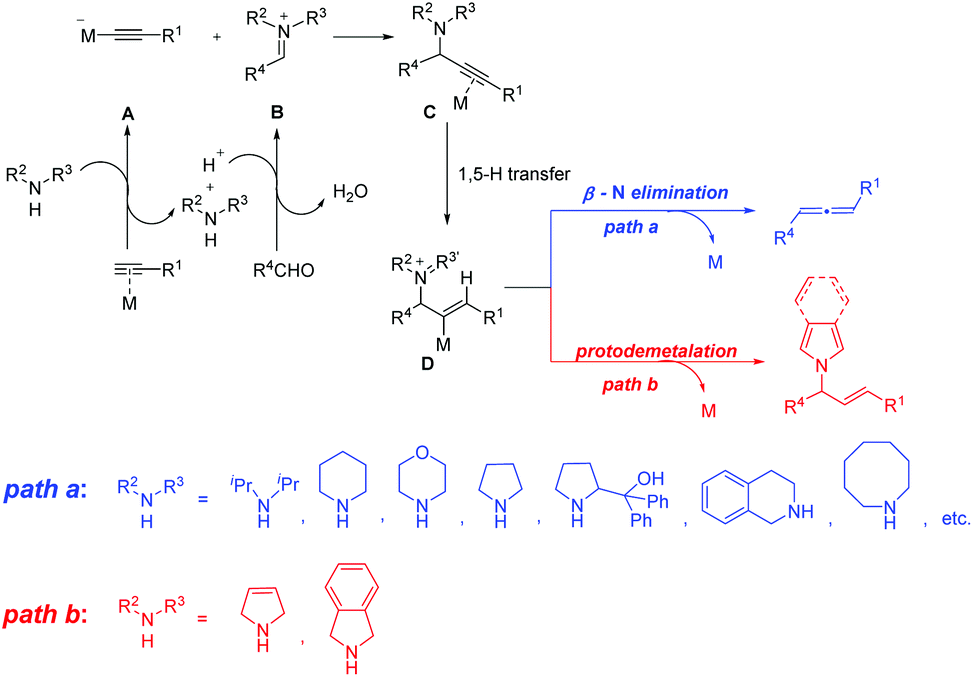
Metal-catalyzed three-component coupling of an alkyne, an aldehyde and an amine (A3-coupling) is a straightforward protocol for preparing propargylic amines,1 which serve as the key intermediates for the synthesis of many organic compounds.2 Mechanistically, the A3-coupling involves the reaction of the in situ generated metal alkynylide species A with the iminium ion B that is in situ generated from the condensation of an aldehyde with an amine, resulting in the formation of the metal-coordinated propargylic amine C, which undergoes 1,5-H transfer to afford the iminium intermediate D (Scheme 1). Subsequent β-N elimination would produce the allene product (path a in Scheme 1).3 This mechanism has been supported by our detailed theoretical studies.3c It is worth noting that the amine in these reactions can be diisopropylamine,2f dicyclohexylamine,2g morpholine,2h pyrrolidine,2i,k,n diphenylprolinol,2j,m tetrahydroisoquinoline,2l azocane,2o and so on. Interestingly, E-allylic amines were obtained unexpectedly when terminal alkynes and aldehydes were treated with 3-pyrroline or isoindoline in the presence of copper(i) (path b in Scheme 1).4 We presumed that the E-allylic amine is afforded through the protodemetalation process after the generation of the iminium intermediate D. Deuterium-labeling experiments have been performed to confirm that the β-position proton of E-allylic amines came from the azacycle and the α-position proton came from the proton in the ambient environment (Scheme 2).4 Here, we sought to investigate the detailed reaction mechanism of this Cu(i)-catalyzed5 reductive A3-coupling reaction, as well as the crucial role of 3-pyrroline or isoindoline. The origin of the different chemoselectivity of the afforded allylic amine or allene was also discussed.

DFT calculations were carried out using the Gaussian 09 software.6 M06-2X,7 including Grimme's D3 dispersion corrections, was employed to performed geometry optimizations. The LANL2DZ8 basis set in conjunction with the LANL2DZ pseudopotential9 was used for Cu and Br atoms, while the 6-31G(d,p)10 basis set was used for the other atoms. Geometry optimizations were fully carried out. Harmonic vibration frequency calculations were conducted at the same level of theory to verify the stationary points to be the minima (no imaginary frequency) or saddle points (one imaginary frequency). Intrinsic reaction coordinate (IRC)11 calculations were performed to confirm the connection of the transition structures with their corresponding reactants and products. The single point energies and solvent effects in toluene (ε = 2.37) were computed with M06-2X-D3/SDD12-6-311++G(d,p) basis sets by using the SMD13 solvation model. The solution-phase Gibbs free energy was used in the present discussions. The calculations were carried out on the exact compounds applied experimentally: 2-methybut-3-yn-2-ol (1), cyclohexanaldehyde (2) and 3-pyrroline (3) with copper(i) bromide as the catalyst (Scheme 3).

