
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification, cloning, expression and functional interrogation of the biosynthetic pathway of the polychlorinated triphenyls ambigol A–C from Fischerella ambigua 108b†
Elke R.
Duell‡
 af,
Tobias M.
Milzarek‡
ab,
Mustafa
El Omari
c,
Luis J.
Linares-Otoya
de,
Till F.
Schäberle
de,
Gabrielle M.
König
c and
Tobias A. M.
Gulder
*ab
af,
Tobias M.
Milzarek‡
ab,
Mustafa
El Omari
c,
Luis J.
Linares-Otoya
de,
Till F.
Schäberle
de,
Gabrielle M.
König
c and
Tobias A. M.
Gulder
*ab
aBiosystems Chemistry, Department of Chemistry and Center for Integrated Protein Science Munich (CIPSM), Technical University of Munich, Lichtenbergstraße 4, 85748 Garching, Germany. E-mail: tobias.gulder@ch.tum.de
bChair of Technical Biochemistry, Technische Universität Dresden, Bergstraße 66, 01062 Dresden, Germany. E-mail: tobias.gulder@tu-dresden.de
cInstitute for Pharmaceutical Biology, University of Bonn, Nußallee 6, 53115 Bonn, Germany
dInstitute for Insect Biotechnology, Justus Liebig University of Giessen, Heinrich-Buff-Ring 26–32, 35392 Giessen, Germany
eDepartment of Bioresources, Fraunhofer Institute for Molecular Biology and Applied Ecology, Giessen, Germany
fInstitute for Infectious Diseases and Zoonoses, Ludwig-Maximilians-University Munich, Veterinärstraße 13, 80539 Munich, Germany
First published on 20th August 2020
Abstract
The terrestrial cyanobacterium Fischerella ambigua 108b produces the three polychlorinated triphenyls ambigol A–C that exhibit interesting antimicrobial, antiviral and cytotoxic activities. They are structurally related to polybrominated diphenylethers synthesized by diverse marine bacteria that are known to be highly toxic and are bioaccumulating in natural food webs. All ambigols display unusual connectivities: Ambigols A and B exhibit chlorination and ambigol C biaryl-ether bonds in the relative meta position at the central phenol unit, which is flanked by two 2,4-dichlorophenol units in all three compounds. Here we report on the identification of the biosynthetic gene cluster (BGC) reponsible for ambigol production in F. ambigua. After bioinformatic discovery of a putative ambigol BGC (ab) containing 10 genes, we cloned and heterologously expressed this cluster in Synechococcus elongatus PCC 7942 using Direct Pathway Cloning (DiPaC). In vivo and in vitro characterization of the two cytochrome P450 enzymes present in the ab BGC revealed complementary selectivity for either biaryl-ether bond (Ab2) or biaryl formation (Ab3) and provided a biosynthetic route to the ambigols.
Introduction
Polyhalogenated aromatics can be found almost everywhere in Nature and are often associated with negative biological effects on human and animal life, especially as they are known to be highly persistent and bioaccumulative in natural food webs. Many toxic representatives of this class of molecules have an anthropogenic source and their incautious distribution in the environment is clearly caused by humankind. Amongst these substances are the polychlorinated biphenyls (PCBs, 1), formally used, e.g., as industrial coolants and plasticizers, and the polybrominated diphenyl ethers (PBDEs, 2a/b), utilized as flame retardants (Fig. 1A). These compounds are now restricted from commercial use due to their toxicity and aggregation in the environment. Polyhalogenated dibenzo-p-dioxines (PHDDs), e.g., 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, 3), which is a side product in the synthesis of herbicides such as 2,4,5-trichlorophenoxyacetic acid (trioxone), is one of the most toxic and environmentally persistent man-made pollutants known today.1–3 The structurally related substance triclosan (4), which was developed as disinfectant, can still be found in toothpastes, soaps and detergents. However, recent studies suggest it to be very likely toxic for animals and humans,4–6 especially due to formation of PHDDs from 4via its photodegradation.7,8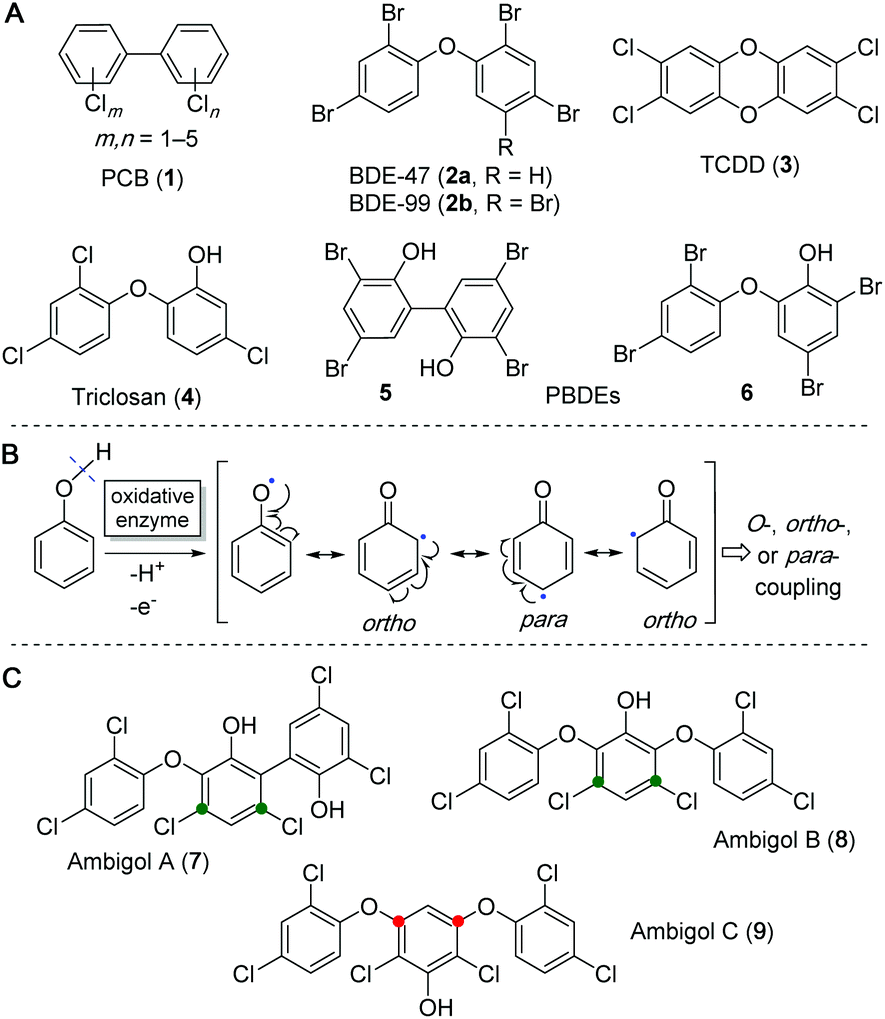 | ||
| Fig. 1 (A) Structures of selected synthetic (1–4) and naturally occurring (5, 6) polyhalogenated aromatic compounds. (B) The radical mechanism underlying oxidative coupling of phenols allows reactions only at the phenol oxygen or in relative ortho and para position to this activating group. The identical positions are activated for nucleophilic halogenation by flavin-dependent halogenases. (C) Ambigols A (7) and B (8) show chlorination in meta position (green dots), ambigol C (9) contains two biaryl-ether-bonds in meta position (red dots) within the central phenol building block. | ||
