DOI:
10.1039/D0QO00559B
(Research Article)
Org. Chem. Front., 2020,
7, 2916-2924
An enzymatic acetal/hemiacetal conversion for the physiological temperature activation of the alkoxyamine C–ON bond homolysis†
Received
9th May 2020
, Accepted 21st July 2020
First published on 30th July 2020
Abstract
The potential of alkoxyamines as theranostic agents has been recently promoted by our groups. The success of such an approach relies on the switch upon enzymatic triggering between highly stable precursor alkoxyamines and activated alkoxyamines exhibiting fast homolysis of the C–ON bond. Hence, at 37 °C in water, benzyl 2-(2,2,6,6-tetramethylpiperidin-N-oxy)-3-ethoxy-3-acetoxypropanoate and benzyl 2-ditert-butylaminoxy-3-ethoxy-3-acetoxy propanoate afford tmax of 2000 s (35% conversion) and 500 s (60% conversion), respectively, for the C–ON bond homolysis in the presence of Subtilisin A whereas t1/2 of ca. 42 thousand millenniums and 330 years are expected accordingly to Ea values in n-propanol. These results nicely highlight the on/off switch, provided that an enzymatic activity controls the C–ON bond homolysis.
Introduction
Labile alkoxyamines were first1–4 applied, in 1986, as controller/initiator agents for Nitroxide Mediated Polymerization (NMP).5–11 During the last decade, several variants – Enhanced Spin Capture Polymerization (ESCP),12 Nitroxide Mediated PhotoPolymerization (NMP2),13–15 Coordination Initiated-NMP (CI-NMP),16 Surface Initiated-NMP (SI-NMP),17 Spin Label NMP (SL-NMP),18,19 Plasmon Initiated NMP (PI-NMP)20 – have emerged, which extend the versatility of this radical polymerization technique. Recently, the lability of the C–ON bond has been used to develop new applications of alkoxyamines, e.g., self-healing polymers,21,22 dynamic microcrystal assemblies,23 and information-encoding polymers.24–26 For a few years,27 our groups have promoted the therapeutic application of alkoxyamines as drugs against cancer.28–30 For such an application, we aim to combine four antagonistic features: a highly stable precursor (or pro-drug), a highly reactive drug, random reactivity, and specific addressing. The aim is to circumvent drug-resistant cancer by using highly reactive and unselective drugs, and to improve the patient welfare by using highly selective pro-drugs via specific addressing, which is a step towards personalized medicine. For several years, a keen interest in using radical species (mainly ROS) as drugs has been observed,31 although two challenges have arisen: the bio-distribution of the drug and the generation of radicals at physiological temperature. Radical generation at physiological temperature has been mainly achieved by radiochemistry and by PDT, with the use of photosensitizers. Nevertheless, it has a strong impact on the welfare of the patient and it is not always efficient. A few years ago,27 we proposed the concept of smart alkoxyamines, i.e., stable alkoxyamines that are transformed into labile alkoxyamines upon chemical triggering. Several chemical activation events – protonation,32 alkylation,33,34 oxidation,33,34 coordination35,36 – have been applied to both nitroxyl and alkyl fragments.37–39 To overcome the aforementioned bottlenecks, the specific addressing and activation events are ensured by the persistent and specific activities of protease present in the tumor environment, thus highly labile alkoxyamines in the vicinity of tumors are released.27,40 Hence, the proteases must hydrolyze specific peptides to release highly reactive alkyl radicals in a handful of seconds to react in the vicinity of the tumor, i.e., the issues of drug bio-distribution and release location are circumvented. Recently, we reported the triggering of the C–ON bond homolysis due to specific enzymatic activity but the large half-life time t1/2 of 111 min observed at 37 °C and the insufficient stability of the pro-drug impeded any application of alkoxyamines as drug.40 Consequently, our research is now focused on the development of switches able to transform a stable alkoxyamine into a highly labile alkoxyamine upon specific enzymatic activity.
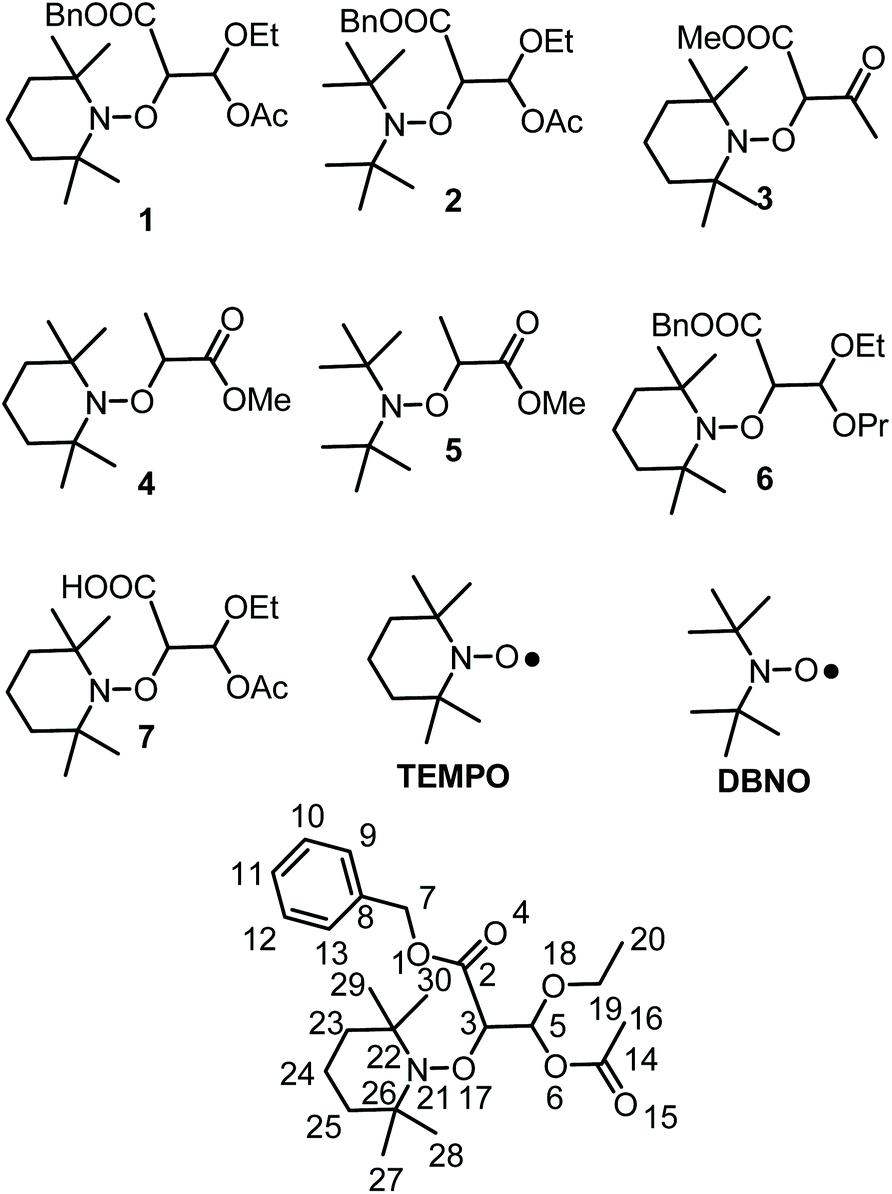
To this end, alkoxyamines 1 and 2 (Chart 1) carrying a malonic-like alkyl fragment with a carbonyl function hidden as an unsymmetrical acetal were prepared and investigated in various conditions of temperatures, solvents, and enzymes. Using Subtilisin A as trigger, t1/2 values of a few hundred seconds were observed for 1 and 2, whereas several centuries are expected in the absence of activation.