The A3-coupling reaction proceeds initially from the sp C–H activation of the terminal alkyne 1 by the metal catalyst and the condensation of cyclohexanaldehyde 2 with 3-pyrroline 3, affording the ion pair Int3, which consists of copper alkynylide anion Int1 and iminium cation Int2. This process leads to an energy increase of 4.0 kcal mol−1, accompanied by the loss of one molecule of water. The subsequent electrophilic attack of iminium Int2 on the copper attached C1viaTS1 requires an energy barrier of 10.1 kcal mol−1, providing the propargyl amine–CuBr complex Int4. Subsequent isomerization of Int4 to Int5, affording the precursor for 1,5-H transfer, in which CuBr and the amino group are in the trans position, is endergonic by 7.4 kcal mol−1 relative to Int4.
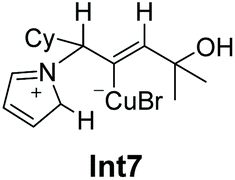
With the envelope conformation of the pyrrolidine moiety in Int5, there are two axial hydrogen atoms (H1 and H2 in Fig. 1) in perfect orientation for transfer to the C2 atom. The six-membered cyclic transition state for the 1,5-H transfer has been located as TS2_1 and TS2_2 for the transfer of H1 and H2 atoms, respectively. The free energy barrier for 1,5-H1 transfer is calculated to be 27.7 kcal mol−1 (TS2_1 relative to Int4), which is lower by 1.8 kcal mol−1 than that for the 1,5-H2 transfer (TS2_2), due to the fact that H1 is closer than H2 to the C2 atom (2.80 Å vs. 3.03 Å). Hence, TS2_1 is more favorable, leading to the formation of N-allylic iminium–CuBr intermediate Int6_1, during which a σ bond between Cu(i) and C1 is formed. Subsequent isomerization of Int6_1 by single-bond rotation produces the more stable species Int7, which is exergonic by 3.6 kcal mol−1 relative to Int6_1. The 1,5-H transfer step features the highest free energy barrier of the whole profile (27.7 kcal mol−1, TS2_1), and may, thus, be rate determining.

From iminium intermediate Int7, there are two alternative pathways: one is β-N elimination to produce the allene product (path a in Fig. 2), and the other is the protodemetalation process to afford the allylic amine (path b in Fig. 2). The optimized transition structure for path a has been located as TS3 and TS4, in which the C–N and C–Cu bonds break simultaneously. The cis elimination from Int7 proceeds through TS3, which requires an energy barrier of 24.9 kcal mol−1 (TS3 relative to Int7). However, the trans elimination through TS4 is much easier with a barrier of 14.3 kcal mol−1. The precursor for TS4 is Int8, which is provided through the single-bond rotation of Int7. Thus, we consider that the β-N elimination should be in a trans manner through TS4, which needs to overcome a free energy barrier of 14.3 kcal mol−1 to afford Int9. Finally, allene 5 is released from Int9, with an overall exergonicity of 19.2 kcal mol−1 (relative to Int7).