However, not all harmful polyhalogenated aromatic substances are of anthropogenic origin. A wide range of PBDEs, such as 5 and 6, has been isolated from marine invertebrates9–11 and only recently it was shown that they are produced by a variety of marine bacteria such as γ-proteobacteria of the genus Pseudoalteromonas and the cyanobacterial Hormoscilla spongeliae spp.12,13 These naturally occurring PBDEs, including their hydroxylated (OH-BDEs) and dioxine-like derivatives, can be found in all trophic levels of marine life, are accumulating in the food chain via marine plants14 and algae,15–17 and are ultimately detectable in marine animals18–20 and even human tissue.21–24 Similar to their man-made relatives, they exhibit toxic effects on mammalian hormone mediated signalling pathways and essential enzymatic reactions.24–28 The biosynthesis of these PBDEs of natural origin has been extensively studied by pioneering work of the Moore laboratory over the last years, leading to the discovery of the encoding biosynthetic gene cluster (BGC) bmp, which can be found in varying genetic compositions in all bacterial strains associated with PBDE biosynthesis.12,13 The biosynthesis of PBDEs utilizes the primary metabolite chorismic acid (10), which is converted to 4-hydroxybenzoic acid (4-HBA, 11) by the chorismate lyase Bmp6 (cf.Fig. 3).29,30 A so far unprecedented decarboxylative phenol bromination mechanism, catalyzed by the halogenase Bmp5, uses 11 as a substrate to form 2,4-dibromophenol (2,4-DBP).12 The cytochrome P450 enzyme Bmp7 is then able to couple two 2,4-DBP units to form a variety of different dimeric PBDEs.12,13,30 These biaryl- and biaryl-ether-coupling reactions between aromatic rings follow the common principle of phenol oxidative coupling, where an initial hydroxyl radical is formed by abstraction of one electron and one proton (Fig. 1B).31 Due to mesomeric delocalization of the free electron on the benzene ring in relative ortho and para position to the oxygen functionality, a large diversity of C–C- and C–O–C-bond containing coupling products can be biosynthesized.32
Not only marine but also terrestrial cyanobacteria produce polyhalogenated phenols. Due to its abundance in soil, chlorine is generally favoured over bromine for incorporation into halogenated natural products of terrestrial origin. In 1993 and in 2005, the three polychlorinated ambigols A–C (7–9, Fig. 1C) were discovered from the soil-dwelling filamentous cyanobacterium Fischerella ambigua 108b. Ambigol A (7) exhibits strong antimicrobial, antifungal and virostatic activities and moderate cytotoxicity whereas ambigol C (9) shows exclusive, potent activity against Gram positive bacteria and fungi and ambigol B (8) generally displays only weak activity in biological assays.33–35 At first glance, 7–9 are structurally related to the marine PBDEs as they also contain two halogen atoms per phenol and are linked either by biaryl- or biaryl-ether-bonds. Unlike the PBDEs, 7–9 incorporate a third dihalophenol unit. In addition, either the chlorine substituents (in 7 and 8) or the biaryl ether structural motif (in 9) are not located at the typical relative ortho or para position to the activating phenol function in the central aromatic portion, but rather situated in meta position (Fig. 1C). Owing to the interesting biological activities of the ambigols 7–9 and these structural peculiarities pointing at unusual biosynthetic transformations, we investigated ambigol biosynthesis within this work, with a focus on the installation of the biaryl structural features.
Results and discussion
Bioinformatic analysis of the ab BGC
To identify the ambigol BGC, the natural producer Fischerella ambigua 108b was submitted to 454 genome sequencing (see ESI†). Bioinformatic screening for the presence of halogenases allowed for the identification of a putative ambigol BGC ab (14.3 kb) containing 10 genes (Table 1), among them a number of genes predicted to be required for ambigol biosynthesis (Fig. 2). All 10 putative ab genes appear to be located on a single transcript. Upstream of ab1 a non-coding DNA region is followed by a gene encoding a protein kinase while ab10 is neighbored by genes encoding two small hypothetical proteins and two transposases, thus indicating the likely borders of the ab BGC. Besides genes encoding halogenating enzymes, additional similarities to the gene clusters encoding the structurally related PBDEs were expected. Indeed, ab contains a gene encoding a chorismate lyase ab5 that is highly homologous to bmp6 found in H. spongeliae.13 In analogy to the bmp pathway, Ab5 thus likely recruits building blocks from the pool of primary metabolites for ambigol biosynthesis by formation of 4-HBA (11) from chorismic acid (10). Furthermore, ab contains a gene encoding a 3-deoxy-7-phosphoheptulonate (DAHP) synthase (ab7), an enzyme catalyzing the first dedicated step in the shikimic acid biosynthetic pathway towards 10 (see Fig. 3).| Gene | Size (bp) | Proposed function | Closest xBLAST hit | Identity (protein) | GenBank acc. number |
