
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards the synthesis of calotropin and related cardenolides from 3-epiandrosterone: A-ring related modifications†
Vanessa
Koch
a,
Martin
Nieger
b and
Stefan
Bräse
 *ac
*ac
aInstitute of Organic Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: braese@kit.edu
bDepartment of Chemistry, University of Helsinki, P.O. Box 55, 00014 Helsinki, Finland
cInstitute for Biological and Chemical Systems – Functional Molecular Systems (IBCS-FMS), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 14th May 2020
Abstract
Calotropin and related cardiac glycosides isolated from plants such as calotropis gigantea represent an interesting target for biological investigations and are based on a cardiac steroid that is doubly connected to a sugar moiety. This naturally occurring family of cardiac glycosides was not only reported to have similar cardiac properties as the drugs digitoxin and digoxin, but also show cytotoxic activity against several cancer cell lines. Herein, the first synthetic access to these molecules is reported highlighting the required transformations of the A-ring of the steroid when starting from commercially available and inexpensive 3-epiandrosterone. Our strategy is based on a regioselective C–H oxidation of the methyl group at C-17 delivering the 2α,3β-trans-diol moiety at the same time and ensuring its connection to the sugar unit.
Introduction
Cardiac glycosides were already known to the ancient Egyptians over 3000 years ago and have been always used in folk medicine.1 Since the 18th century, cardiac glycosides from the digitalis plant typified by digitoxin (1) or digoxin (2) were successfully applied by Withering for the treatment of congestive heart failure.2,3 This was later attributed to the so called “cardiotonic effect” which is correlated to an inhibitory interaction with the sodium pump (Na+/K+-ATPase) leading to an increased intercellular sodium and calcium ion concentration. The inhibition finally results in a more powerful and faster contraction of the cardiomyocytes explaining the successful application of these drugs.4,5Cardiac glycosides show several characteristic features if compared to other classes of steroids. For example, they possess a tertiary hydroxyl group at C-14 and a β-oriented butenolide substituent at C-17 (Fig. 1).6 The A/B and C/D rings of cardiac glycosides are usually both cis-fused whereas cardiac glycosides isolated from the milkweed family Asclepiadaceae differ in having a trans-junction of the A/B rings.7 While digitoxin (1), digoxin (2) and ouabain (3) are attached via their 3β-OH group to the sugar moiety, the cardenolides calotropin (4) and their related cardenolides (5–10) are connected by the 2α- and 3β-position to the sugar unit forming a 1,4-dioxane ring.8–15 This double linkage of the steroidal pattern to the carbohydrate explains the unusual stability towards acids16 and reduction.17
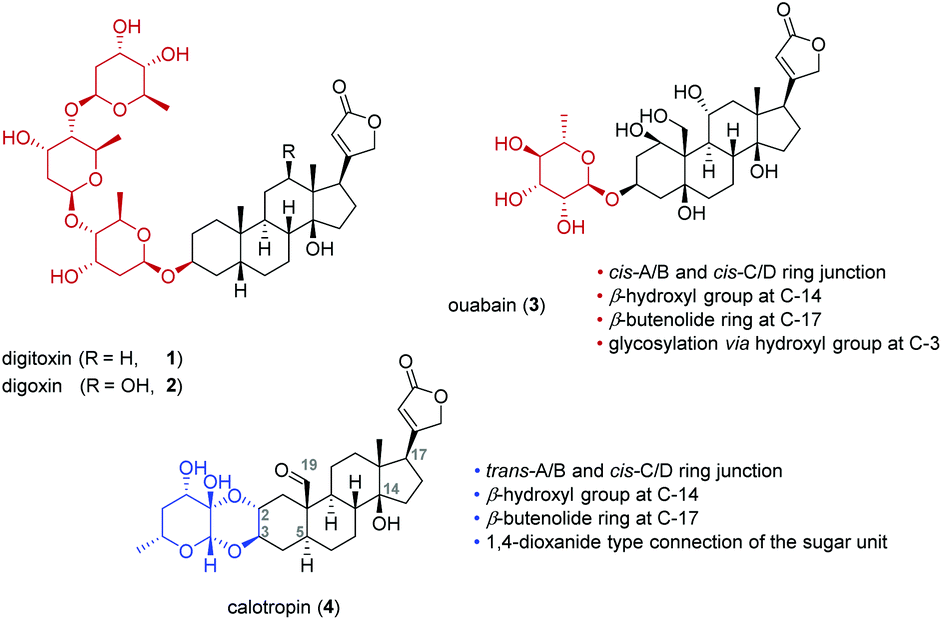 | ||
Fig. 1 Different cardiac glycosides and their connection of the steroidal aglycon to the sugar fragment. Calotropin (4) is doubly attached to the sugar moiety forming a 1,4-dioxane ring, while digitoxin (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H, 1), digoxin (ROH, 2) and ouabain (3) are linked to the sugar unit via the 3-OH group. H, 1), digoxin (ROH, 2) and ouabain (3) are linked to the sugar unit via the 3-OH group. | ||
Calotropin (4) and related cardenolides (5–10) bear moreover an aldehyde functionality at C-1916,18–20 that distinguishes them from gomphoside derivatives and occurs occasionally in some cardenolide classes, for instance also in k-strophanthin. Most cardiac glycosides contain one to four sugar residues attached to the genin inducing both the water solubility and the ability to bind the heart muscle.7,21 In the case of the Calotropin family, many different substitution patterns were found at the sugar moiety (Fig. 2). Modifications occur mostly at C-3′ including the acetylation22 or epimerization9,10,15,23 of the hydroxyl group, but also its oxidation.10,11,14,15 Moreover, an attachment of a thiazolidine9–11,13,15 or a dihydrothiazolidine11,14,15 moiety was discovered at C-3′ as a frequent modification of the sugar building block demonstrating the broad diversity of the presented cardenolide class.