An alternative pathway from allyl iminium–CuBr intermediate Int7 is the protodemetalation process to afford the allylic amine (path b in Fig. 2), which is the only product obtained experimentally. The direct intramolecular protodemetalation of Int7 proceeds through a concerted five-membered cyclic transition structure (TS5), in which the deprotonation of the alkyl hydrogen (H3) adjacent to the N atom and protodemetalation of CuBr take place simultaneously, leading to the CuBr-coordinated allylic amine Int11. This process is computed to be exergonic (ΔG = −40.8 kcal mol−1) and requires a 14.2 kcal mol−1 activation barrier (TS5 relative to Int7). However, Int7 can isomerize to a less stable species Int12 (1.0 kcal mol−1 higher in energy than Int7) through single-bond rotation with the hydroxyl group coordinating to the Cu atom (d(Cu⋯O) = 2.41 Å). Int12 may serve as the precursor for the protodemetalation process as well, which also proceeds through a concerted five-membered cyclic transition structure (TS6). TS6 needs to overcome a free energy barrier of 13.5 kcal mol−1, which is lower in energy by 0.7 kcal mol−1 than TS5 (14.2 kcal mol−1), leading to Int11 as well. Thus, we consider that TS6 is preferable to TS5 for the direct intramolecular protodemetalation of Int7. The final step of this process involves the release of the product allylic amine 4 through CuBr coordinated to another 3-pyrroline molecule, which is calculated to be exergonic by 10.4 kcal mol−1 (relative to Int11) due to the strong coordination ability of nitrogen with CuBr.
The abovementioned computational results show that the direct protodemetalation is preferred over β-N elimination both kinetically and thermodynamically, indicating that an allylic amine should be obtained as the major product. However, in the experiment, allylic amines are obtained as the only products, and allenes are not observed at all. We do not think that the free energy difference of 0.8 kcal mol−1 could account for the exclusive formation of allylic amines. In addition, deuterium-labeling experiments confirm that the β-position proton of allylic amines may come from not only the azacycle but also the moisture in the ambient environment (Scheme 2).4 Hence, the stepwise protodemetalation assisted by other molecules is taken into account. When one molecule of water is taken into consideration,14 the single bond C2–C3 in Int7 rotates to facilitate the hydrogen bond formation between the hydroxyl group and the H4 in water, leading to the formation of the complex Int14, in which three hydrogen bonds take shape with water [d(O2⋯H3) = 2.23 Å, d(O1⋯H4) = 1.96 Å, d(Br⋯H5) = 2.73 Å] (Fig. 3). The deprotonation process of Int14 involves the abstraction of the proton H3 from the azacycle by water and the subsequent protonation of the hydroxyl group, affording the intermediate Int15, in which the protonated hydroxyl group is stabilized by the hydrogen bond formed with water. This deprotonation process, accompanied by the aromatization of the azacycle of Int14, requires a free energy barrier of 8.1 kcal mol−1 (TS7, relative to Int7). Isomerization of Int15 to Int16 facilitates the subsequent protodemetalation process, which takes place intramolecularly through a concerted five-membered cyclic transition state (TS8). TS8 is calculated to be higher in electronic energy by only 1.8 kcal mol−1 than Int16. However, when the thermal correction and the solvent effect are taken into account, TS8 is slightly lower in free energy than intermediate Int16, indicating that the water-assisted protodemetalation could proceed with almost no barrier to afford the CuBr-coordinated allylic amine Int17. Finally, the product allylic amine 4 can be obtained by the coordination of CuBr to another 3-pyrroline molecule.

When the water cluster, modeled by three molecules of water, is taken into account, a similar energy profile is obtained, which is shown in Fig. 4. The water cluster assisted deprotonation requires to overcome a lower energy barrier of 5.2 kcal mol−1. Different from the one water molecule assisted pathway, the subsequent protodemetalation takes place in an intermolecular way with an energy barrier of 4.2 kcal mol−1.

Calculations show that the presence of water (or water cluster) turns the direct concerted protodemetalation into a reversible stepwise deprotonation–protodemetalation process, which requires a much lower free energy barrier in total. Obviously, water (or the water cluster) may act as a proton shuttle in this process, making the protodemetalation process more kinetically favorable.
Interestingly, when an additional 3-pyrroline was taken into account as the proton shuttle, another kinetically favorable pathway of the stepwise deprotonation–protodemetalation process was discovered (Fig. 5). All efforts to locate the hydrogen-bonding complex of 3-pyrroline with Int7 were unsuccessful. When the N atom of 3-pyrroline approaches the proton H3 of the azacycle of Int7, the abstraction of H3 takes place immediately, indicating that the deprotonation process of Int7 requires no barrier in the presence of 3-pyrroline. The abstraction of the proton H3 from Int7 produces Int21, which is exergonic by 24.5 kcal mol−1, accompanied by the aromatization of the azacycle. The subsequent protodemetalation takes place intermolecularly by the protonated 3-pyrroline through TS11 with an energy barrier of 2.3 kcal mol−1, affording the more stable CuBr-coordinated allyl amine Int22. Finally, the coordination of CuBr with 3-pyrroline releases allylic amine 4 to complete the whole process. Thus, an extra 3-pyrroline molecule could also act as the proton shuttle to change the concerted protodemetalation process to a stepwise deprotonation–protodemetalation process. But different from the water (or water cluster) involved pathway, the rate-limiting step of the 3-pyrroline assisted process is the protodemetalation step with an activation barrier of only 2.3 kcal mol−1.