|---|---|---|---|---|---|
| ab1 | 1452 | Precursor chlorination | NAD(P)/FAD-dependent oxidoceductase [Scytonema hofmanii] | 281/486 (58%) | WP_017744817.1 |
| ab2 | 1464 | Biaryl coupling | Cytochrome P450 [Hormoscilla spongeliae GUM020] | 337/485 (69%) | AQU14168.1 |
| ab3 | 1476 | Biaryl coupling | Cytochrome P450 [Hormoscilla spongeliae GUM020] | 301/474 (64%) | AQU14168.1 |
| ab4 | 615 | Unknown | Cyclase [Nostoc punctiforme NIES-2108] | 123/180 (68%) | RCJ31428.1 |
| ab5 | 624 | Synthesis of 11 from 10 | Chorismate lyase [Hormoscilla spongeliae GUM096] | 106/177 (60%) | AQU14179.1 |
| ab6 | 1536 | Acyl activation | Acyl-CoA synthetase [filamentous cyanobacterium CCP3] | 410/511 (80%) | WP_106922238.1 |
| ab7 | 1131 | Synthesis of DAHP | 3-Deoxy-7-phosphoheptulonate synthase [Nostoc minutum NIES-26] | 296/374 (79%) | RCJ32495.1 |
| ab8 | 273 | PKS (ACP) | Acyl carrier protein [Chroogloeocystis siderophilia] | 49/81 (60%) | WP_073551326.1 |
| ab9 | 2631 | NRPS (C-PCP-TE) | non-ribosomal peptide syntethase [Moorea producens] | 519/870 (60%) | WP_008188544.1 |
| ab10 | 1725 | Precursor chlorination | FAD-dependent oxidoreductase [Scytonema sp. HK-05] | 430/573 (75%) | WP_073630058.1 |
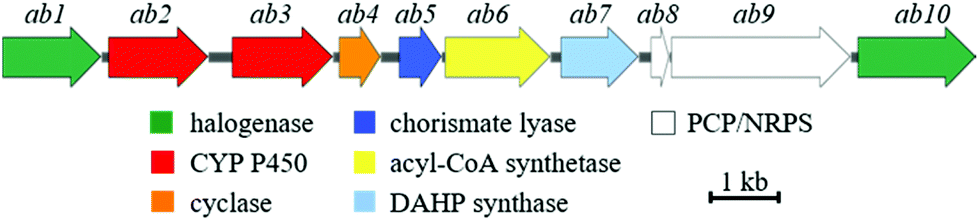 | ||
| Fig. 2 Schematic illustration of the genetic organization within the ab BGC from F. ambigua containing ten genes. | ||
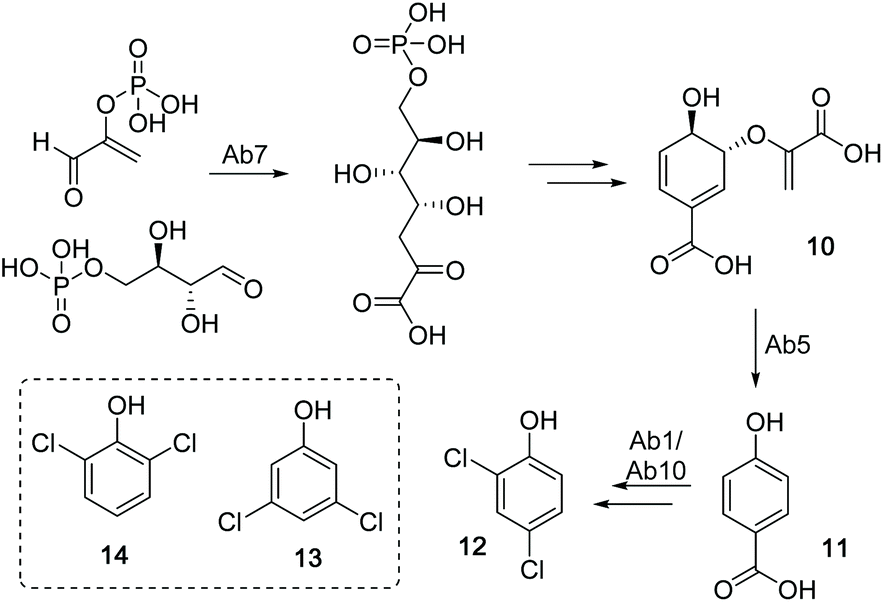 | ||
| Fig. 3 Biosynthetic route to the ambigol precursor 12 from chorismic aicd (10) via 4-HBA (11). Box: alternative chlorinated precursors 13 and 14. | ||
The entire ab BGC is framed by two genes encoding FAD-dependent halogenases, ab1 and ab10. As in PBDE biosynthesis, 11 has to be chlorinated twice to give 2,4-dichlorophenol (12) as monomeric building block found in all ambigol structures. Formation of monomeric precursors in case of PDBE biosynthesis was shown to be catalyzed by a single halogenase, Bmp5, which not only performs an ortho bromination, but also a highly unusual second decarboxylative halogenation, thus replacing the acid function in para position to the phenol in 11 by a bromine substituent.12 In contrast, the ab BGC contains genes encoding two FAD-dependent halogenases ab1 and ab10. The underlying mechanistically different halogenation reactions towards 12 therefore might require two separate halogenases in the case of ambigol biosynthesis. Interestingly, neither ab1 nor ab10 show a particularly strong homology to bmp5. Alternatively, one of the halogenases might be involved in the assembly of 3,5-dichlorophenol (13) or 2,6-dichlorophenol (14), as potential further required central ambigol biosynthetic building blocks (Fig. 3). Three additional genes located in ab, i.e., ab6, ab8, and ab9, resemble elements of PKS/NPRS biosynthesis for precursor activation (ab6) and covalent tethering (ab8, ab9). The corresponding proteins might thus likewise be involved in enzymatic halogenation, as PCP-tethering is known to be required by a number of FAD-dependent halogenases.36–39
However, despite considerable attempts to obtain soluble halogenases Ab1 and Ab10 (data not shown), we did not yet succeed in recombinant production of these enzymes in a soluble form and thus could not further probe their functions. In case of PDBEs, cross-coupling of the phenolic building blocks typically occurs in the mechanistically permitted ortho and para positions relative to an activating phenol function (cf.Fig. 1B). The bmp BGC in γ-proteobacteria only contains a single cytochrome P450 encoding gene, bmp7. The corresponding protein catalyzes the required biaryl bond formations between two units of 2,4-dibromophenol.13 In contrast, the ab cluster harbours two genes encoding the closely related CYP P450 homologs Ab2 and Ab3. It was thus tempting to speculate that one of these enzymes catalyzes biaryl bond formation, while the other homolog is involved in biaryl ether assembly. Such a complementary set of C–C- and C–O–C- coupling enzymes would be a valuable tool for the biocatalytic synthesis of biaryl structures. We thus set out to firmly link the putative ambigol BGC to ambigol production and to characterize the function of the cytochrome P450 enzymes Ab2 and Ab3.