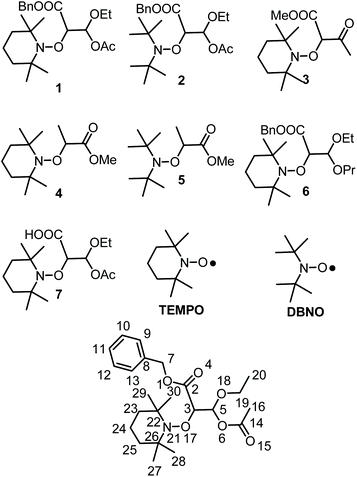 |
| | Chart 1 Alkoxyamines and nitroxides discussed in the article. | |
Results
Synthesis of 1 and 2
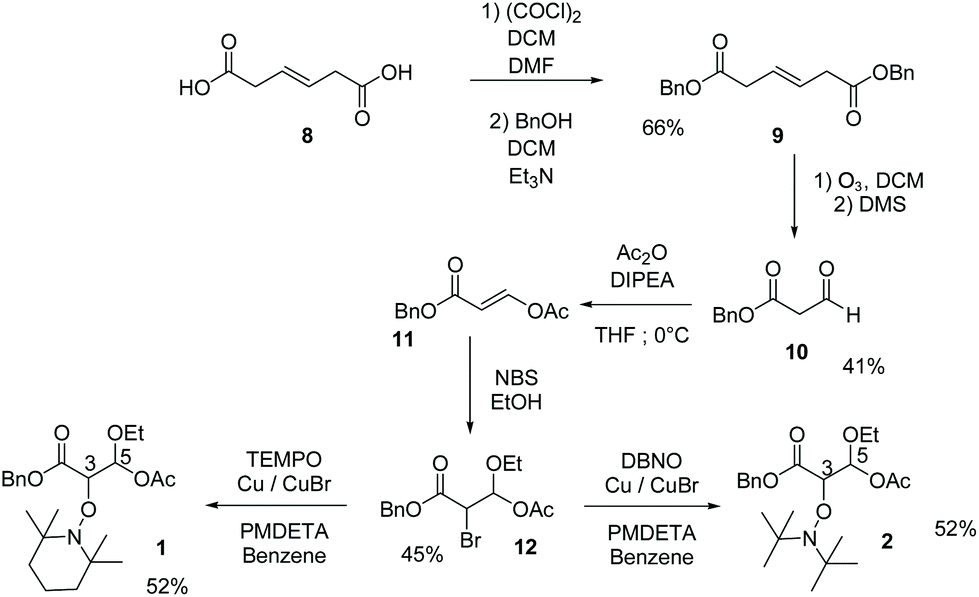
With the adapted reported procedure, trans-beta-hydromucanic acid 8 was esterified with benzyl alcohol to yield 9,41 which was oxidized by ozonolysis to afford 10. The enolate of 10 was scavenged by acetic anhydride to yield the E-enol 11. The ethanol solution of 11 in the presence of NBS (generation of an intermediate bromonium) afforded bromoacetal 12.42 Using the ATRA procedure, in the presence of TEMPO and DBNO,43 bromide 12 yielded alkoxyamines 1 and 2, respectively (Scheme 1 and ESI†).
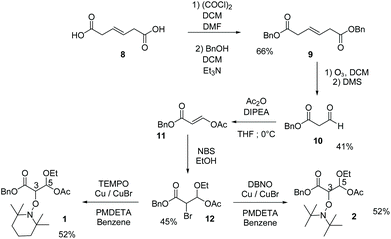 |
| | Scheme 1 Preparation of 1 and 2 (see ESI†). | |
Because of the presence of stereocenters in the alkyl fragment, two diastereoisomers are observed for 1 and 2. Taking into account that enzymes are dramatically sensitive to the configuration of the stereocenters, enantiomers were separated by chiral HPLC (see ESI†).
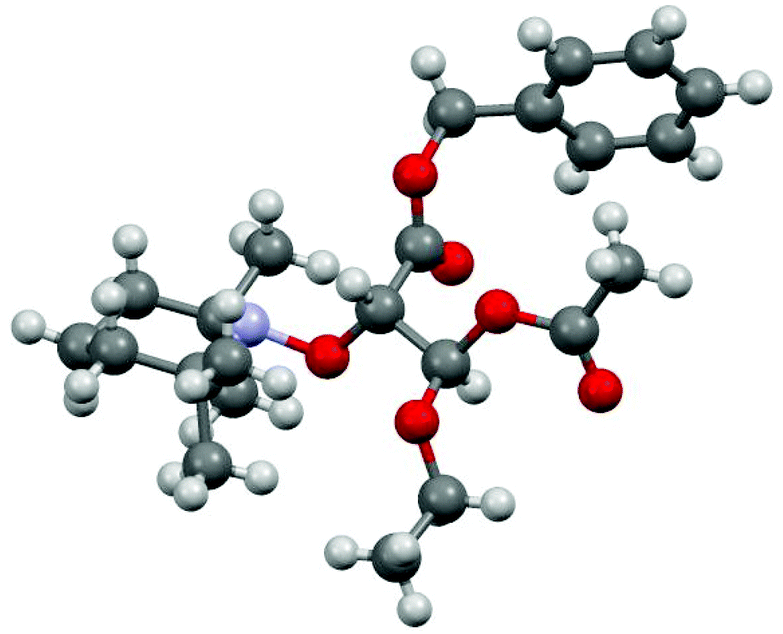
Isomers were noted A and B, and (+) and (−) signs were given according to chromatographic retention order and to the deviation of the polarized light, respectively. Colourless crystal plates of enantiomer 1A(−)‡ were obtained from diethyl ether (slow evaporation at 4 °C) and provided configurations S and R (Fig. 1) for carbon atoms 3 and 5, respectively (Fig. 1 and Chart 1). The geometrical parameters of 1A(−) were not very different from those reported for similar models (alkoxyamines carrying an ester group) except for shorter O17–C3 bond and N21⋯C3 distance by 0.02 Å and 0.01 Å, respectively. Hence, configurations 3S5R and 3R5S are ascribed to isomers 1A(−) and 1A(+), respectively, and the relative configuration RR/SS to isomers 1B(+) and 1B(−). As no XRD data are available for 2, the assessment of absolute and/or relative configurations is not possible.
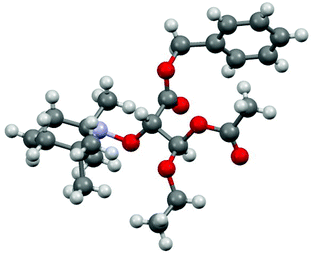 |
| | Fig. 1 XRD structure (see ESI†) of 3S5R-1 aka 1A(−). | |
Synthesis of 6
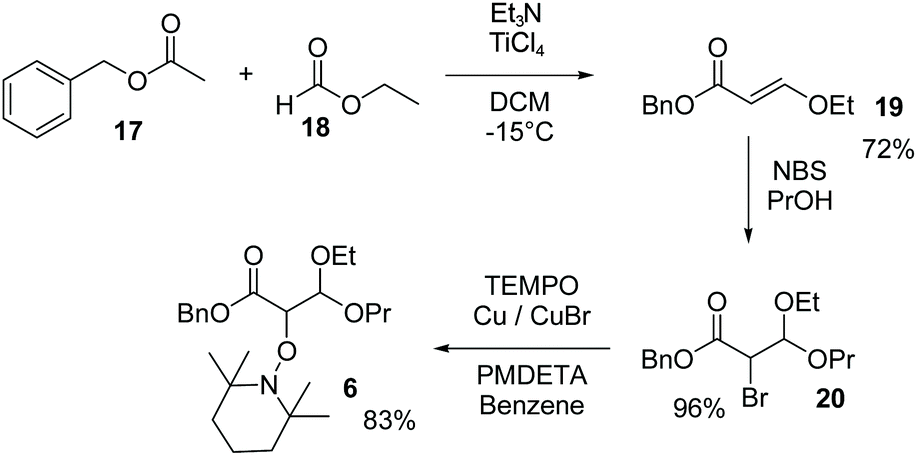
The enol 19 was synthesised by a condensation of benzyl acetate 17 and ethyl formate 18 activated by TiCl4 as reported.44 Propanol solution of 19 in the presence of NBS afforded bromoacetal 20.42 Using ATRA procedure,43 in the presence of TEMPO, bromide 20 yielded alkoxyamine 6 (see Scheme 2 and ESI†).
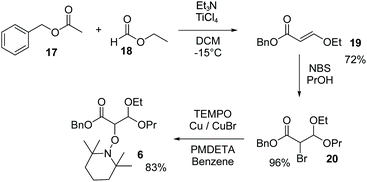 |
| | Scheme 2 Preparation of 6 (see ESI†). | |
Enzyme-free kinetics
Kinetics of 1 and 2 were measured as previously reported,45 using either the plateau method (eqn (1), [nitroxide]∞ = [alkoxyamine]0) or the initial slope method (eqn (2), [nitroxide]∞ = [alkoxyamine]0). The homolysis of all the isomers was investigated in t-BuPh and in n-PrOH. Taking into account the high Ea observed in t-BuPh, n-PrOH (bp = 97 °C) was selected as solvent to mimic water as much as possible.| | 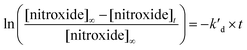 | (1) |
| | 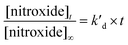 | (2) |
As expected, the pairs of enantiomers display the same values of kd (Table 1) and the small differences in Ea between diastereoisomers are in the range (1–2 kJ mol−1) of those already reported in the literature and do not deserve more comments.46
Table 1 Homolysis rate constants k′d at different temperatures T, in various conditions, and the subsequent activation energies Ea as well as the re-estimated kd values at 120 °C and the half-life time t1/2 at 37 °C
| |
|
Conditions |
T (°C)a |
k
d (s−1)b |
E
a (kJ mol−1)c |
k
d (120 °C, s−1) |
t
1/2 (37 °C, year)d |
|
Error is ±1 °C.