The propargylic alcohol 1, as one of the reactants, may also act as a proton shuttle to assist the protodemetalation process (Fig. 6). The computed potential energy surface is qualitatively similar to that of the one water molecule assisted pathway, but with more energy demanded in the deprotonation step, which needs to overcome an activation barrier of 10.1 kcal mol−1 (TS12). The following protodemetalation proceeds intramolecularly with an energy barrier of 1.8 kcal mol−1.

The normal terminal alkynes with no hydroxy group present relatively lower reactivities in the experimental studies.4 For example, when using 1-octyne under the standard reaction conditions, the E-allylic amine was also formed with a lower yield (20% NMR yield), accompanied by 70% NMR yield of the propargylic amine.4 The effect of the hydroxyl group in 2-methybut-3-yn-2-ol (1) was further rationalized by DFT calculations on comparison with the 1-octyne-participated system.15 The computed potential energy surfaces are qualitatively similar to those of the 2-methybut-3-yn-2-ol (1)-involved system, but with more free energy demanded in the rate-limiting 1,5-H transfer step [28.8 kcal mol−1 (TS15_1) vs. 27.7 (TS2_1)], which is in accordance with the experimental observations. As shown in Fig. 7, the hydrogen bond, formed between the hydroxyl group and the migrating hydrogen [d(O⋯H) = 2.51 Å] in TS2_1, facilitates the 1,5-H transfer, resulting in the enhanced stability of TS2_1.

The activation energies for the stepwise deprotonation–protodemetalation process of Int7 assisted by different species are presented separately in Table 1, together with the energy barrier of the β-N elimination process. It is obvious that the direct intramolecular pathway without the aid of other species is unfavorable, owing to the high energy demand of 13.5 kcal mol−1. The presence of different assisting species makes the concerted intramolecular process an irreversible stepwise deprotonation–protodemetalation process, reducing the activation barrier significantly. Among the four assisting species, 3-pyrroline shows the highest activity in the deprotonation step owing to the relatively strong basicity of the N atom. Once the rate-limiting 1,5-H transfer completes, the deprotonation–protodemetalation process takes place immediately with the aid of 3-pyrroline to afford the final allylic amine product. It is noteworthy that without 3-pyrroline (or isoindoline) in the reaction system, other species, such as water cluster, could also act as the proton shuttle to assist the protodemetalation, which is in agreement with the observation in deuterium-labeling experiments (Scheme 2). Significantly, the aromatization of the azacycle is the essential driving force for the protodemetalation process.
The pathway selectivity is determined by the activation energy difference between β-N elimination and the protodemetalation from the intermediate Int7 formed via the 1,5-H transfer. Consistent with the experimental observations, with 3-pyrroline, protodemetalation is favored over β-N elimination by 12.0 kcal mol−1 (14.3 kcal mol−1vs. 2.3 kcal mol−1), leading to the overwhelming formation of the allylic amine product. However, all the other amines listed in Scheme 1, other than 3-pyrroline and isoindoline, completely reverse the pathway selectivity, providing allenes as the final products. Due to the lack of the driving force arising from the aromatization of the azacycle, the protodemetalation of the intermediate D (Scheme 1) is rather difficult. Therefore, β-N elimination becomes the favorable pathway for the classical ATA reactions using the amines except 3-pyrroline or isoindoline.
In summary, the mechanism of this Cu(i)-catalyzed reductive A3-coupling reaction affording E-allylic amines was investigated by DFT calculations in detail. The calculations disclose that 3-pyrroline (or isoindoline) serves not only as a reactant but also as a shuttle in a proton relay to assist the protodemetalation step, resulting in the production of an allylic amine other than an allene. Importantly, the calculations reveal the factors governing the selectivity: the aromatization of the azacycle (formed from 3-pyrroline or isoindoline) is the essential driving force for the preference of the allylic amine products. Furthermore, we also discovered that formation of a hydrogen bond with the migrating hydrogen to facilitate the rate-determining 1,5-H transfer step is the key role of the hydroxyl group in the terminal alkyne substrate in the reactivity.
There are no conflicts to declare.
Financial support from the Natural Science Foundation of Shanghai (19ZR1468500) and the National Natural Science Foundation of China (Grant No. 21690063 for S. Ma) is greatly appreciated.
This journal is © the Partner Organisations 2020