Cloning and expression of the ab BGC in E. coli and S. elongatus
To unambiguously link the putative ambigol BGC ab to ambigol biosynthesis, we first aimed at recombinant production of the target molecules. Mutation free cloning of the whole ab cluster into pET-28b-SUMO as well as subsequent addition of a gfp gene downstream of the ab BGC as reporter for successful transcription of the entire pathway40 was achieved using our DiPaC strategy.40–42 Initial expression tests with the resulting construct in E. coli BAP1 harbouring pET-28b-SUMO::ab-gfp did not show production of any new compounds when compared to empty cells as controls, nor of Gfp-mediated fluorescence. The T7 promoter was therefore exchanged by ptetO, as this promoter is known to efficiently upregulate larger transcripts.43 While the resulting system pET28b-ptetO::ab-gfp facilitated the expression of Gfp, as indicted by significant fluorescence of the recombinant E. coli BAP1 host cells (see ESI Fig. S5†) thus clearly also confirming complete transcription of all ab genes, again no production of ambigols or potential intermediates was observable. The reason for this is likely the inability of E. coli to translate and/or fold one or more enzymes of the ab BGC into their catalytically active forms. Related problems were observed by Moore et al. when expressing selected genes of the bmp BGC of cyanobacterial origin,13 while the bmp genes from γ-proteobacteria, by contrast, were expressible in E. coli.12,30,39,44 We thus focused on the recombinant production of ambigols in a phylogenetically more closely related species when compared to F. ambigua, i.e., the cyanobacterial heterologous host Synechococcus elongatus PCC 794245,46 previously utilized for the production of PBDEs13 and industrially important compounds such as butyrate,47 2-deoxy-scyllo-inose48 and propanediol.48,49 Due to the size of the ab BGC and the known difficulties in introducing large DNA fragments into S. elongatus by natural transformation, we decided to split the ab BGC in two halves of approx. 7–8 kb and to sequentially integrate these parts into two independent genomic sites of S. elongatus PCC 7942. Mutation free integration of ab1–5 into pAM505146 and ab6–10 into pCV009413 as well as the integration of ab6–10 into the chromosomal site NSII of S. elongatus by natural transformation was successful (see ESI Fig. S3†). Generation of double transformants additionally carrying ab1–5 in all copies of NSI was subsequently verified by PCR. To the best of our knowledge, this is the first introduction of a >10 kb BGC into the S. elongatus host system. More importantly, heterologous production of the encoded metabolites was observable using this system (Fig. 4): production of a dichlorophenol-dimer with m/z 323.1 [M − H]−, later identified to be 2,4-dichloro-6-(2,4-dichlorophenoxy)-phenol (36, see Fig. 5 and ESI Fig. S7†), and one trimeric molecule with m/z 482.7 [M − H]− corresponding to the ambigol molecular mass was observed at the retention time of ambigol A (7, cf. Fig. 7D). In addition, two dichlorophenol-trimers each lacking one Cl-atom with m/z 449.1 [M − H]− were produced. As no dichlorophenol monomer was detectable, we assume that this intermediate is rapidly consumed by the downstream enzymes to give the observed dimeric and trimeric products. Each of the observed products displayed the expected isotopic pattern of multi-chlorinated substances (Fig. 4). The corresponding signals were not present in wildtype S. elongatus control experiments, thus firmly linking ambigol production to the identified ab BGC. Unfortunately, the recombinant production system proved to be unstable, likely due to the metabolic stress exerted on the recombinant S. elongatus host system resulting from integration of two genetic elements requiring cultivation of the highly sensitive host system in the presence of multiple antibiotics. In general, the double mutant S. elongatus cells took significantly longer to grow when compared to wild type or single mutant cells and were visibly less vital.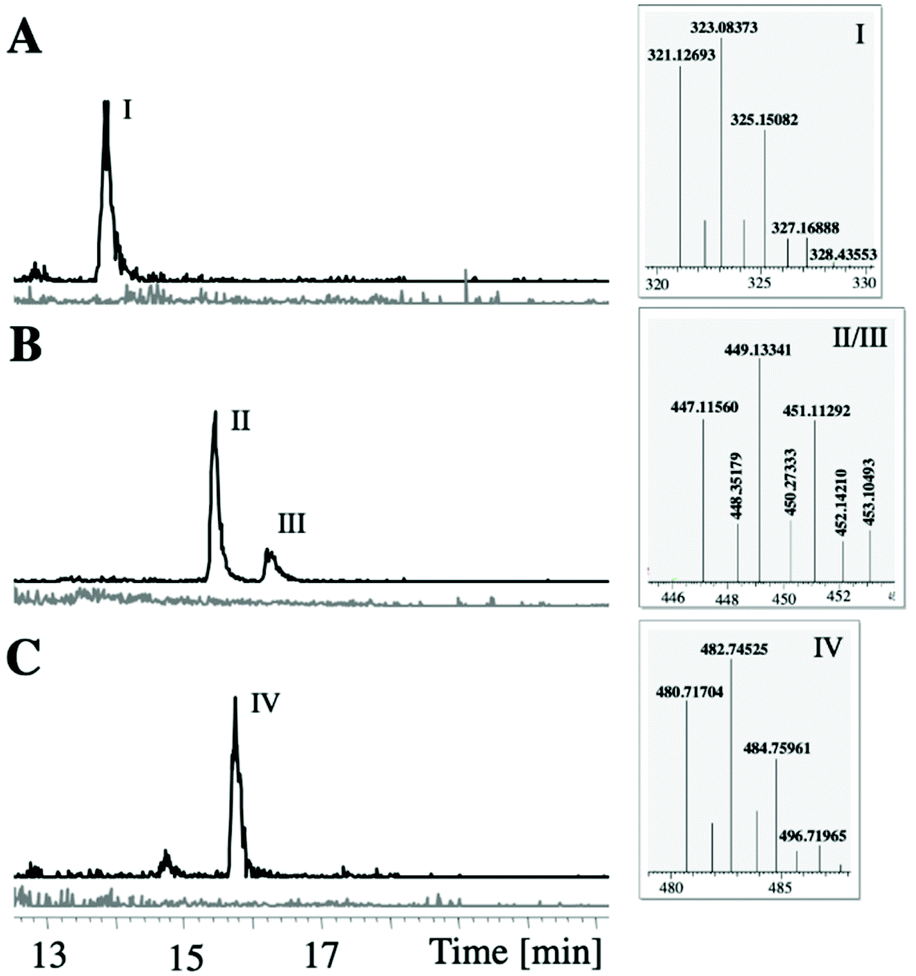 | ||
| Fig. 4 LC-MS analysis of pellet extracts of S. elongatus WT (grey) and S. elongatus simultaneously carrying ab1–5 in NSI and ab6–10 in NSII (black). The production of a DCP-dimer I (A, detected at m/z = 322.5–323,5), two trimers-Cl II and III (B, detected at m/z = 448.0–450.0) and one ambigol trimer IV (C, detected at m/z = 482.0–483.0, retention time corresponding to ambigol A (7), cf. Fig. 7) showing the characteristic Cl-isotopic pattern (see boxes) can only be observed in the presence of the ab cluster. | ||