Error <5%.
Activation energies Ea were estimated using a frequency factor A of 2.4 × 1014 s−1, as reported in ref. 9. Errors in Ea are around 1–2 kJ mol−1.
Unless otherwise mentioned.
k
d values determined using eqn (1).
4 isomer mixture.
k
d values determined using eqn (2) and [alkoxyamine]0 = 0.1 M.
k
d values determined using eqn (2) and [alkoxyamine]0 = 10−4 M.
k
d values determined using eqn (2) and [alkoxyamine]0 ≈ 10−2 M.
See ref. 45.
Not given.
See ref. 53.
|
| 1 |
A+e |
t-BuPh |
164 |
6.0 × 10−4 |
147.3 |
8.0 × 10−6 |
433 |
| A−e |
t-BuPh |
163 |
8.3 × 10−4 |
146.0 |
1.1 × 10−5 |
294 |
| B+e |
t-BuPh |
155 |
3.3 × 10−4 |
146.5 |
9.3 × 10−6 |
371 |
| B−e |
t-BuPh |
164 |
3.7 × 10−4 |
149.2 |
1.0 × 10−5 |
353 |
|
,
|
n-PrOH |
85 |
4.4 × 10−10 |
162.6 |
6.0 × 10−8 |
231![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| A+h |
n-PrOH/H2O:2/8 |
96 |
4.8 × 10−11 |
174.0 |
1.8 × 10−9 |
18.5 × 106 |
| A−h |
n-PrOH/H2O:2/8 |
96 |
2.8 × 10−11 |
176.0 |
9.9 × 10−10 |
41.5 × 106 |
| B−h |
n-PrOH/H2O:2/8 |
96 |
5.9 × 10−11 |
173.4 |
2.2 × 10−9 |
14.7 × 106 |
| B+h |
n-PrOH/H2O:2/8 |
96 |
7.5 × 10−11 |
172.0 |
3.4 × 10−9 |
8.5 × 106 |
| 2 |
A+i |
t-BuPh |
100.6 |
1.5 × 10−5 |
137.4 |
1.3 × 10−4 |
13 |
| A−i |
t-BuPh |
100.7 |
1.9 × 10−5 |
136.6 |
1.7 × 10−4 |
9 |
| B+i |
t-BuPh |
101.9 |
2.0 × 10−5 |
138.6 |
9.2 × 10−5 |
20 |
| B−i |
t-BuPh |
101.5 |
1.8 × 10−5 |
137.2 |
1.4 × 10−4 |
12 |
|
,
|
n-PrOH |
70.2 |
1.6 × 10−8 |
145.8 |
1.0 × 10−5 |
330 |
|
,
|
n-PrOH |
89.5 |
2.8 × 10−7 |
145.4 |
1.2 × 10−5 |
281 |
| 3 |
|
t-BuPhj |
|
|
139.3 |
7.3 × 10−5 |
27 |
|
|
Water/MeOHj |
|
|
121.6 |
0.016 |
248 hours |
| 4 |
|
t-BuPhl |
|
|
141.8 |
3.4 × 10−5 |
72 |
| 5 |
|
t-BuPhl |
|
|
128.2 |
0.002 |
133 days |
| 6 |
|
t-BuPh |
158 |
5.8 × 10−4 |
145.8 |
1.1 × 10−5 |
296 |
Using linear free energy relationships reported in the literature,47,48Ea of 1§ and 2¶ are estimated as 137.0 kJ mol−1 and 126.5 kJ mol−1,45 respectively. Polar, steric, stabilization and conformational effects of the nitroxyl fragment TEMPO and DBNO in 1 and 2 are the same as those reported in the literature, as Ea of 2 is ca. 10 kJ mol−1 lower than Ea of 1. On the other hand, Ea for 1 and 2 are clearly higher by 10 kJ mol−1 than expected from the literature,46 denoting an unexpected effect (see Discussion), as parameters used to describe steric, polar, and stabilization effects in the alkyl fragment are rather reliable (see ESI†). The clear increase in Ea of 1 and 2 due to solvent effects, i.e., by 15 kJ mol−1 (ca. 200-fold) and 8 kJ mol−1 (ca. 10-fold) from t-BuPh to n-PrOH, respectively, is in sharp contrast to the slight decrease in Ea reported when changing from non-polar to polar solvents.49,50 As claimed in a previous article,45 this striking solvent effect is likely due to a change in conformation in the alkyl fragment depending on the dipole moment of the molecule, which is strongly sensitive to the solvation cage related both to the polarity and the structuredness/stiffness of the molecules of solvent. This is nicely supported by the 30-fold decrease in kd of 1 from bulk n-PrOH to 2:8 n-PrOH:water (Table 1).
Enzymatic kinetics
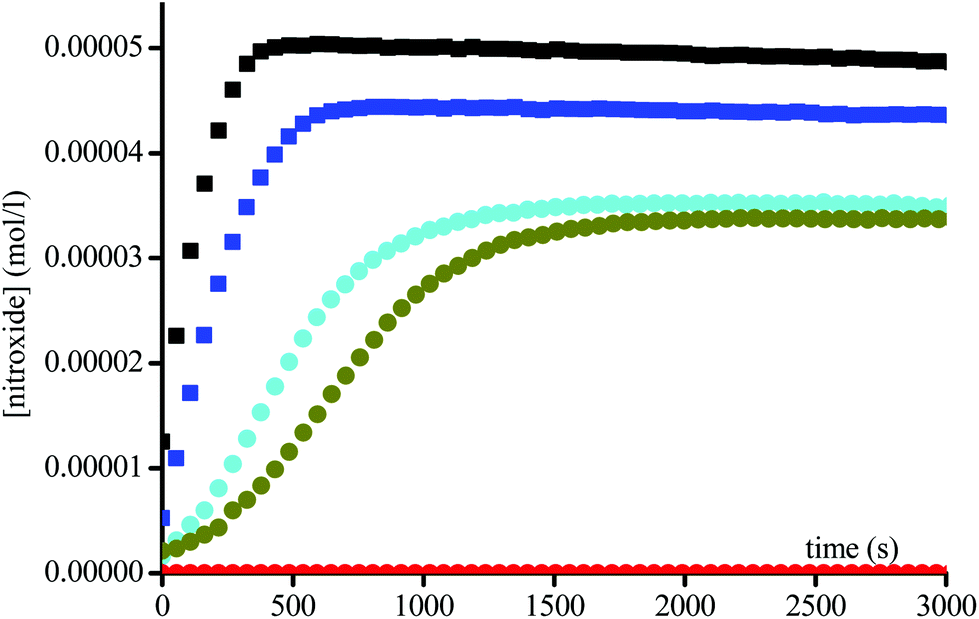
Since an acetyl leaving group is not a favoured substrate for this enzyme, i.e., the Michaelis constant Km is very large, very high concentrations up to 6 × 10−3 M of the enzyme were used to afford the fast activation of the alkoxyamines. Fig. 2 and 3 display two groups of isomers: a fast reacting one (Fig. 2) and a slow reacting one (Fig. 3).