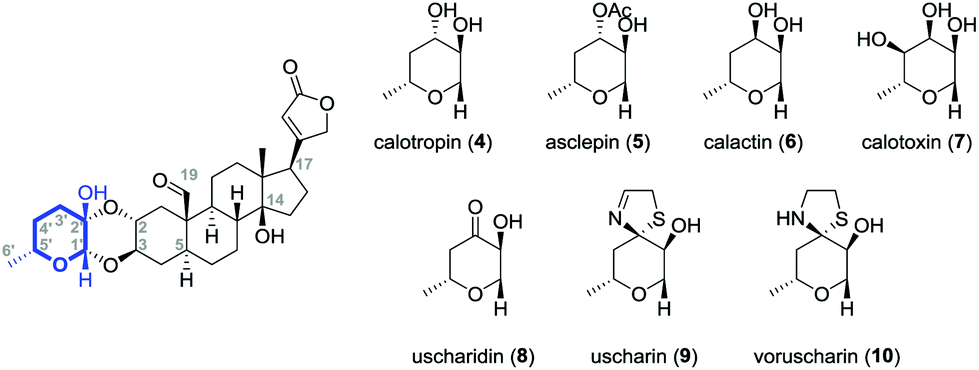 | ||
| Fig. 2 Calotropin (4) and selected cardenolide glycosides (5–10) isolated from the milkweed family Asclepiadaceae. | ||
While cardiac glycosides were used by the ancient Egyptians and Romans as heart tonic, emetic and diueretic,24 natives in Africa took advantage of their toxic effects by applying calotropis plants as arrow poison.25,26 In lower doses, extracts of these plants exhibit a wide range of biological activities including anti-inflammatory,27–30 analgesic,31,32 anti-microbial27,33 or wound-healing properties.34,35 In the last years, cytotoxic and antitumor effects of cardenolides glycosides have been reported more frequently.36–42 The mode of action is still under investigation leading to several conclusions. For instance, Ishibashi and coworkers showed that calotropin (4) and related compounds inhibit the wnt signaling pathway in a dose-dependent manner.43 Although these cardenolides have been studied more than one century8 and are reported to have similar cardiac properties to well-established cardiac glycosides, they remain less investigated.40,44
Motivated by the interesting biological properties of the cardenolide family, we envisioned a modular semisynthesis from easy available starting materials that enables the modification of the lead structure on all parts of the molecule in order to gain insight into the structure activity relationship. A retrosynthetic analysis divides calotropin (4) into two fragments (Scheme 1), one of which is based on a sugar unit, while the other gives calotropagenin (11) as steroidal building block.
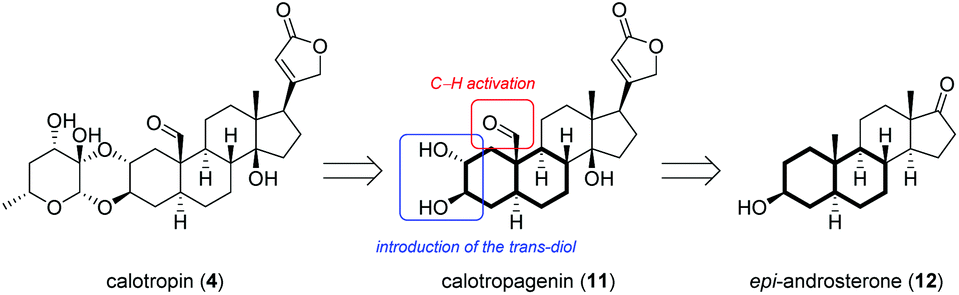 | ||
| Scheme 1 Retrosynthetic analysis of calotropin (4) gives calotropagenin (11) as steroidal fragment. Highlighted are the desired transformations for the A/B-ring system with 3-epiandrosterone (12) as starting material. This includes the installation of the trans-diol at C-2/C-3 and the oxidation to an aldehyde at C-19. | ||
As steroidal precursor, we chose commercially available 3-epiandrosterone (12) possessing already six of the eight required stereogenic centers. Nevertheless a couple of modifications at the A-ring of the steroid are required including the installation of the 2,3-trans-diol and the oxidation of C-19. Herein, we wish to report on the introduction of the oxygen moiety at C-19 allowing later on the generation of the aldehyde as well as the installation of the 2α,3β-diol which allows the attachment to the sugar unit according to Lichtenthaler's glycosylation.45
Results and discussion
Our approach for the synthesis of calotropin (4) is based on the idea to introduce the 2,3-trans-diol and the oxygen functionality at C-19 via a remote intramolecular free radical C–H oxidation giving the key intermediate 13. A Lewis-acid promoted ring-opening of the A-ring bridging ether in 13 should deliver the desired trans-diol as well as the desired oxygen functionality at C-19. As suitable precursor for the intended remote C–H oxidation, the C-3 acetylated cis-diol 14 was chosen (Scheme 2).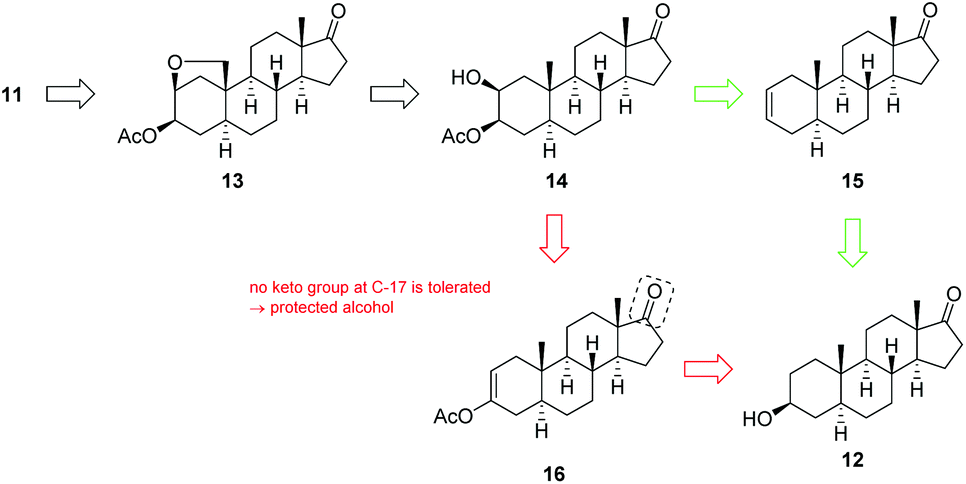 | ||