 | ||
| Fig. 5 Developed synthetic routes for the formation of C–C-coupled dimers 33–35 and of C–O–C-coupled dimers 36–41. | ||
 | ||
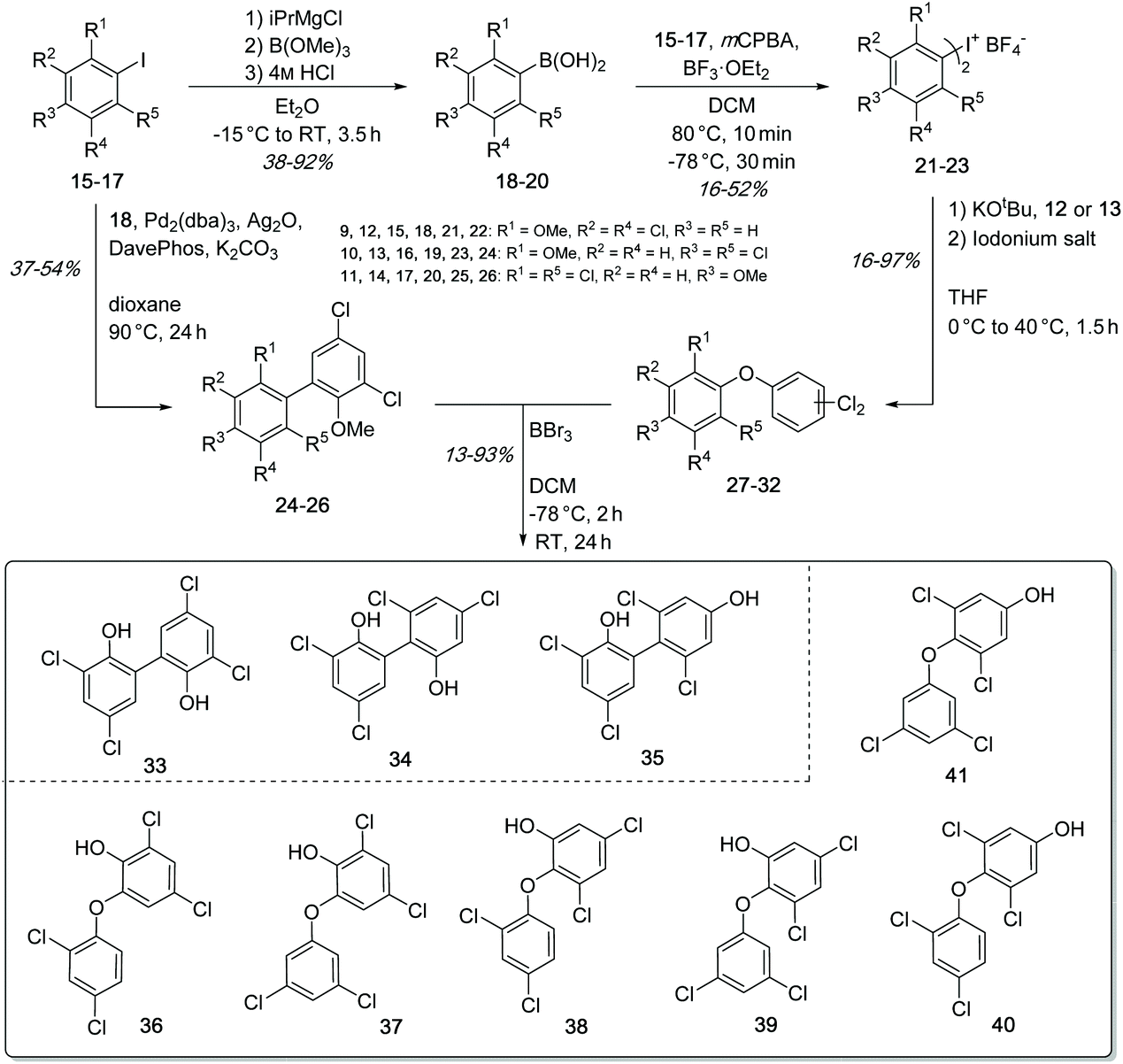
| Fig. 6 In vivo and in vitro catalytic activity of Ab2 and Ab3 when incubated with 12. Ab2 predominantly catalyzed the formation of the C–O–C coupled dimer 36 whereas Ab3 selectively produced the C–C coupled dimer 33. EIC (ESI-) at m/z = 322.5–323.5. I = injection peak. | ||
 | ||
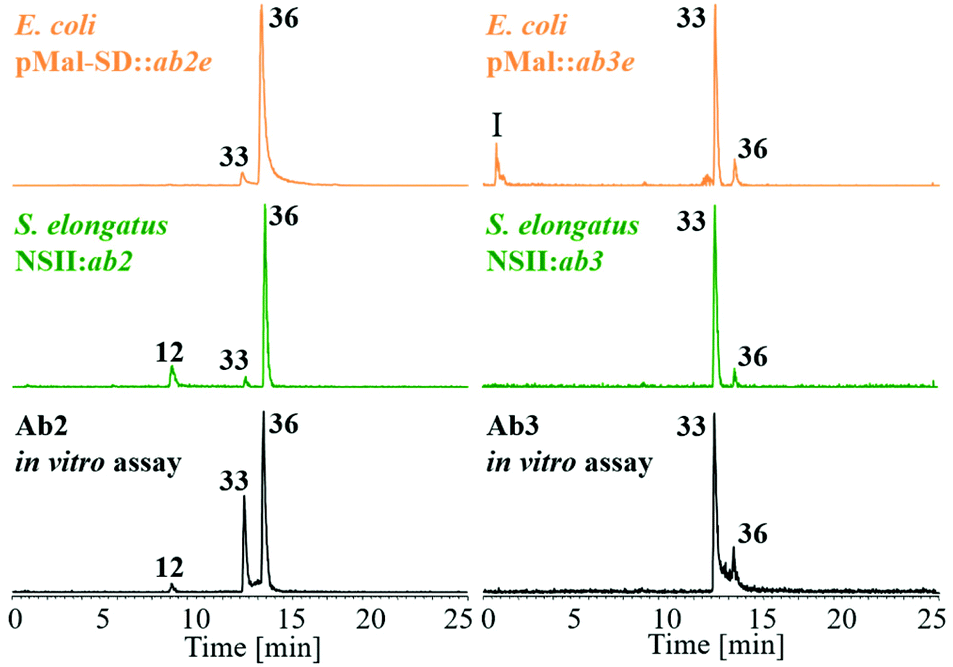
| Fig. 7 (A)–(C) HPLC-MS analysis at m/z = 322.5–323.5 of the product spectrum of Ab2 (A), Ab3 (B) and a combination of Ab2 and Ab3. (C) In vitro (black trace) and in vivo (brown trace) when simultaneously incubated with 12 and 13 as substrates compared to synthetic standards of 33 (green), 35 (blue) and 36 (olive). (D) In vitro assembly of ambigol A (7) from 12 and 35 catalyzed by Ab2 (black) compared to an authentic standard of 7 (red). | ||
Cloning, expression and purification of the CYP P450 enzymes Ab2 and Ab3
We next turned our attention to the in vivo and in vitro characterization of the enzymes Ab2 and Ab3 putatively involved in biaryl formation. For the in vivo experiments, we again utilized S. elongatus as well as E. coli BL21 as host organisms. Mutation free integration of the native sequences of ab2 and/or ab3 into pCV0092 was confirmed by sequencing. Successful natural transformation of S. elongatus with these constructs was verified by PCR screening (see ESI Fig. S4†). As this heterologous system is not capable of producing high amounts of proteins after induction, the transformed cells were only used for in vivo feeding experiments. For in vitro work, E. coli codon optimized versions of ab2 and ab3 were successfully integrated into the pMal expression plasmid containing an N-terminal MBP-tag. Heterologous overexpression in E. coli BL21 and purification via MBP-tag based affinity chromatography led to soluble protein, resulting in a slightly yellow enzyme solution (see ESI Fig. S6†). Before characterizing the enzymatic transformation catalyzed by these CYPs, we set out to synthesize all feasible C–C and C–O–C coupling dimers as analytical standards to allow for an unambiguous structural assignment, as very similar chromatographic and spectroscropic properties were to be expected for these structurally highly related compounds.Chemical synthesis of standards and functional characterization of the CYP P450 enzymes in vitro and in vivo
The synthesis of the biaryls 33–35 and biaryl-ethers 36–41 was performed by an improved palladium-catalyzed Suzuki cross-coupling reaction as well as a nucleophilic aromatic substitution (SNAr) using symmetric hypervalent iodine(III) salts (Fig. 5). The basic building blocks, 2,4-dichlorophenol (12) and 3,5-dichlorophenol (13) were transformed into methoxy-protected aryl iodides 15–17 (see ESI†), which were used to synthesize the respective boronic acids 18–20 through metal-halogen exchange in 38–92% yield. By Suzuki coupling, the aryl iodides 15–17 and (3,5-dichloro-2-methoxy-phenyl)boronic acid (18) were coupled to give the protected biaryls 24–26 in 37–54% yield. The symmetric iodonium salts were synthesized using the same aryl iodides 15–17 and the corresponding boronic acids 18–20 under oxidative conditions.50 The subsequent conversion in the SNAr reaction led to the methoxy-protected biaryl-ethers 27–32 in yields of 16–97%.51 In order to obtain the desired dimers 33–41, the methoxy protecting groups in 24–32 were cleaved off using boron tribromide. The deprotection gave the expected products 33–41 in 13–93% yield. It should be noted that the total yield of all executed reactions is highly dependent on the steric hindrance of the building blocks. In summary, we have developed a short and fast synthetic route for the formation of all required C–C and C–O–C dimers, using functionalized aromatic building blocks.LC-MS analysis of both in vivo (feeding experiments) and in vitro assays of Ab2 with substrate 12 showed the predominant formation of one coupling product with m/z 323.0 [M − H]− (Fig. 6). NMR analysis of the isolated product suggested the production of 2,4-dichloro-6-(2,4-dichlorophenoxy)phenol (36). Comparison of HPLC retention times and 1H-NMR spectra of biochemically produced dimer with the synthetic standards indeed confirmed the identity of 36 (see ESI Fig. S7, S12 and 13†). A formation of trimeric products was not observed.