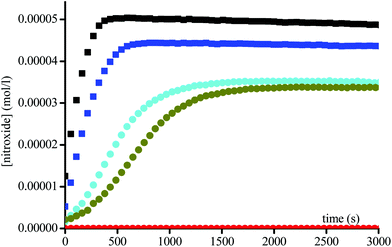 |
| | Fig. 2 Growth of TEMPO and DBNO released by the homolysis of 1A(−) ( ), 1B(+) ( ), 1B(+) ( ), 2A(−) (■), and 2B(+) ( ), 2A(−) (■), and 2B(+) ( ) against time at 37 °C in buffer (pH = 7.4) and in the presence of Subtilisin A (1.5 × 10−3 M), and of 1A(−) ( ) against time at 37 °C in buffer (pH = 7.4) and in the presence of Subtilisin A (1.5 × 10−3 M), and of 1A(−) ( ) in the absence of Subtilisin A. Initial concentrations in alkoxyamines are 0.1 mM. ) in the absence of Subtilisin A. Initial concentrations in alkoxyamines are 0.1 mM. | |
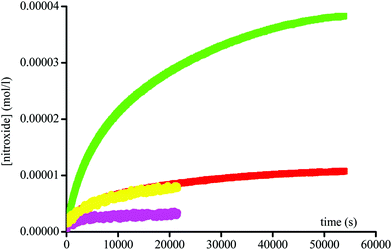 |
| | Fig. 3 Growth of TEMPO and DBNO released by the homolysis of 1A(+) ( ), 1B(−) ( ), 1B(−) ( ), 2A(+) ( ), 2A(+) ( ), and 2B(−) ( ), and 2B(−) ( ) against time at 37 °C in buffer (pH = 7.4) and in the presence of Subtilisin A (1.5 × 10−3 M). Initial concentration in alkoxyamines are 0.1 mM. ) against time at 37 °C in buffer (pH = 7.4) and in the presence of Subtilisin A (1.5 × 10−3 M). Initial concentration in alkoxyamines are 0.1 mM. | |
Interestingly, two isomers of 1 and two isomers of 2 are present in each group, suggesting an enantiomeric selection by the enzyme. In the absence of enzyme no homolysis events were detected.
By varying the enzyme concentration, an optimum is observed around 3 × 10−3 M, affording the fastest homolysis rate and the highest conversion (Fig. 4). Surprisingly, for all enantiomers the maximal conversion for 1 and 2 is of 40% and 50%, respectively, and not the full conversion as expected. Furthermore the conversion varies with the enzyme concentration (Fig. 4).
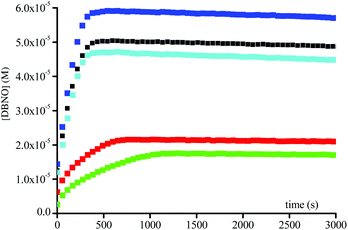 |
| | Fig. 4 Plots of DBNO released during the homolysis of 0.1 mM of 2A(−)vs. time in buffer (pH = 7.42) at 37 °C and in the presence of various amounts of Subtilisin A ( ) 4.0 × 10−4 M, ( ) 4.0 × 10−4 M, ( ) 8.3 × 10−4 M, (■) 1.6 × 10−3 M, ( ) 8.3 × 10−4 M, (■) 1.6 × 10−3 M, ( ) 3.3 × 10−3 M, and ( ) 3.3 × 10−3 M, and ( ) 6.6 × 10−3 M. ) 6.6 × 10−3 M. | |
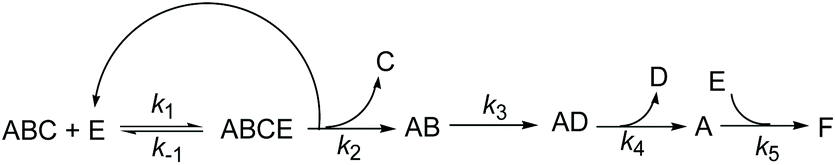
In fact, Subtilisin A contains some residues, e.g., cysteine or tyrosine residues, capable of reducing the nitroxide, as highlighted in Fig. 5, impeding the increase in concentration of the nitroxide up to completion. This loss of nitroxide is only clearly detected at high concentrations of the enzyme. Thus, when using Subtilisin A in large excess,|| the values of k5 are determined as kT5 = 3.0 × 10−6 s−1 for TEMPO and kD5 = 1.1 × 10−5 s−1 for DBNO,** assuming a pseudo first-order reaction to occur between the enzyme and the nitroxide, as shown in Scheme 3 and given by eqn (3).
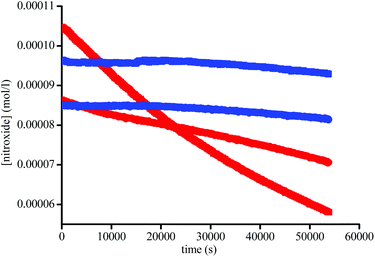 |
| | Fig. 5 Plot of [1] (●) and [2] (■) vs. time in the absence (blue) and in the presence of 1.5 × 10−3 M of Subtilisin A (red) at 37 °C in buffer (pH = 7.4). | |
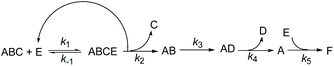 |
| | Scheme 3 Kinetic scheme for the decay of alkoxyamine ABC in the presence of enzyme E (Subtilisin A). ABCE is the enzymatic complex, AB the hemiacetal alkoxyamine (see Scheme 4), AD the activated aldehyde alkoxyamine (see Scheme 4), A and D the nitroxyl and alkyl radicals, respectively, and F the degradation product of the reaction between A and E. | |
Because the kinetics of enzymatically catalyzed reactions are often complicated,51 because the enzyme plays both the role of catalyst and reducing agent for the nitroxide, because intermediates have not yet been clearly identified, the kinetic equations derived from Scheme 3 cannot be developed in a simple way and would not provide more information on the homolysis process.
Discussion
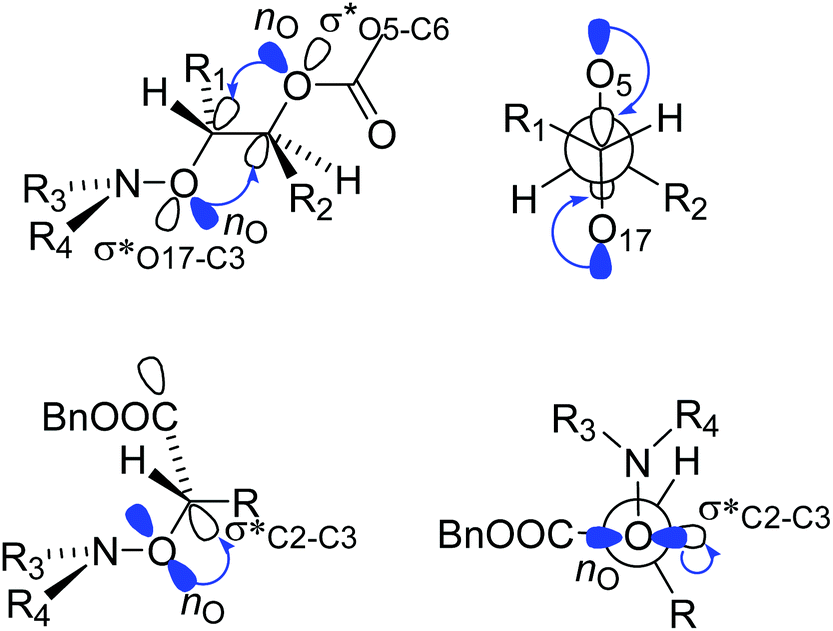
Alkoxyamines 1 and 2 exhibit Ea values (Table 1) in the range of those of their simple models 4 and 5,52 respectively, meaning that, as reported in literature,46 conventional polar, steric and stabilization effects are ruling the C–ON bond homolysis. These differences of 5–10 kJ mol−1 in Ea are ascribed to a homoanomeric and hyperconjugation effects stabilizing the starting materials. As mentioned above, the conventional geometrical parameters of 1A(−) are very close to those reported for ester based alkoxyamines,48 except that alkoxyamines carrying acetal groups have never been reported. Interestingly, the dihedral angle <O17C3C5O6> exhibits a value of 180°, meaning that the two C–O bonds have an antiperiplanar conformation, favouring the hyperconjugation effect (Fig. 1 and 6). Thus, the donations from the oxygen lone pairs nO of atoms O6 and O17 into the antibonding orbitals σ* of O17–C3 and C5–O6 bonds, respectively, occur and this is known as the Plough effect.53†† Moreover, combination of the presence of electron withdrawing ester function at C2 and the dihedral angle <N21O17C3C2> close to 90° (Fig. 6) support the hyperconjugation interaction 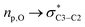 via the electron donation from p pure lone pair of O18 into the antibonding orbital of the C2–C3 bond The anomeric (hyperconjugation) effect,54 intramolecular H-bonding55 and intramolecular coordination56 between the alkyl and nitroxyl fragments are well known for stabilizing starting materials and leading to stronger C–ON bond, i.e., in all cases it is like cleaving an extra bond.
via the electron donation from p pure lone pair of O18 into the antibonding orbital of the C2–C3 bond The anomeric (hyperconjugation) effect,54 intramolecular H-bonding55 and intramolecular coordination56 between the alkyl and nitroxyl fragments are well known for stabilizing starting materials and leading to stronger C–ON bond, i.e., in all cases it is like cleaving an extra bond.