| Scheme 2 Overview of the two concepts for the installation of the trans-diol moiety at C-2/C-3 and the aldehyde at C-19 starting from 3-epiandrosterone (12). While the first generation approach (red) is based on an epoxidation of the acetyl enol ether 16, the second generation approach (green) relies on a diastereoselective Sharpless dihydroxyation of 15 followed by a regioselective acetylation of C-3's hydroxyl group. | ||
The key intermediate 14 was initially synthesized according to typical steroidal transformation leading to the first generation approach (Scheme 2, highlighted in red). This approach is based on the epoxidation and ring-opening of the acetyl enol ether 16. Since this route is not compatible with the keto group at C-17, the latter was reduced and the obtained alcohol was protected with either an acetyl (14-Ac) or a benzyl protecting group (14-Bn). 3-Epiandrosterone (12) was therefore transformed to the keto compound 17 bearing (a) a TBS-protected or (b) a benzyl-protected alcohol at C-17 (Scheme 3). Treatment of 17 with Ac2O and catalytic amounts of HClO4 produces regioselectively the acetyl enol ether 16 whereby the acidic conditions replace the TBS-group by an acetyl group. After a substrate-induced stereoselective epoxidation from the α-face, the corresponding 2,3-epoxides 18 were opened with pyridine in toluene to give the 3-oxo 2β-acetate 19 in good yields. Under basic conditions, 19 rearranged to the 2-oxo 3β-acetate 20 by 1,2-carbonyl transposition. The isomerization proceeds by an enolisation followed by an acyl group migration presumably via the cyclic intermediate I that has been reported first by Fieser et al.46–48 The structure was determined by 2D NMR experiments and X-ray crystallography (the crystal structure of 20-Ac can be found in the ESI†) confirming the presumed configuration of the acetoxy group. Side reactions such as the cleavage of the acetyl protecting group at the A-ring or epimerization could not be completely suppressed by modifications in reaction time or the choice of the base and solvent. The reduction of the 2-keto group with NaBH4 provided the desired precursors 14-Ac and 14-Bn in satisfying yields and good diastereoselectivities. An X-ray crystal structure was obtained from 14-Ac confirming the molecular structure and the configuration at C-2 and C-3 in the solid state. In addition, NOESY experiments in combination with further 2D NMR experiments verified the cis-configuration of the hydroxyl and acetoxy group. The first generation approach including ten classical steroidal transformation steps delivered the precursor with an acetyl protecting group at C-17 14-Ac with 13% overall yield, while the corresponding benzyl protected precursor 14-Bn was delivered with 6.5% yield. It is noteworthy that this route requires two additional steps including the cleavage of the protecting group and reoxidation of C-17 for being applicable for the synthesis of calotropin since the installation of the butenolide moiety demands a keto functionality at C-17 as it was previously shown by us.49