Ab3 catalyzed the formation of a major product with identical mass but different retention time when compared to the Ab2 product in both in vitro and in vivo experiments (Fig. 6). Comparison to the synthetic standards revealed the dimer to be 3,3′,5,5′-tetrachloro-[1,1′-biphenyl]-2,2′-diol (33) resulting from a biaryl-coupling of two units of 12 in relative ortho position to the phenol functions (see ESI Fig. S9 and 14†). Particularly in in vitro assays, Ab2 and Ab3 were found to also catalyze the formation of small amounts of the dimer characteristic of the respective other CYP P450 enzyme. In addition to these products, small amounts of two DCP trimers at m/z 449.1 [M − H]− and m/z 449.0 [M − H]−, thus lacking one chlorine substituent, were likewise produced both in vitro and in vivo in the presence of Ab3 (see ESI Fig. S11†), in agreement with the observed product spectrum in the S. elongatus recombinant ab expression host (see above). The combination of both Ab2 and Ab3 in one assay did not lead to the formation of any additional, new products. In vivo and in vitro assays with 2,4-DBP instead of 12 showed that both Ab2 and Ab3 can accept and couple this substrate as well, following the same site selectivity. On the basis of these experimental results, we identified two cytochrome P450 enzymes that complementarily perform either biaryl- or biaryl-ether bond formation with 12 as substrate.
To obtain further insights into the biosynthesis of ambigol precursor molecules, Ab2 and Ab3 were simultaneously probed with 12 and 13, both in vivo in E. coli (Fig. 7; brown chromatogram) and in vitro (Fig. 7; black). Compared to incubation with 12 alone, Ab2 retained identical selectivity for production of 36in vivo (cf.Fig. 6) and also showed similar product distribution of 33versus36in vitro (Fig. 7A). However, the in vitro experiment additionally revealed the formation of 35, the central building block of ambigol A (7), the major ambigol congener in F. ambigua.
For Ab3, by contrast, a significantly altered product spectrum was observed. While only small amounts of dimer 33 were still detectable, Ab3 predominantly produced biaryl 35 both in vivo and in vitro (Fig. 7B), thus showing pronounced selectivity for the production of the mixed cross-coupling product of 12 and 13. Combination of Ab2 and Ab3 in a single assay delivered the result expected for combining the reaction outcomes of the individual enzymatic transformations (Fig. 7C).
Given the selectivity of Ab3 to form the C–C-coupled precursor 35 of ambigol A (7) and the previously observed selectivity of Ab2 to perform C–O–C-bond formation (cf.Fig. 6), we next incubated Ab2 with 35 and 12, the missing building block to conclude ambigol A (7) assembly via C–O–C-coupling. This indeed led to the selective formation of 7, as evidenced by HPLC-MS analysis and comparison with an authentic standard of 7 isolated from F. ambigua (Fig. 7D). Traces of 7 were also obtained upon simultaneous incubation of Ab2 and Ab3 with both 12 and 13. These experiments thus unambiguously proof that Ab2 and Ab3 perform step-wise phenol-oxidative cross-coupling of 12 and 13 to yield ambigol A (7).
Conclusions
Within this work, we set out to identify the biosynthetic machinery for the production of the polyhalogenated ambigols. A putative ambigol BGC ab was identified by sequencing of the natural producer F. ambigua 108b. Using the model cyanobacterial heterologous expression system S. elongatus, we firmly linked ambigol A (7) production to the identified ab BGC containing ten genes. To our knowledge, this study is the first example for a successful construction of S. elongatus double mutants by integrating in total more than 14 kb foreign DNA into the two neutral sites NSI and NSII.To shed light on the biaryl coupling biochemistry involved in ambigol biosynthesis, the function of the two cytochrome P450 enzymes, Ab2 and Ab3, encoded in ab were probed both in vivo (in E. coli and S. elongatus) and in vitro using purified recombinant protein produced in E. coli. Using 12 as a substrate, all these experiments consistently revealed Ab2 to selectively form the O,ortho-coupled biaryl ether 36, whereas Ab3 produces the ortho,ortho-linked, C–C-coupled biaryl 33. The identity of these products was unambiguously established by comparison with all feasible coupling products 33–41 that were prepared by chemical synthesis. Given the structural simplicity of the joint precursor molecule 2,4-dichlorophenol (12), the exceptional site selectivity of Ab2 and Ab3 is highly remarkable and warrants future studies on the underlying structural basis of this selectivity in these biaryl coupling enzymes.
Key to fully uncover the biosynthetic assembly of 7 were in vitro experiments using the two bis-halogenated monomeric phenols 12 and 13 as substrates for Ab2 and Ab3. These experiments revealed the production of the mixed C–C-coupled 35, a central precursor to 7, particularly by Ab3. Incubation of 35 and 12 with Ab2 selectively delivered ambigol A (7), revealing the stepwise assembly of this polyhalogenated polyphenol by an interplay of the two cytochrome P450 enzymes Ab2 and Ab3 encoded in the ambigol BGC ab.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Dr James W. Golden and Dr Arnaud Taton for sharing their S. elongatus PCC 7942 based expression system including the plasmids pAM5054 and pCV0094 and for their helpful advice. We furthermore thank Stefan Ernst and Prof. Dr Aymelt Itzen (UKE Hamburg) for providing the pMal expression plasmid. We also thank Prof. Dr Timo Niedermeyer (University of Halle) for providing isolated standards of the ambigols and the group of Prof. Dr Stephan A. Sieber (TU Munich) for measuring HRMS data. E.R.D. thanks the Deutsche Bundesstiftung Umwelt (DBU) for her Ph.D. scholarship and CIPSM-Women for funding. T.M.M. thanks the Stiftung der Deutschen Wirtschaft (sdw) for funding. We thank the Max Buchner Research Foundation and the DFG for generous financial support of the work in our laboratory (Emmy Noether program (GU 1233/1-1) and Center for Integrated Protein Science Munich CIPSM).Notes and references
- R. A. Hites, Dioxins: An Overview and History, Environ. Sci. Technol., 2011, 45, 16–20 CrossRef CAS.
- M. Van den Berg, L. Birnbaum, A. T. Bosveld, B. Brunstorm, P. Cook, M. Feeley, J. P. Giesy, A. Hanberg, R. Hasegawa, S. W. Kennedy, T. Kubiak, J. C. Larsen, F. X. van Leeuwen, A. K. Liem, C. Nolt, R. E. Peterson, L. Poellinger, S. Safe, D. Schrenk, D. Tillitt, M. Tysklind, M. Younes, F. Waern and T. Zacharewski, Toxic equivalency factors (TEFs) for PCBs, PCDDs, PCDFs for humans and wildlife, Environ. Health Perspect., 1998, 106, 775–792 CrossRef CAS.
- M. Van den Berg, L. S. Birnbaum, M. Denison, M. De Vito, W. Farland, M. Feeley, H. Fiedler, H. Hakansson, A. Hanberg, L. Haws, M. Rose, S. Safe, D. Schrenk, C. Tohyama, A. Tritscher, J. Tuomisto, M. Tysklind, N. Walker and R. E. Peterson, The 2005 World Health Organization Reevaluationof Human and Mammalian Toxic Equivalency Factors for Dioxins and Dioxin-Like Compounds, Toxicol. Sci., 2006, 93, 223–241 CrossRef CAS.
- J. L. Wu, F. Ji, H. Zhang, C. Hu, M. H. Wong, D. Hu and Z. Cai, Formation of dioxins from triclosan with active chlorine: A potential risk assessement, J. Hazard. Mater., 2018, 367, 128–136 CrossRef.
- H. Yang, W. Wang, K. A. Romano, M. Gu, K. Z. Sanidad, D. Kim, J. Yang, B. Schmidt, D. Panigraphy, R. Pei, D. A. Martin, E. I. Ozay, Y. Wang, M. Song, B. W. Bollinh, H. Xiao, L. M. Minter, G. Y. Yang, Z. Liu, F. E. Rey and G. Zhang, A common antimicrobial additive increases colonic inflammation and colitis-associated colon tumorigensis in mice, Sci. Transl. Med., 2018, 10 Search PubMed.