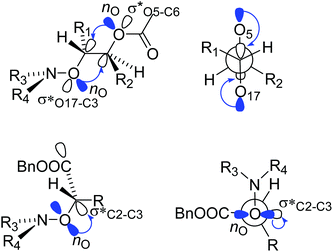 |
| | Fig. 6 Homoanomeric Plough (top) and hyperconjugation (bottom) effects in alkoxyamines 1 and 2. | |
Indeed, the Plough effect due to the 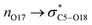 interaction affords an interaction between the nitroxyl fragment with O17 and the alkyl fragment with C5–O18 bond, which may be considered as an extra weak bond to be cleaved for homolysis to occur. It is assumed that these comments hold for all other isomers of 1 (as well as for the isomers of 2), as they exhibit very similar Ea that of 1A(−).
interaction affords an interaction between the nitroxyl fragment with O17 and the alkyl fragment with C5–O18 bond, which may be considered as an extra weak bond to be cleaved for homolysis to occur. It is assumed that these comments hold for all other isomers of 1 (as well as for the isomers of 2), as they exhibit very similar Ea that of 1A(−).
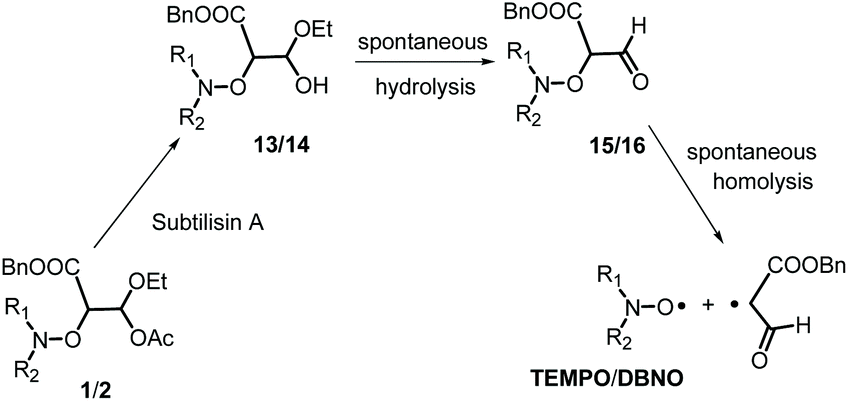
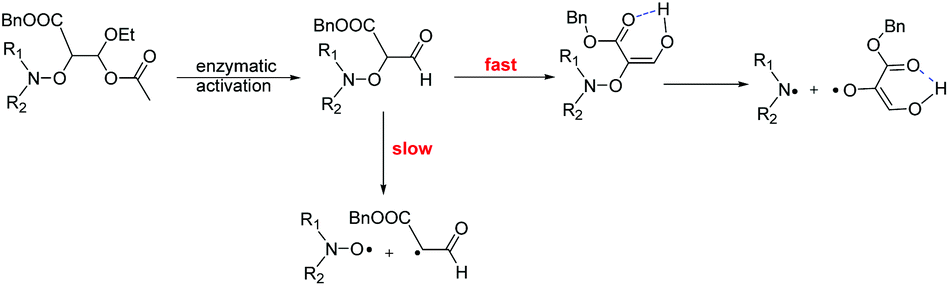
Although Subtilisin A (4.0 × 10−4 to 6.6 × 10−3 M) is not used in sub-stœchiometric amounts as expected in enzymatic catalysis, it still plays the role of a catalyst, as in its absence no homolysis occurs whereas in its presence a maximal apparent conversion of 50% and 30% (Fig. 2–4) is reached for 2A(−) and 1A(−), respectively, in less than 500 s and 1500 s, respectively. Remarkably, less than 10−16 M of 1 and 2 are decomposed during this time in the absence of enzyme according to their Ea values! The dramatic effect of Subtilisin A on the stability of 1 and 2 is accounted for only by a dramatic change in structures of 1 and 2. Indeed, the Subtilisin A enzyme is known as a poorly selective protease capable of hydrolyzing an ester bond, the acetoxy group in 1 and in 2 is thus “readily” hydrolysed (Scheme 4) to afford hemiacetal 13 and 14, respectively. The latter are not stable in aqueous media and collapse into aldehydes 15 and 16 respectively, which are prone to homolyze into the corresponding TEMPO and DBNO and the malonic-like alkyl radicals (Scheme 4). This sequence of events is supported by the high values of Ea for 6 (Ea = 145 kJ mol−1, Table 1) and 7 (Ea = 146 kJ mol−1),47‡‡ which leads to discard the replacement of the acetoxy group of 1 by the propyloxy group in 6 and the hydrolysis of the benzyloxycarbonyl moiety of 1 into the carboxylic/carboxylate function in 7.
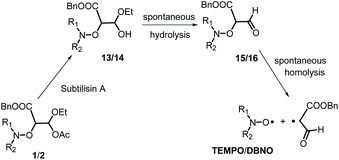 |
| | Scheme 4 Tentative activation pathway for the homolysis of 1 and 2 induced by Subtilisin A. | |
Moreover, as 3 experiences a dramatic solvent effect from tBuPh to water/MeOH with a decrease in the Ea by 18 kJ mol−1, the same effect is expected for 15, with a conformational effect likely stronger, as the oxo group in 3 is replaced by a formyl group in 13, which may favour the required conformation at TS, and hence, afford a striking increase in kd. Therefore, the enzymatic hydrolysis of 1 affords the hemiacetal which is spontaneously hydrolized into formyl-based alkoxyamine 15, which accounts for the spontaneous generation of TEMPO.††,§§¶¶ Unfortunately, despite several attempts it was not possible to prepare 13via conventional methods, meaning either the formyl derivative 9 is less stable than the oxo derivative 3 or the intermediates do not react as expected. It is assumed that these comments hold for 2.
Because of a side-reaction between nitroxides and Subtilisin A (Fig. 6 and Scheme 3) and a selective efficiency of the enzyme for the hydrolysis of the acetoxy group, the steady state in the nitroxide is reached between 5% to 50% conversion (Fig. 2 and 3). Indeed, for 1 and 2, Subtilisin A shows a selectivity in favour of enantiomers A(−) and B(+) against enantiomers A(+) and B(−). Taking into account that 1 and 2 exhibit very different homolysis rate constants and that TEMPO and DBNO react at different rates with Subtilisin A, the role of the nitroxyl fragment on the selectivity of Subtilisin A is unclear.
As mentioned above, in enzymatic catalysis, the use of a large excess of enzyme is rather unconventional. The rate of DBNO generation and the yield of the reaction vary with the enzyme concentration (Fig. 4), suggesting that for such a simple acetoxy leaving group the enzymatic activation is rate-limiting. The bell-shaped curve of the maximal concentration in nitroxide vs. the amount of Subtilisin A (Fig. 7) shows a maximal concentration of ca. 70% conversion in nitroxide for ca. 4 10−3 M in enzyme. Interestingly, this bell-shaped curve is divided in 3 regions: region I for sub-stœchiometric enzyme concentrations, for which a different regime (Fig. 7) is observed for the generation of nitroxide, because the non-reversible nitroxide and enzyme reaction competes with the enzyme-intermediate formation (ABCE in Scheme 3), affording lower and lower concentrations in enzyme with time, slowing down the formation of intermediate and, hence, the release of 15 and 16; region II for an excess of enzyme for which the Ostwald assumption holds, and then the release of nitroxide is favoured over its degradation by the enzyme; and region III for which the excess of enzyme is so large that the decay of nitroxide by reaction with the enzyme becomes the favoured reaction.
