
An exocyclic π-system extension of the phenanthriporphyrin framework: towards azaaceneporphyrinoids†
Bartosz
Szyszko,  *a
Sebastian G.
Mucha,
bc
Michał J.
Białek,
a
Katarzyna
Matczyszyn
c
and
Lechosław
Latos-Grażyński
*a
*a
Sebastian G.
Mucha,
bc
Michał J.
Białek,
a
Katarzyna
Matczyszyn
c
and
Lechosław
Latos-Grażyński
*a
*a
Sebastian G.
Mucha,
bc
Michał J.
Białek,
a
Katarzyna
Matczyszyn
c
and
Lechosław
Latos-Grażyński
*a
Abstract
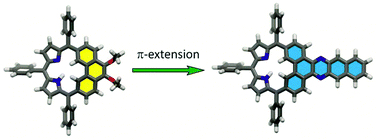
A facile method for the exocyclic π-extension of the carbaporphyrinoid embedding phenanthrene moiety – phenanthriporphyrin – has been described. The reaction between 5,6-dioxophenanthriporphyrin and a series of aliphatic and aromatic amines provided a family of carbaporphyrinoids incorporating azaacene components. The 1H NMR spectroscopic, electrochemical, and optical properties of azaaceneporphyrinoids strongly depend on the structure of the incorporated moiety. The π-system extension of the attached azaacene subunit reduces the macrocyclic antiaromatic character.
 Please wait while we load your content...
Please wait while we load your content...