- M. F. Yueh, K. Taniguchi, S. Chen, R. M. Evans, B. D. Hammock, M. Karin and R. H. Tukey, The commonly used antimicrobial additive triclosan is a liver tumor promoter, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17200–17205 CrossRef CAS.
- R. U. Halden, On the Need and Speed of Regulating Triclosan and Triclocarban in the United States, Environ. Sci. Technol., 2014, 48, 3603–3611 CrossRef CAS.
- R. U. Halden, A. E. Lindeman, A. E. Aiello, D. Andrews, W. A. Arnold, P. Fair, R. E. Fuoco, L. A. Geer, P. I. Johnson, R. Lohmann, K. McNeill, V. P. Sacks, T. Schettler, R. Weber, R. T. Zoeller and A. Blum, The Florence Statement on Triclosan and Triclocarban, Environ. Health Perspect., 2017, 125, 64501 CrossRef.
- M. D. Unson, N. D. Holland and D. J. Faulkner, A brominated secondary metabolite synthesized by the cyanobacterial symbiont of a marine sponge and accumulation of the crystalline metabolite in the sponge tissue, Mar. Biol., 1994, 119, 1–11 CrossRef CAS.
- X. Fu, F. J. Schmitz, M. Govindan, S. A. Abbas, K. M. Hanson, P. A. Horton, P. Crews, M. Laney and R. C. Schatzman, Enzyme Inhibitors: New and Known Polybrominated Phenols and Diphenyl Ethers from Four Indo-Pacific Dysidea Sponges, J. Nat. Prod., 1995, 58, 1384–1391 CrossRef CAS.
- L. Calcul, R. Chow, A. G. Oliver, K. Tenney, K. N. White, A. W. Wood, C. Fiorilla and P. Crews, NMR Strategy for Unraveling Structures of Bioactive Sponge-Derived Oxy-polyhalogenated Diphenyl Ethers, J. Nat. Prod., 2009, 72, 443–449 CrossRef CAS.
- V. Agarwal, A. A. El Gamal, K. Yamanaka, D. Poth, R. D. Kersten, M. Schorn, E. E. Allen and B. S. Moore, Biosynthesis of polybrominated aromatic organic compounds by marine bacteria, Nat. Chem. Biol., 2014, 10, 640–647 CrossRef CAS.
- V. Agarwal, J. M. Blanton, S. Podell, A. Taton, M. A. Schorn, J. Busch, Z. Lin, E. W. Schmidt, P. R. Jensen, V. J. Paul, J. S. Biggs, J. W. Golden, E. E. Allen and B. S. Moore, Metagenomic discovery of polybrominated diphenyl ether biosynthesis by marine sponges, Nat. Chem. Biol., 2017, 13, 537–543 CrossRef CAS.
- S. Gaul, P. Bending, D. Olbrich, N. Reosenfelder, P. Ruff, C. Gaus, J. F. Mueller and W. Vetter, Identification of the natural product 2,3,4,5-tetrabromo-1-methylpyrrole in Pacific biota, passive samplers and seagrass from Queensland, Australia, Mar. Pollut. Bull., 2011, 62, 2463–2468 CrossRef CAS.
- M. Kuniyoshi, K. Yamada and T. Higa, A biologically active diphenyl ether from the green algaCladophora facicularis, Experientia, 1985, 41, 523–524 CrossRef CAS.
- K. Makoto, K. Tomoyuki, N. Yoshikatsu and U. Daisuke, Corallinafuran and Corallinaether, Novel Toxic Compounds from Crustose Coralline Red Algae, Chem. Lett., 2005, 34, 1272–1273 CrossRef.
- A. Malmvärn, Y. Zebühr, L. Kautsky, K. Bergman and L. Asplund, Hydroxylated and methoxylated polybrominated diphenyl ethers and polybrominated dibenzo-p-dioxins in red alga and cyanobacteria living in the Baltic Sea, Chemosphere, 2008, 72, 910–916 CrossRef.
- W. Vetter, E. Stoll, M. J. Garson, S. J. Fahey, C. Gaus and J. F. Müller, Sponge halogenated natural products found at parts-per-million levels in marine mammals, Environ. Toxicol. Chem., 2002, 21, 2014–2019 CrossRef CAS.
- E. L. Teuten, L. Xu and C. M. Reddy, Two Abundant Bioaccumulated Halogenated Compounds Are Natural Products, Science, 2005, 307, 917–920 CrossRef CAS.
- N. J. Shaul, N. G. Dodder, L. I. Aluwihare, S. A. Mackintosh, K. A. Maruya, S. J. Chivers, K. Danil, D. W. Weller and E. Hoh, Nontargeted Biomonitoring of Halogenated Organic Compounds in Two Ecotypes of Bottlenose Dolphins (Tursiops, truncates) from the Southern California Bight, Environ. Sci. Technol., 2015, 49, 1328–1338 CrossRef CAS.
- Y. Wan, K. Choi, S. Kim, K. Ji, H. Chang, S. Wiseman, P. D. Jones, J. S. Khim, S. Park, J. Park, M. H. Lam and J. P. Giesy, Hydroxylated Polybrominated Diphenyl Ethers and Bisphenol A in Pregnant Women and Their Matching Fetuses: Placental Transfer and Potential Risks, Environ. Sci. Technol., 2010, 44, 5233–5239 CrossRef CAS.
- H. S. Wand, Z. J. Chen, K. L. Ho, L. C. Ge, J. Du, M. H. Lam, J. P. Giesy, M. H. Wong and C. K. Wong, Hydroxylated and methoxylated polybrominated diphenyl ethers in blood plasma of humans in Hong Kong, Environ. Int., 2012, 47, 66–72 CrossRef.
- A. Chen, J. S. Park, L. Linderholm, A. Rhee, M. Petra, E. A. DeFranco, K. N. Dietrich and S. M. Ho, Hydroxylated Polybrominated Diphenyl Ethers in Paired Maternal and Cord Sera, Environ. Sci. Technol., 2013, 47, 3902–3908 CrossRef CAS.
- S. B. Wiseman, Y. Wan, H. Chang, X. Zhang, M. Hecker, P. D. Jones and J. P. Giesy, Polybrominated diphenyl ethers and their hydroxylated/methoxylated analogs: Environmental sources, metabolic relationships, and relative toxicities, Mar. Pollut. Bull., 2011, 63, 179–188 CrossRef CAS.
- E. N. Segraves, R. R. Shah, N. L. Segraves, T. A. Johnson, S. Whitman, J. K. Sui, V. A. Kenyon, R. H. Cichewicz, P. Crews and T. R. Holman, Probing the Activity Difference of Simple and Complex Brominated Aryl Compounds against 15-Soybean, 15-Human, and 12-Human Lipoxygenase, J. Med. Chem., 2004, 47, 4060–4065 CAS.
- R. F. Canton, D. E. Scholten, G. Marsh, P. C. de Jong and M. van den Berg, Inhibition of human placental aromatase activity by hydroxylated polybrominated diphenyl ether (OH-PBDEs), Toxicol. Appl. Pharmacol., 2008, 227, 68–75 CrossRef CAS.
- J. A. de la Fuente, S. Manzanaro, M. J. Martín, T. G. de Quesada, I. Reymundo, S. M. Luengo and F. Gago, Synthesis, Activity, and Molecular Modeling Studies of Novel Human Aldose Reductase Inhibitors Based on a Marine Natural Product, J. Med. Chem., 2003, 46, 5208–5221 CrossRef CAS.