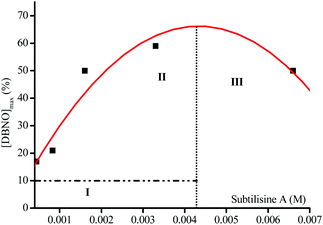 |
| | Fig. 7 Maximal concentration in DBNO (%) for various amounts of Subtilisin A. | |
Conclusions
The enzyme-triggered hydrolysis of acetal into hemiacetal, which, in turn, hydrolyzes spontaneously into aldehyde, is a powerful switch to transform highly stable alkoxyamines into highly labile ones. The results reported here show that the enzymatic reaction is the limiting event, in sharp contrast to the results reported for an alkoxyamine locked with a peptide. Thus, the next step should focused on more targeted peptides including those relevant for the enzymes found in the tumour environment. Despite this limitation, more than 60% conversion of 2A(−) is reached in less than 10 minutes, meaning that the homolysis of peptide-locked alkoxyamines should be faster, and that the C–ON bond can be weakened by changing the nitroxyl fragment, ester and alkoxy functions on the alkyl fragment.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
S. R. A. M., G. A., M. H., N. V., and M. A. are grateful to Aix-Marseille University and CNRS for support. P. M. and V. P. are grateful to University of Bordeaux and INSERM for support. S. R. A. M. and G. A., thank ANR for financial support (ANR-17-CE18-0017). All authors are grateful to Spectropôle (AMU/CNRS) platform for technical support.
Notes and references
-
D. H. Solomon, E. Rizzardo and P. Cacioli, Eur. Pat. Appl., 1985, 135280 Search PubMed;
D. H. Solomon, E. Rizzardo and P. Cacioli, US Pat, 4,581,429, 1986 Search PubMed; D. H. Solomon, E. Rizzardo and P. Cacioli, Chem. Abstr., 1985, 102, 221335q Search PubMed.
- E. Rizzardo, Living free radical polymerisation, Chem. Aust., 1987, 54, 32–33 Search PubMed.
- C. H. J. Johnson, G. Moad, D. H. Solomon, T. H. Spurling and D. J. Vearing, The Application of Supercomputers in Modeling Chemical Reaction Kinetics: Kinetic Simulation of “Quasi-Living” Radical Polymerization, Aust. J. Chem., 1990, 43, 1215–1230 CrossRef CAS.
- G. Moad and E. Rizzardo, Alkoxyamine-Initiated Living Radical Polymerization: Factors Affecting Alkoxyamine Homolysis Rates, Macromolecules, 1995, 28, 8722–8728 CrossRef CAS.
-
G. Moad, D. H. Solomon and G. Moad, The Chemistry of Radical Polymerization, Elsevier, Amsterdam; Boston, 2006, and references cited therein Search PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Nitroxide-Mediated Polymerization, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
-
D. Gigmes and S. R. A. Marque, Encyclopedia of Radicals in Chemistry, Biology, and Materials, ed. C. Chatgilialoglu and A. Studer, Wiley, Chichester, U.K., 2012, vol. 4, pp. 1813–1850 Search PubMed.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagci and K. Matyjaszewski, Photomediated Controlled Radical Polymerization, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS.
-
Nitroxide Mediated Polymerization: From Fundamentals to Applications in Materials Science, RSC polymer chemistry series, ed. D. Gigmes, Royal Society of Chemistry, Cambridge, 2016 Search PubMed.
- M. V. Edeleva, E. G. Bagryanskaya and S. R. A. Marque, Imidazoline and Imidazolidine Nitroxides as Controlling Agents in Nitroxide-Mediated Pseudoliving Radical Polymerization, Russ. Chem. Rev., 2018, 87(4), 328–349 CrossRef CAS.
- M. Destarac, Controlled Radical Polymerization: Industrial Stakes, Obstacles and Achievements, Macromol. React. Eng., 2010, 4, 165–179 CrossRef CAS.
- E. H. H. Wong, T. Junkers and C. Barner-Kowollik, Nitrones in Synthetic Polymer Chemistry, Polym. Chem., 2011, 2, 1008–1010 RSC.
- Y. Guillaneuf, D. Bertin, D. Gigmes, D. L. Versace, J. Lalevee and J. P. Fouassier, Toward Nitroxide-Mediated Photopolymerization, Macromolecules, 2010, 43, 2204–2212 CrossRef CAS.
- D. L. Versace, J. Lalevee, J. P. Fouassier, D. Gigmes, Y. Guillaneuf and D. Bertin, Photosensitized Alkoxyamines as Bicomponent Radical Photoinitiators, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2910–2915 CrossRef CAS.
- E. Yoshida, Nitroxide-Mediated Photo-Living Radical Polymerization of Methyl Methacrylate in Solution, Colloid Polym. Sci., 2010, 288, 1639–1643 CrossRef CAS.
- G. Audran, E. Bagryanskaya, M. Edeleva, S. R. A. Marque, D. Parkhomenko, E. Tretyakov and S. Zhivetyeva, Coordination-Initiated Nitroxide-Mediated Polymerization (CI-NMP), Aust. J. Chem., 2018, 71, 334–340 CrossRef CAS.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H. Klok, Surface-Initiated Controlled Radical Polymerization: State-of-the-Art, Opportunities, and Challenges in Surface and Interface Engineering with Polymer Brushes, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS PubMed.
- G. Audran, E. Bagryanskaya, I. Bagryanskaya, P. Brémond, M. Edeleva, S. R. A. Marque, D. Parkhomenko, O. Yu. Rogozhnikova, V. M. Tormyshev, E. V. Tretyakov, D. V. Trukhin and S. I. Zhivetyeva, Trityl-Based Alkoxyamines as NMP Controllers and Spin-Labels, Polym. Chem., 2016, 7, 6490–6499 RSC.
- M. V. Edeleva, S. R. A. Marque, O. Yu. Rogozhnikova, V. M. Tormyshev, T. I. Troitskaya and E. G. Bagryanskaya, Radical Polymerization of Radical-Labeled Monomers: The Triarylmethyl-Based Radical Monomer as an Example, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2656–2664 CrossRef CAS.
- O. Guselnikova, S. R. A. Marque, E. Tretyakov, D. Mares, V. Jerabek, G. Audran, J.-P. Joly, M. Trusova, V. Švorčik, O. Lyutakov and P. Postnikov, Unprecedented Plasmon-Induced Nitroxide-Mediated Polymerization (PI-NMP): A Method for Preparation of Functional Surfaces, J. Mater. Chem. A, 2019, 7(20), 12414–12419 RSC.
- C. Yuan, M. Z. Rong, M. Q. Zhang, Z. P. Zhang and Y. C. Yuan, Self-Healing of Polymers via Synchronous Covalent Bond Fission/Radical Recombination, Chem. Mater., 2011, 23, 5076–5081 CrossRef CAS.
- Z. P. Zhang, M. Z. Rong, M. Q. Zhang and C. Yuan, Alkoxyamine with Reduced Homolysis Temperature and Its Application in Repeated Autonomous Self-Healing of Stiff Polymers, Polym. Chem., 2013, 4, 4648–4654 RSC.
- B. Schulte, M. Tsotsalas, M. Becker, A. Studer and L. De Cola, Dynamic Microcrystal Assembly by Nitroxide Exchange Reactions, Angew. Chem., Int. Ed., 2010, 49, 6881–6884 CrossRef CAS PubMed.
- L. Charles, C. Laure, J.-F. Lutz and R. K. Roy, MS/MS Sequencing of Digitally Encoded Poly(alkoxyamine Amide)s, Macromolecules, 2015, 48, 4319–4328 CrossRef CAS.
- R. K. Roy, A. Meszynska, C. E. Laure, L. Charles, C. Verchin and J.-F. Lutz, Design and Synthesis of Digitally Encoded Polymers That Can Be Decoded and Erased, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- G. Cavallo, A. Al Ouahabi, L. Oswald, L. Charles and J.-F. Lutz, Orthogonal Synthesis of “Easy-to-Read” Information-Containing Polymers Using Phosphoramidite and Radical Coupling Steps, J. Am. Chem. Soc., 2016, 138, 9417–9420 CrossRef CAS PubMed.
- G. Audran, P. Brémond, J.-M. Franconi, S. R. A. Marque, P. Massot, P. Mellet, E. Parzy and E. Thiaudière, Alkoxyamines: A New Family of pro-Drugs against Cancer. Concept for Theranostics, Org. Biomol. Chem., 2014, 12, 719–723 RSC.
- D. Moncelet, P. Voisin, N. Koonjoo, V. Bouchaud, P. Massot, E. Parzy, G. Audran, J.-M. Franconi, E. Thiaudière, S. R. A. Marque, P. Brémond and P. Mellet, Alkoxyamines: Toward a New Family of Theranostic Agents against Cancer, Mol. Pharmaceutics, 2014, 11, 2412–2419 CrossRef CAS PubMed.