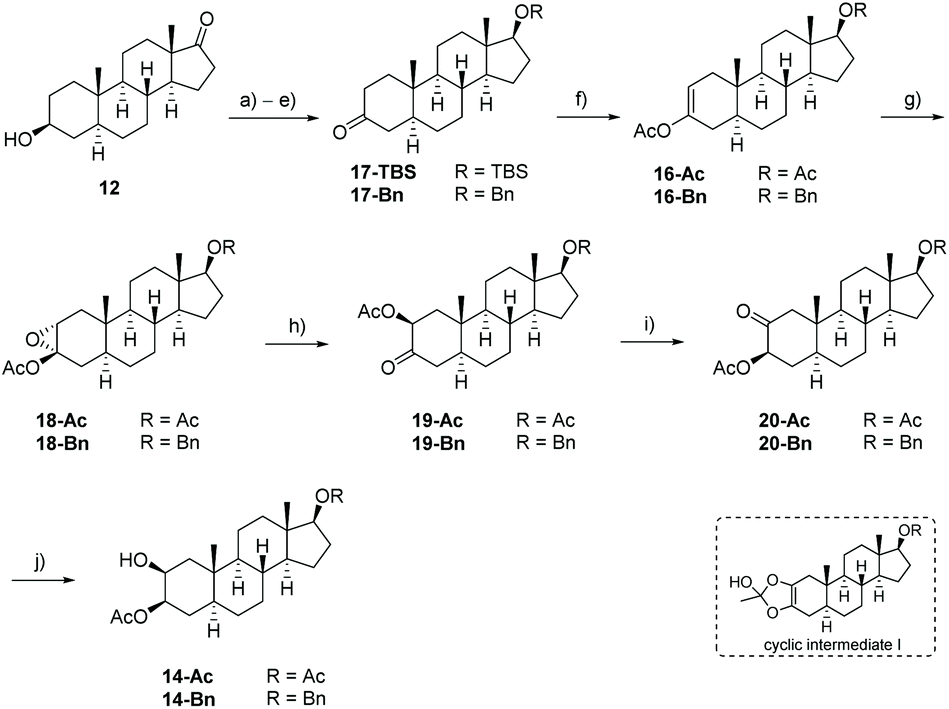 | ||
Scheme 3 Synthesis of the precursor 14-Ac and 14-Bn according to the first generation route. (a) Ac2O, pyridine, r.t., 16 h, 98%; (b) NaBH4, MeOH, −20 °C → 0 °C, 3 h, 98%; (c) TBSCl, imidazole, DMF, r.t., 16 h, 80% or BnCONHCCl3, TfOH, 1,4-dioxane, r.t., 16 h, 82%; (d) KOH, MeOH, Δ, 5 h; (e) NMO, cat. TPAP, CH2Cl2, 0 °C → r.t., 16 h or DMP, CH2Cl2, 0 °C → r.t., 16 h, 90% (17-TBS)/79% (17-Bn) over two steps; (f) HClO4, Ac2O, EtOAc, r.t., 3 h, 69% (16-Ac)/77% (16-Bn); (g) m-CPBA, CH2Cl2, 0 °C → r.t., 16 h, 70% (18-Ac)/47% (18-Bn); (h) pyridine/toluene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10), Δ, 16 h, 86% (19-Ac)/75% (19-Bn); (i) iPrOH, K2CO3, H2O, r.t., 16 h, 58% (20-Ac)/48% (20-Bn); (j) NaBH4, MeOH, −20 °C → 0 °C, 3 h, 76% (14-Ac)/82% (14-Bn); TBS = tert-butyldimethylsilyl; Bn = benzyl. :10), Δ, 16 h, 86% (19-Ac)/75% (19-Bn); (i) iPrOH, K2CO3, H2O, r.t., 16 h, 58% (20-Ac)/48% (20-Bn); (j) NaBH4, MeOH, −20 °C → 0 °C, 3 h, 76% (14-Ac)/82% (14-Bn); TBS = tert-butyldimethylsilyl; Bn = benzyl. | ||
The obtained poor overall yield of the required precursor along with the number of synthetic steps, encouraged us to search for an alternative route maintaining the oxidation state at C-17 (Scheme 2, green route). The novel approach relies on a Sharpless dihydroxylation of androst-2-ene 15 followed by a regioselective acetylation of the C-3 hydroxyl group. With androst-2-ene 15 as key intermediate, the opportunity of a diversity orientated synthesis was given allowing the synthesis of several 2,3-diols (Scheme 4).
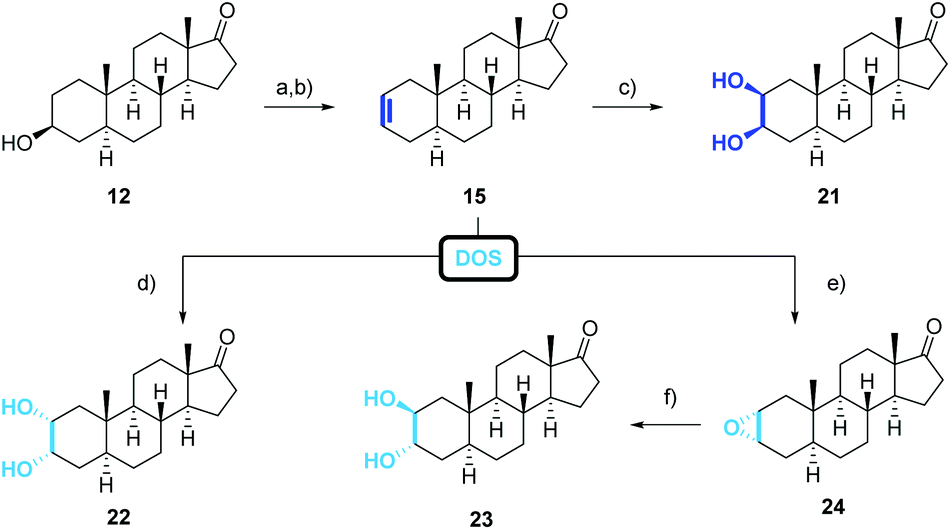 | ||
| Scheme 4 Synthesis of the 2β,3β-cis-diol 21 according to the second generation route. (a) p-TsCl, pyridine, r.t., 16 h, 92%; (b) LiBr, Li2CO3, DMF, 175 °C, 7 h, 95%; (c) α-AD-mix, MeSO2NH2, tBuOH/H2O, r.t., 12 h, 61% (23); (d) β-AD-mix, MeSO2NH2, tBuOH/H2O, r.t., 7 d, 41%; (e) m-CPBA, CH2Cl2, r.t., 14 h, 75%; (f) 2 m H2SO4, THF, r.t., 7 d, 54%; (h) HOCH2CH2OH, p-TsOH, HC(OMe)3, CH2Cl2, r.t., 16 h, 88%; DOS = diversity orientated synthesis. | ||
Androst-2-ene 15 was prepared from 3-epiandrosterone (12) by tosylation followed by an elimination reaction (Scheme 4). From all tested bases comprising inorganic and organic bases such as 2,4,6-collidine or 2,6-lutidine, the combination of LiBr and Li2CO3 gave the best result regarding the yield and the regioselectivity. Unfortunately, the direct conversion with p-toluenesulfonic acid adsorbed on silica gel50 did not afford androst-2-ene 15 in satisfying yield and selectivity. We were also able to obtain single crystals that were suitable for X-ray crystallography (Fig. 5) confirming the stated position of the double bond. The molecular structure shows that due to the sp2 hybridization the A-ring adapts a half chair conformation. When comparing both structures, the steric strain for the androst-3-ene is higher than for its regioisomer explaining the observed regioselectivity of the elimination favoring the more stable androst-2-ene 15.
