, J. Lumin., 2012, 132, 622–628 CrossRef CAS
- F. Geng, L. Zheng, J. Liu, L. Yu and C. Tung, Colloid Polym. Sci., 2009, 287, 1253–1259 CrossRef CAS
- M. Kumari, J. K. Maurya, U. K. Singh, A. B. Khan, M. Ali, P. Singh and R. Patel, Spectrochim. Acta, Part A, 2014, 124, 349–356 CrossRef CAS PubMed
- X. Wang, J. Liu, L. Sun, L. Yu, J. Jiao and R. Wang, J. Phys. Chem. B, 2012, 116, 12479–12488 CrossRef CAS PubMed
- T. Singh, P. Bharmoria, M. Morikawa, N. Kimizuka and A. Kumar, J. Phys. Chem. B, 2012, 116, 11924–11935 CrossRef CAS PubMed
- P. Bharmoria, K. S. Rao, T. J. Trivedi and A. Kumar, J. Phys. Chem. B, 2014, 118, 115–124 CrossRef CAS PubMed
- F. Zhang, F. Roosen-Runge, M. W. A. Skoda, R. M. J. Jacobs, M. Wolf, P. Callow, H. Frielinghaus, V. Pipich, S. Prévostf and F. Schreibera, Phys. Chem. Chem. Phys., 2012, 14, 2483–2493 RSC
- S. Chodankar, V. K. Aswal, J. Kohlbrecher, R. Vavrin and A. G. Wagh, J. Phys.: Condens. Matter, 2007, 19, 326102–326113 CrossRef
- S. H. Chen and J. Teixeira, Phys. Rev. Lett., 1986, 57, 2583–2586 CrossRef CAS PubMed
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039–2044 CrossRef CAS
- R. Kamboj, P. Bharmoria, V. Chauhan, G. Singh, A. Kumar, S. Singh and T. S. Kang, Phys. Chem. Chem. Phys., 2014, 16, 26040–26050 RSC
- K. J. Bijma, B. F. N. Engberts, M. J. Blandamer, P. M. Cullis, P. M. Last, K. D. Irlam and L. G. Soldi, J. Chem. Soc., Faraday Trans., 1997, 93, 1579–1584 RSC
- K. Bouchemal, Drug Discovery Today, 2008, 13, 960–972 CrossRef CAS PubMed
- S. Paula, W. Süs, J. Tuchtenhagen and A. Blume, J. Phys. Chem., 1995, 99, 11742–11751 CrossRef CAS
- H.-H. Cai, X. Zhong, P.-H. Yang, W. Wei, J. Chen and J. Cai, Colloids Surf., A, 2010, 372, 35–40 CrossRef CAS
- C. Hetényi and D. V. D. Spoel, Protein Sci., 2002, 11, 1729–1737 CrossRef PubMed
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS
- M. M. Jaghoori, B. Bleijlevens and S. D. Olabarriaga, J. Comput.-Aided Mol. Des., 2016, 30, 237–249 CrossRef CAS PubMed
- K. Onodera, K. Satou and H. Hirota, J. Chem. Inf. Model., 2007, 47, 1609–1618 CrossRef CAS PubMed
- A. Penzkofer and W. Leupacher, J. Lumin., 1987, 37, 61–72 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05549e |
| This journal is © The Royal Society of Chemistry 2020 |