- N. A. Popova, G. M. Sysoeva, V. P. Nicolin, V. I. Kaledin, E. V. Tretyakov, M. V. Edeleva, S. M. Balakhnin, E. L. Louschnikova, G. Audran and S. Mark, Comparative Study of Toxicity of Alkoxyamines In Vitro and In Vivo, Bull. Exp. Biol. Med., 2017, 164(1), 49–53 CrossRef CAS PubMed.
- T. Yamasaki, D. Buric, Ch. Chacon, G. Audran, D. Braguer, S. R. A. Marque, M. Carré and P. Brémond, Chemical Modifications of Imidazole-Containing Alkoxyamines Increase C–ON Bond Homolysis Rate: Effects on Their Cytotoxic Properties in Glioblastoma Cells, Bioorg. Med. Chem., 2019, 27, 1942–1951 CrossRef CAS PubMed.
- J. Wang and J. Yi, Cancer Cell Killing via ROS: To Increase or Decrease, That Is the Question, Cancer Biol. Ther., 2008, 7, 1875–1884 CrossRef CAS PubMed.
- P. Brémond and S. R. A. Marque, First Proton Triggered C–ON Bond Homolysis in Alkoxyamines, Chem. Commun., 2011, 47(14), 4291–4293 RSC.
- P. Brémond, A. Koïta, S. R. A. Marque, V. Pesce, V. Roubaud and D. Siri, Chemically Triggered C–ON Bond Homolysis of Alkoxyamines. Quaternization of the Alkyl Fragment, Org. Lett., 2012, 14(1), 358–361 CrossRef PubMed.
- G. Audran, L. Bosco, P. Brémond, S. R. A. Marque, V. Roubaud and D. Siri, Chemically Triggered C–ON Bond Homolysis of Alkoxyamines. 8. Quaternization and Steric Effects, J. Org. Chem., 2013, 78, 9914–9920 CrossRef CAS PubMed.
- G. Audran, E. Bagryanskaya, I. Bagryanskaya, P. Brémond, M. Edeleva, S. R. A. Marque, D. Parkhomenko, E. Tretyakov and S. Zhivetyeva, C–ON Bond Homolysis of Alkoxyamines Triggered by Paramagnetic Copper(II) Salts, Inorg. Chem. Front., 2016, 3(11), 1464–1472 RSC.
- G. Audran, E. Bagryanskaya, I. Bagryanskaya, M. Edeleva, S. R. A. Marque, D. Parkhomenko, E. Tretyakov and S. Zhivetyeva, Zinc(II) Hexafluoroacetylacetonate Complexes of Alkoxyamines: NMR and Kinetic Investigations. First Step for a New Way to Prepare Hybrid Materials, ChemistrySelect, 2017, 2, 3584–3593 CrossRef CAS.
- M. V. Edeleva, I. A. Kirilyuk, I. F. Zhurko, D. A. Parkhomenko, Y. P. Tsentalovich and E. G. Bagryanskaya, pH-Sensitive C–ON Bond Homolysis of Alkoxyamines of Imidazoline Series with Multiple Ionizable Groups As an Approach for Control of Nitroxide Mediated Polymerization, J. Org. Chem., 2011, 76, 5558–5573 CrossRef CAS PubMed.
- G. Audran, E. Bagryanskaya, I. Bagryanskaya, M. Edeleva, P. Klina, S. R. A. Marque, D. Parkhomenko, E. Tretyakov and S. Zhivetyeva, The Effect of the Oxophilic Tb(III) Cation on C ON Bond Homolysis in Alkoxyamines, Inorg. Chem. Commun., 2018, 91, 5–7 CrossRef CAS.
- G. Audran, P. Brémond and S. R. A. Marque, Labile Alkoxyamines: Past, Present, and Future, Chem. Commun., 2014, 50(59), 7921–7928 RSC.
- G. Audran, L. Bosco, P. Brémond, N. Jugniot, S. R. A. Marque, P. Massot, P. Mellet, T. Moussounda Moussounda Koumba, E. Parzy, A. Rivot, E. Thiaudière, P. Voisin, C. Wedl and T. Yamasaki, Enzymatic Triggering of C–ON Bond Homolysis of Alkoxyamines, Org. Chem. Front., 2019, 6, 3663–3672 RSC.
- S. Edvinsson, S. Johansson and A. Larsson, An Efficient Procedure for the Synthesis of Formylacetic Esters, Tetrahedron Lett., 2012, 53, 6819–6821 CrossRef CAS.
- J. H. A. Babler, A Facile Route to Glyoxal Mono(diethyl Acetal): A Versatile Synthon for Preparation of A,β-Unsaturated Aldehydes, Synth. Commun., 1987, 17, 77–84 CrossRef CAS.
- K. Matyjaszewski, B. E. Woodworth, X. Zhang, S. G. Gaynor and Z. Metzner, Simple and Efficient Synthesis of Various Alkoxyamines for Stable Free Radical Polymerization, Macromolecules, 1998, 31, 5955–5957 CrossRef CAS.
- J. M. Álvarez-Calero, Z. D. Jorge and G. M. Massanet, TiCl 4 /Et 3 N-Mediated Condensation of Acetate and Formate Esters: Direct Access to β-Alkoxy- and β-Aryloxyacrylates, Org. Lett., 2016, 18, 6344–6347 CrossRef PubMed.
- P. Nkolo, G. Audran, R. Bikanga, P. Brémond, S. R. A. Marque and V. Roubaud, C–ON Bond Homolysis of Alkoxyamines: When Too High Polarity Is Detrimental, Org. Biomol. Chem., 2017, 15, 6167–6176 RSC.
-
E. G. Bagryanskaya and S. R. A. Marque, Kinetic Aspects of Nitroxide Mediated Polymerization, in Polymer Chemistry Series, ed. D. Gigmes, Royal Society of Chemistry, Cambridge, 2015, pp. 45–113 Search PubMed.
- D. Bertin, D. Gigmes, S. R. A. Marque and P. Tordo, Polar, Steric,
and Stabilization Effects in Alkoxyamines C−ON Bond Homolysis: A Multiparameter Analysis, Macromolecules, 2005, 38(7), 2638–2650 CrossRef CAS.
- E. Beaudoin, D. Bertin, D. Gigmes, S. R. A. Marque, D. Siri and P. Tordo, Alkoxyamine C–ON Bond Homolysis: Stereoelectronic Effects, Eur. J. Org. Chem., 2006, 1755–1768 CrossRef CAS.
- G. Audran, P. Brémond, S. R. A. Marque and G. Obame, Chemically Triggered C–ON Bond Homolysis of Alkoxyamines. Part 4: Solvent Effect, Polym. Chem., 2012, 3, 2901–2908 RSC.
- G. Audran, P. Brémond, S. R. A. Marque and G. Obame, Chemically Triggered C–ON Bond Homolysis of Alkoxyamines. 5. Cybotactic Effect, J. Org. Chem., 2012, 77(21), 9634–9640 CrossRef CAS PubMed.
-
K. A. Connors, Chemical Kinetics: The Study of Reaction Rates in Solution, Wiley-VCH, Nachdr., New York, NJ, 1990 Search PubMed.
- S. Marque, C. Le Mercier, P. Tordo and H. Fischer, Factors Influencing the C−O−Bond Homolysis of Trialkylhydroxylamines, Macromolecules, 2000, 33(12), 4403–4410 CrossRef CAS.
-
I. V. Alabugin, Stereoelectronic Effects: A Bridge between Structure and Reactivity, Wiley, Chichester, West Sussex, UK; Hoboken, NJ, USA, 2016 Search PubMed.
- A. Gaudel-Siri, D. Siri and P. Tordo, Homolysis of N-Alkoxyamines: A Computational Study, ChemPhysChem, 2006, 7, 430–438 CrossRef CAS PubMed.
- G. Audran, R. Bikanga, P. Brémond, M. Edeleva, S. R. A. Marque, P. Nkolo and V. Roubaud, How Intramolecular Hydrogen Bonding (IHB) Controls the C–ON Bond Homolysis in Alkoxyamines, Org. Biomol. Chem., 2017, 15, 8425–8439 RSC.