As previously reported for comparable examples,51 the treatment with catalytic amounts of OsO4 and NMO as stoichiometric oxidant favors in our case the formation of the undesired 2α,3α-cis-diol 22, whereas the corresponding 2β,3β-cis-diols are reported to be prepared by the addition of iodine followed by a nucleophilic displacement with acetate in the presence of water and the hydrolysis of the intermediately formed ester using AgOAc,52,53 Tl(OAc)354 or CuOAc.55 Driven by the idea of a direct conversion of androst-2-ene to the corresponding 2,3-cis-diol, we contemplated a Sharpless dihydroxylation adjusting the side of attack by the choice of the ligand.56,57 While the AD-mix β introduces the hydroxyl groups at the sterically more accessible α-side to build the 2α,3α-cis-diol 22, the use of AD-mix α mainly forms the desired, but sterically more demanding 2β,3β-cis-diol 21. Despite the reported accelerating effect of methanesulfonamide,58 long reaction times of 7 days were required with commercially available AD-mix α. While the extra addition of OsO4 or K2[OsO2(OH)4] did not lead to a faster conversion of the starting material, 2.5 equiv. of “self-made” reduces luckily the reaction time to only 12 h even on 0.1 mmol scale (please see ESI† for further information). However, the corresponding 2,3-cis-diols were obtained in good yields and selectivities.
Different C-17 derivatives bearing a ketal (27) or TBS/Bn protected OH-group (18-OH, 18-TBS, 18-Bn) at C-17 were synthesized and subjected to the developed reaction conditions generating the corresponding 2β,3β-cis-diols (29 and 30) in good yields. Unfortunately, the purification via column chromatography on silica turned challenging resulting in rather low isolated yields (please see ESI†).
When androst-2-ene 15 was reacted with m-CPBA followed by an acid-catalyzed epoxide ring-opening, the 2β,3α-trans-diol was obtained. Once more, the reagents attack occurred from the sterically less hindered α-side releasing the 2α,3α-epoxide 24 with 75% yield. The epoxide was opened by simple acidic treatment to afford with its diaxial 2β,3α-substituents the least stable 2,3-diol.
Even though 2α,3β-trans-diol 27 is not accessible from the precursor 15, the missing trans-diol can be synthesized by a five-step procedure from 3-epiandrosterone (12) (Scheme 5). The synthesis starts with the protection of the keto functionality at C-17 followed by the oxidation of the hydroxyl group at C-3 enabling the stereo- and regioselective introduction of an α-hydroxyl group at C-2 by a Rubottom oxidation. This was accomplished by the conversion of 25 to the corresponding TMS-enol ether, its epoxidation with m-CPBA and a consecutive ring-opening followed by a silyl migration and finally the cleavage of the silyl group to give 26. By reducing the reaction time for the cleavage with oxalic acid or by replacing it by TBAF, the 2α-hydroxylated androstan-3-one was formed with improved yields of about 56%, 66% respectively. Diastereoselective reduction from the sterically less hindered side and removal of the acetal protecting group with p-PTSA result in the formation of the 2α,3β-trans-diol 27 which shows the same configuration at C-2 and C-3 as calotropagenin (11).
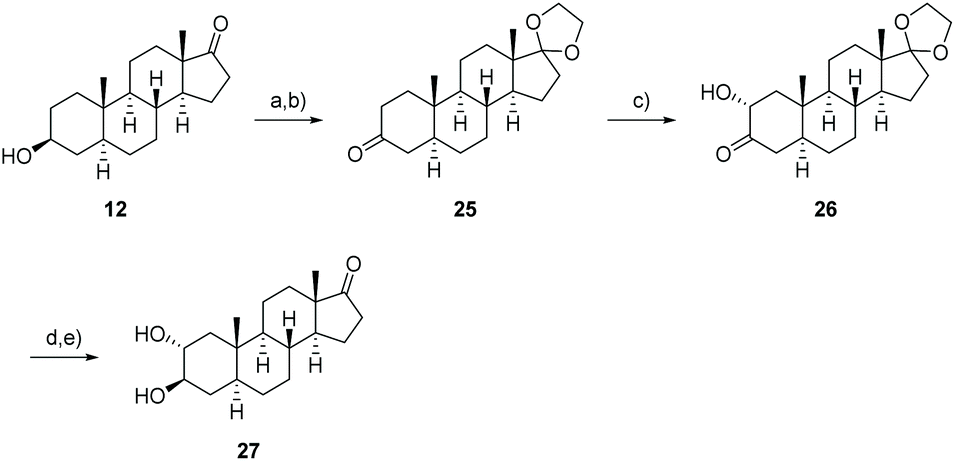 | ||
| Scheme 5 Synthesis of the 2α,3β-trans-diol 27 starting from 3-epiandrosterone (12). (a) HOCH2CH2OH, p-TsOH, HC(OMe)3, CH2Cl2, r.t., 16 h, 88%; (b) DMP, CH2Cl2, 0 °C → r.t., 16 h, 80% (25); (c) I. TMSOTf, NEt3, CH2Cl2, 0 °C, 1 h; II. m-CPBA, NaHCO3, n-hexane, 0 °C, 2 h; III. C2H2O4, MeOH, r.t., 5 min; 56% (26) or III. TBAF, THF, r.t., 10 min; 66% (26); (d) NaBH4, MeOH, CH2Cl2, −20 → 0 °C, 4 h, 66% (33); (e) p-PTSA, acetone, r.t., 3 h, 88% (27). | ||
We were able to obtain suitable single crystals from the four diastereomeric 2,3-diols which are depicted in Fig. 5. The stated molecular structures and orientation of the substituents could be confirmed. The naturally occurring 2α,3β-diol represents with its diequatorial substituents the thermodynamically preferred and therefore most stable diastereomer, while the other trans-diol possesses two axial hydroxyl groups causing an increased transannular strain. The 1,3-diaxial interaction explain also the preferred formation of the 2α,3α-cis-diol 22 over the 2β,3β-cis-diol 21 since the latter owns an axial hydroxyl group at C-2 which conflicts with the 19-methylgroup while in case of the other diastereomer the hydroxyl group at C-2 is equatorially orientated reducing the overall transannular strain.