- J. Legradi, A. K. Dahlberg, P. Cenijn, G. Marsh, L. Asplund, A. Bergman and J. Legler, Disruption of Oxidative Phosphorylation (OXPHOS) by Hydroxylated Polybrominated Diphenyl Ethers (OH-PBDEs) Present in the Marine Environment, Environ. Sci. Technol., 2014, 48, 14703–14711 CrossRef CAS.
- U. Hanefeld, H. G. Floss and H. Laatsch, Biosynthesis of the marine antibiotic pentabromopseudilin. Part 1. The benzene ring, J. Org. Chem., 1994, 59, 3604–3608 CrossRef CAS.
- V. Agarwal and B. S. Moore, Enzymatic Synthesis of Polybrominated Dioxins from the Marine Environment, ACS Chem. Biol., 2014, 9, 1980–1984 CrossRef CAS.
- H. Aldemir, R. Richarz and T. A. M. Gulder, The Biocatalytic Repertoire of Natural Biaryl Formation, Angew. Chem., Int. Ed., 2014, 53, 8286–8293 CrossRef CAS.
- V. Agarwal, J. Li, I. Rahman, M. Borgen, L. I. Aluwihare, J. S. Biggs, V. J. Paul and B. S. Moore, Complexity of Naturally Produced Polybrominated Diphenyl Ethers Revealed via Mass Spectrometry, Environ. Sci. Technol., 2015, 49, 1339–1346 CrossRef CAS.
- B. S. Falch, G. M. König, A. D. Wright, O. Sticher, H. Ruegger and G. Bernardinelli, Ambigol A and B: New Biologically Active Polychlorinated Aromatic Compounds from the Terrestrial Blue-Green Alga Fischerella ambigua, J. Org. Chem., 1993, 58, 6570–6575 CrossRef CAS.
- B. S. Falch, G. M. König, A. D. Wright, O. Sticher, C. K. Angerhofer, J. M. Pezzuto and H. Bachmann, Biological Activities of Cyanobacteria: Evaluation of Extracts and Pure Compounds, Planta Med., 1995, 61, 321–328 CAS.
- A. D. Wright, O. Pappendorf and G. M. König, Ambigol C and 2,4-Dichlorobenzoic Acid, Natural Products Produced by the Terrestrial Cyanobacterium Fischerella ambigua, J. Nat. Prod., 2005, 68, 459–461 CrossRef CAS.
- P. C. Dorrestein, E. Yeh, S. Garneau-Tsodikova, N. L. Kelleher and C. T. Walsh, Dichlorination of a pyrrolyl-S-carrier protein by FADH2-dependent halogenase PltA during pyoluteorin biosynthesis, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13843–13848 CrossRef CAS.
- S. Lin, S. G. Van Lanen and B. Shen, Regiospecific Chlorination of (S)-β-Tyrosyl-S-Carrier Protein Catalyzed by SgcC3 in the Biosynthesis of the Enediyne Antitumor Antibiotic C-1027, J. Am. Chem. Soc., 2007, 129, 12432–12438 CAS.
- T. Kittilä, C. Knittel, J. Tailhades, D. Butz, M. Schoppet, A. Büttner, R. J. A. Goode, R. B. Schittenhelm, K. H. van Pee, R. D. Süssmuth, W. Wohlleben, M. J. Cryle and E. Stegmann, Halogenation of glycopeptide antibiotics occurs at the amino acid level during non-ribosomal peptide synthesis, Chem. Sci., 2017, 8, 5992–6004 RSC.
- A. El Gamal, V. Agarwal, S. Diethelm, I. Rahman, M. A. Schorn, J. M. Sneed, G. V. Louie, K. L. Whalen, T. J. Mincer, J. P. Noel, V. J. Paul and B. S. Moore, Biosynthesis of coral settlement cue tetrabromopyrrole in marine bacteria by a uniquely adapted brominase-thioesterase enzyme pair, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3797–3802 CrossRef CAS.
- E. R. Duell, P. M. D'Agostino, N. Shapiro, T. Woyke, T. M. Fuchs and T. A. M. Gulder, Direct pathway cloning of the sodorifen biosynthetic gene cluster and recombinant generation of its product in E. coli, Microb. Cell Fact., 2019, 18, 32 CrossRef.
- C. Greunke, E. R. Duell, P. M. D'Agostino, A. Glöckle, K. Lamm and T. A. M. Gulder, Direct Pathway Cloning (DiPaC) to unlock natural product biosynthetic potential, Metab. Eng., 2018, 47, 334–345 CrossRef CAS.
- P. M. D'Agostino and T. A. M. Gulder, Direct Pathway Cloning Combined with Sequence- and Ligation-Independent Cloning for Fast Biosynthetic Gene Cluster Refactoring and Heterologous Expression, ACS Synth. Biol., 2018, 7, 1702–1708 CrossRef.
- S. E. Ongley, X. Bian, Y. Zhang, R. Chau, W. H. Gerwick, R. Müller and B. A. Neilan, High-Titer Heterologous Production in E. coli of Lyngbyatoxin, a Protein Kinase C Activator from an Uncultured Marine Cyanobacterium, ACS Chem. Biol., 2013, 8, 1888–1893 CrossRef CAS.
- A. El Gamal, V. Agarwal, I. Rahman and B. S. Moore, Enzymatic Reductive Dehalogenation Controls the Biosynthesis of Marine Bacterial Pyrroles, J. Am. Chem. Soc., 2016, 138, 13167–13170 CrossRef CAS.
- E. M. Clerico, J. L. Ditty and S. S. Golden, Specialized Techniques for Site-Directed Mutagenesis in Cyanobacteria, Humana Press, Totowa, NJ, 2007, pp. 155–171 Search PubMed.
- A. T. Ma, C. M. Schmitdt and J. W. Golden, Regulation of Gene Expression in Diverse Cyanobacterial Species by Using Theophylline-Responsive Riboswitches, Appl. Environ. Microbiol., 2014, 80, 6704–6713 CrossRef.
- M. J. Lai and E. I. Lan, Photoautotrophic synthesis of butyrate by metabolically engineered cyanobacteria, Biotechnol. Bioeng., 2019, 116, 893–903 CAS.
- S. Watanabe, H. Ozawa, H. Kato, K. Nimura-Matsune, T. Hirayama, F. Kudo, T. Eguchi, K. Kakinuma and H. Yoshikawa, Carbon-free production of 2-deoxy-scyllo-inosose (DOI) in cyanobacterium Synechococcus elongatus PCC 7942, Biosci. Biotechnol. Biochem., 2018, 82, 161–165 CrossRef CAS.
- H. Li and J. C. Liao, Engineering a cyanobacterium as the catalyst for the photosynthetic conversion of CO2 to 1,2-propanediol, Microb. Cell Fact., 2013, 12, 4 CrossRef CAS.
- M. Bielawski, D. Aili and B. Olofsson, Regiospecific One-Pot Synthesis of Diaryliodonium Tetrafluoroborates from Arylboronic Acids and Aryl Iodides, J. Org. Chem., 2008, 73, 4602–4607 CrossRef CAS.
- N. Jalalian, E. E. Ishikawa, L. F. Silva and B. Olofsson, Room Temperature, Metal-Free Synthesis of Diaryl Ethers with Use of Diaryliodonium Salts, Org. Lett., 2011, 13, 1552–1555 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: All experimental data. See DOI: 10.1039/d0qo00707b |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2020 |