- G. Audran, E. Bagryanskaya, I. Bagryanskaya, M. Edeleva, J.-P. Joly, S. R. A. Marque, A. Iurchenkova, P. Kaletina, S. Cherkasov, T. H. Tung, E. Tretyakov and S. Zhivetyeva, How Intramolecular Coordination Bonding (ICB) Controls the Homolysis of the C–ON Bond in Alkoxyamines, RSC Adv., 2019, 9, 2576–25789 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, analysis data, kinetics, LFER analysis and XRD data are reported as ESI. CCDC 1989155. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qo00559b |
| ‡ CCDC 1989155. |
| § For 1: σRS = 0.18, σI = 0.16 and υ = 0.83. More details are provided in ESI.† |
| ¶ For 2, Ea is given by applying the increment −10.5 kJ mol−1 to Ea of 1. See ref. 53. |
| || Values of k4 are given to a factor depending on the number of cysteine and tyrosine on the enzyme. |
| ** For DBNO alone, k5 = 5.3 10−7 s−1. |
| †† The difference between Plough effect and W effect lies in the position of the donating lone pair which is parallel to the bond onto which donating and accepting group are attached. |
| ‡‡ Private communication of authors. Data for 6 will be reported in a forthcoming article. |
§§ The occurrence of a charged intermediate during the enzymatic hydrolysis cannot be straightforwardly disregarded as it would dramatically increase the C–ON bond homolysis rate.
|
¶¶ A reviewer mentioned the possible formation of enol which would dramatically favoured the N–O bond homolysis over the C–O bond homolysis. However, this possibility is discarded because using model 3 and several homologues (ref. 45), basic catalysis with various bases, e.g., KH, LiH, NaH, HMDS, OH−, and acidic catalysis, e.g., H+, Lewis acids and several traps, the formation of enol was never observed and starting materials always recovered.
|
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Philippe
Mellet
*b,
Philippe
Mellet


 ), 1B(+) (
), 1B(+) ( ), 2A(−) (■), and 2B(+) (
), 2A(−) (■), and 2B(+) ( ) against time at 37 °C in buffer (pH = 7.4) and in the presence of Subtilisin A (1.5 × 10−3 M), and of 1A(−) (
) against time at 37 °C in buffer (pH = 7.4) and in the presence of Subtilisin A (1.5 × 10−3 M), and of 1A(−) ( ) in the absence of Subtilisin A. Initial concentrations in alkoxyamines are 0.1 mM.
) in the absence of Subtilisin A. Initial concentrations in alkoxyamines are 0.1 mM.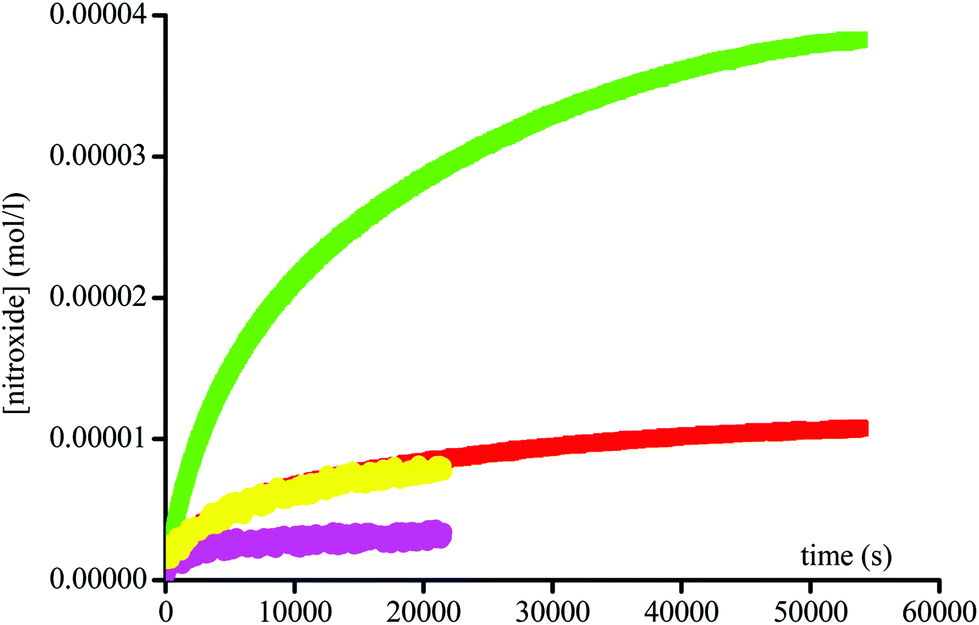
 ), 1B(−) (
), 1B(−) ( ), 2A(+) (
), 2A(+) ( ), and 2B(−) (
), and 2B(−) ( ) against time at 37 °C in buffer (pH = 7.4) and in the presence of Subtilisin A (1.5 × 10−3 M). Initial concentration in alkoxyamines are 0.1 mM.
) against time at 37 °C in buffer (pH = 7.4) and in the presence of Subtilisin A (1.5 × 10−3 M). Initial concentration in alkoxyamines are 0.1 mM.
 ) 4.0 × 10−4 M, (
) 4.0 × 10−4 M, ( ) 8.3 × 10−4 M, (■) 1.6 × 10−3 M, (
) 8.3 × 10−4 M, (■) 1.6 × 10−3 M, ( ) 3.3 × 10−3 M, and (
) 3.3 × 10−3 M, and ( ) 6.6 × 10−3 M.
) 6.6 × 10−3 M.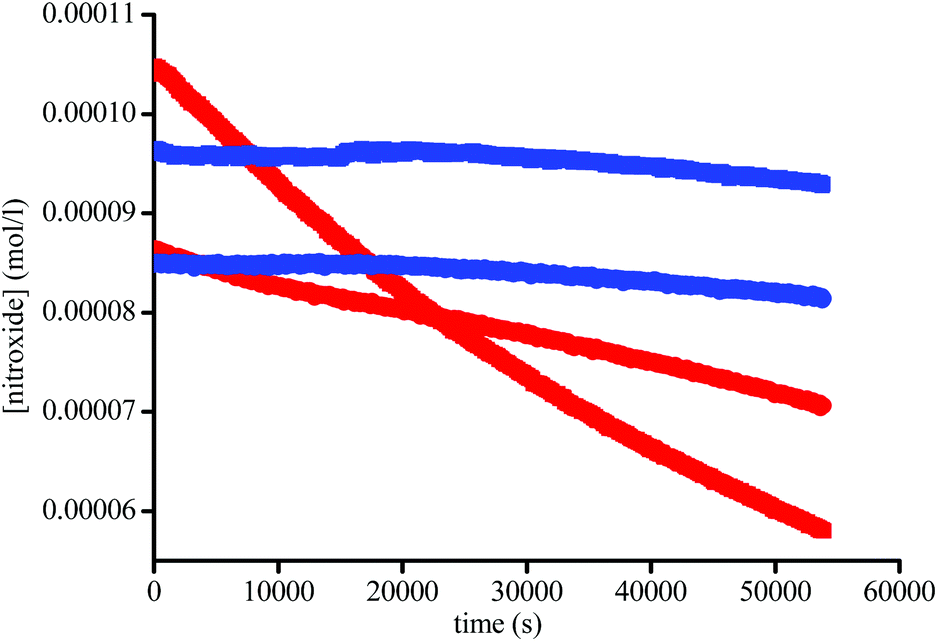
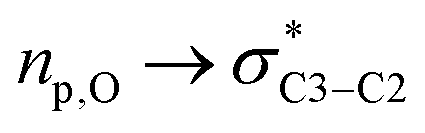 via the electron donation from p pure lone pair of O18 into the antibonding orbital of the C2–C3 bond The anomeric (hyperconjugation) effect,54 intramolecular H-bonding55 and intramolecular coordination56 between the alkyl and nitroxyl fragments are well known for stabilizing starting materials and leading to stronger C–ON bond, i.e., in all cases it is like cleaving an extra bond.
via the electron donation from p pure lone pair of O18 into the antibonding orbital of the C2–C3 bond The anomeric (hyperconjugation) effect,54 intramolecular H-bonding55 and intramolecular coordination56 between the alkyl and nitroxyl fragments are well known for stabilizing starting materials and leading to stronger C–ON bond, i.e., in all cases it is like cleaving an extra bond.
 interaction affords an interaction between the nitroxyl fragment with O17 and the alkyl fragment with C5–O18 bond, which may be considered as an extra weak bond to be cleaved for homolysis to occur. It is assumed that these comments hold for all other isomers of 1 (as well as for the isomers of 2), as they exhibit very similar Ea that of 1A(−).
interaction affords an interaction between the nitroxyl fragment with O17 and the alkyl fragment with C5–O18 bond, which may be considered as an extra weak bond to be cleaved for homolysis to occur. It is assumed that these comments hold for all other isomers of 1 (as well as for the isomers of 2), as they exhibit very similar Ea that of 1A(−).