With 2β,3β-cis-diol 21 in hands, we demonstrated the need of a hydroxyl group protected at C-3 in the directed sp3 C–H oxidation reaction (Scheme 6). Therefore, we exposed the 2β,3β-cis-diol 21 to the later developed reaction conditions of the C–H oxidation reaction. Although full conversion of the 2β,3β-cis-diol 21 was observed, no formation of the desired product was detected. Instead, the dialdehyde 28 was isolated in 48% yield generated by glycol cleavage affirming the relevance of the protection group in order to avoid glycol cleavage.
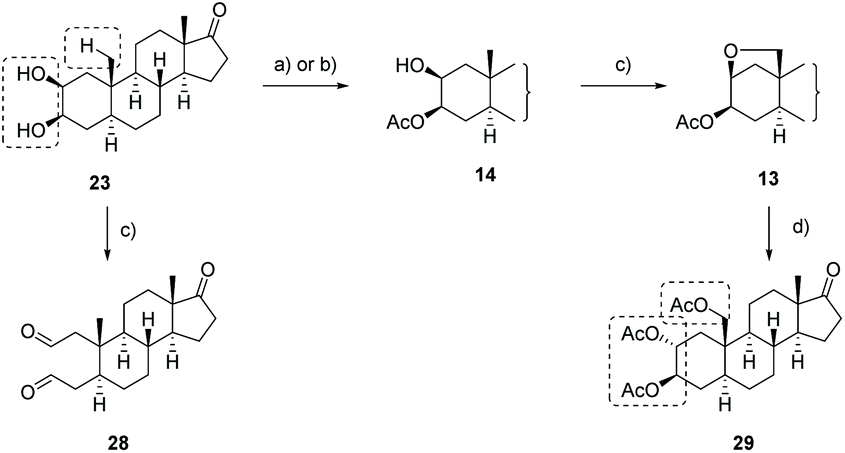 | ||
| Scheme 6 Transformation of the 2β,3β-cis-diol 21 to 2α,3β-trans-19-triacetate 29. (a) Candida rugosa, vinyl acetate, toluene, r.t., 2 d, >77% (14) or (b) AcCl (1.05 equiv.), pyridine, 40 h, r.t., 39% (14); (c) DIB, I2, cyclohexane, C6H6, 30–39 °C, 80 min, sonication (∼), 48% (28)/72% (13); (d) Zn(OTf)2, Ac2O, 45 °C, 16 h → 60 °C, 2 h, 68% (29). | ||
In order to furnish a regioselective protection of the 3-OH group,59–62 several lipases comprising different yeast lipases (C. cylindracea, C. rugosa, C. antarctica), a mold lipase (Mucor miehei), one porcine pankrease and a lipase extracted from the Gram-negative bacterium pseudomonas fluorescens, were evaluated. The esterification reactions were run in toluene or tert-butanol as solvent, all at 36 °C using vinyl acetate as acetyl donor whereby the esterification progress was monitored via TLC. Three of the tested lipases showed partial or complete conversion in toluene within 2 d: in the case of C. cylindracea and pseudomonas fluorescens the formation of a unknown byproduct was observed, whereby C. rugosa delivered only one product which was determined as the desired 3-acetylated 2β,3β-diol 14. By lowering the reaction temperature to r.t. and modifying the reaction experimental setup, an upscale on multigram scale was succeeded providing the 3-acetylated 2β,3β-diol 14 in yields higher than 77%. For comparison, we conducted a kinetically controlled acetylation of 2β,3β-diol 21 with 1.05 equiv. of acetyl chloride in pyridine giving the desired product 14 in only 39% yield.
In order to oxidize the methyl group at C-19, a remote intramolecular free radical C–H oxidation was envisioned exploiting the rigidity of the steroid and the close proximity of the OH-group at C-2 and the C-19 methyl group. The obtained crystal structure of 3-acetylated 2β,3β-diol 14 confirms the stated structure and allows the determination of the distance between the oxygen of the hydroxyl group at C-2 and C-19 or its proton in the solid state to 2.928 Å and 2.278 Å, respectively. In accordance with the literature,63–66 this proximity allows the transfer of a radical from the alkoxy group to the methyl group at C-19 via a hypoiodite reaction (Fig. 3). By using lead tetraacetate and iodine in overstoichiometric amounts, androstane67–69 and cholestane70–72 derivatives have been successfully oxidized.
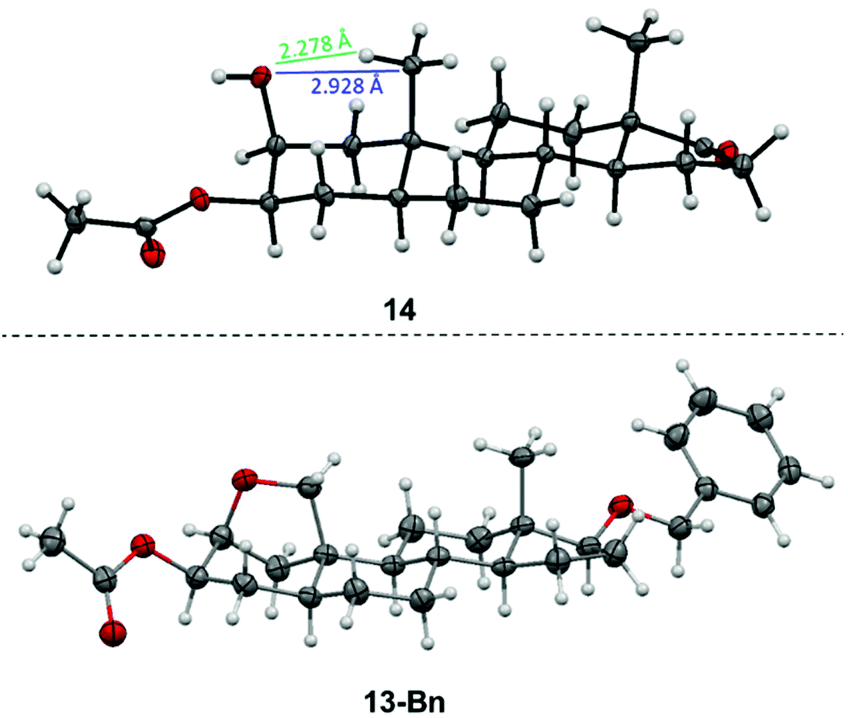 | ||
| Fig. 3 Molecular structure of 14 and 13-Bn (displacement parameters are drawn at 50% probability level) including distance measurements from oxygen at C-2 to the hydrogen at C-19 of 14. Characteristic angles (relative deviations from the standard angle of 111.4°) of 13-Bn: C1–C2–C3: 109.50(16)° (−1.7%), C2–C3–C4: 110.53(14)° (−0.79%), C3–C4–C5: 113.39(15)° (+1.8%), C4–C5–C10: 111.74(15)° (+0.31%), C5–C10–C1: 106.72(14)° (−4.2%), C10–C1–C2: 100.00(14)° (−10%). | ||
Driven by the idea to replace toxic Pb(OAc)4 with DIB and encouraged by the fundamental work of Suárez,73,74 we initiated our studies by probing various reaction parameters such as the amounts of reagents, the concentration, the solvent, reaction time and temperature for the envisioned hypoiodite reaction using 3-acetylated 2β,3β-diol 14 as substrate (Table 1). Preliminary studies revealed problems with reproducibility when a tungsten lamp or LED lamp were deployed. Therefore, we report exclusively on a visible-light free alternative and the usage of a sonication bath to produce the required iodine radicals. In this case, we experienced results that are more consistent and reliable. We noticed the formation of four products including the desired A-ring bridging ether 13, the iodide 30, the overoxidized iodide 31 and the overoxidation product 32 (Table 1). The iodides 30 and 31 can be converted to 13 or respectively 32 by a nucleophilic substitution induced by silver acetate in acetone. All products can be differentiated via1H NMR spectroscopy, whereby the spectra of 30 and 13 show some similarities (Fig. 4). For the iodide 30 the proton at C-2 is shifted downfield, while the proton at C-3 and the more shielded protons of the diastereotopic methylene group at C-16 are shifted upfield compared to the signals of ether 13.
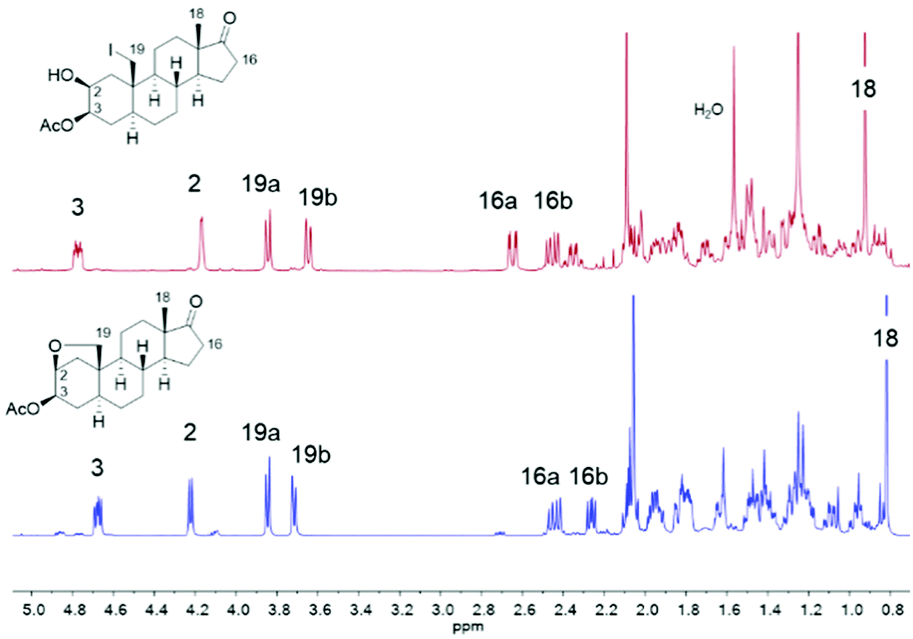 | ||
| Fig. 4 1H NMR spectra (500 MHz, CDCl3, r.t.) of the iodide 30 (red) and the desired ether 13 (blue). | ||
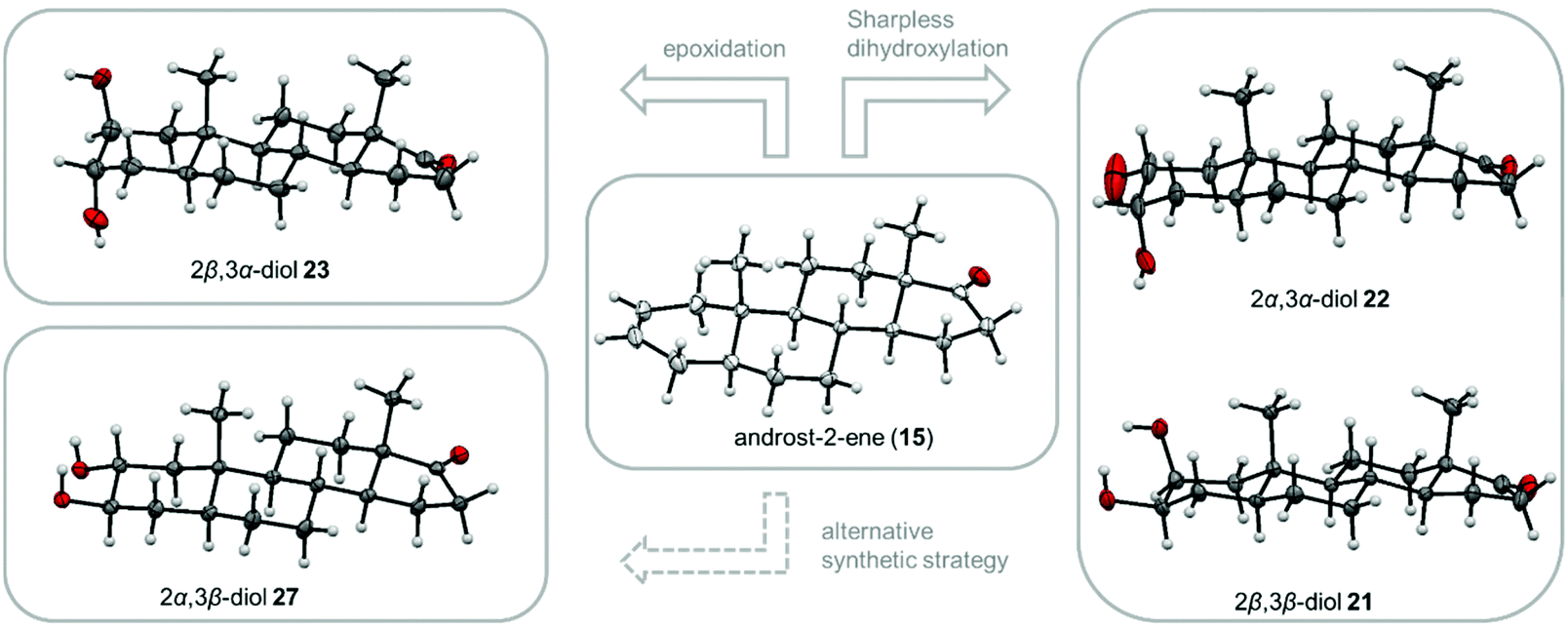 | ||
| Fig. 5 Overview of the synthetic strategies for the four possible 2,3-diol diastereomers starting from androst-2-ene 15 (middle) and their molecular structures (displacement parameters are drawn at 50% probability level). The cis-2,3-diol alcohols 22 and 21 (right) were obtained by a Sharpless dihydroxylation, whereas the trans-diol 23 (left) was accessible via an epoxidation followed by ring-opening. 27 (left) was synthesized from 3-epiandrosterone (12) via a 5-step procedure. | ||
|
|
||||
|---|---|---|---|---|
| Entry | DIB [equiv] | I2 [equiv] | Reaction conditionsa | Isolated yield [%] of (x) |
| a AgOAc, acetone, r.t., overnight; cy = cyclohexane. b c = 10 mm. c c = 30 mm. | ||||
| 1 | 1.5 | 1.3 | cyb, 20–30 °C | 34 (13) |
| 120 min | 20 (14) | |||
| 2 | 2.0 | 1.5 | cyb, 20–30 °C | 38 (13) |
| 139 min | 35 (14) | |||
| 3 | 2.2 | 1.5 | cyb, 20–30 °C | 53 (13) |
| 150 min | 16 (14) | |||
| 4 | 2.2 | 1.5 | cy/C6H6c, 20–30 °C | 39 (13) |
| 100 min | 18 (30) | |||
| 5 | 2.2 | 1.5 | cy/C6H6c, 20–30 °C | 39 (13) |
| 120 min | <18 (30) | |||
| 6 | 2.2 | 1.5 | cy/C6H6c, 20–30 °C | 69 (13) |
| 140 min | 21 (32) | |||
| 7 | 2.2 | 1.5 | cy/C6H6c, 30–50 °C | 50 (13) |
| 140 min | 30 (32) | |||
| 8 | 2.0 | 1.4 | cy/C6H6c, 20–25 °C | <16 (30) |
| 140 min | 27 (13) | |||
| 9 | 2.0 | 1.2 | cy/C6H6c, 20–30 °C | 72 (13) |
| 200 min | 14 (32) | |||
| 10 | 4.4 | 3.0 | cy/C6H6c, 20–30 °C | 62 (13) |
| 140 min | 37 (32) | |||
| 11 | 4.4 | 3.0 | cy/C6H6c, 20–30 °C | 58 (13) |
| 200 min | 34 (32) | |||
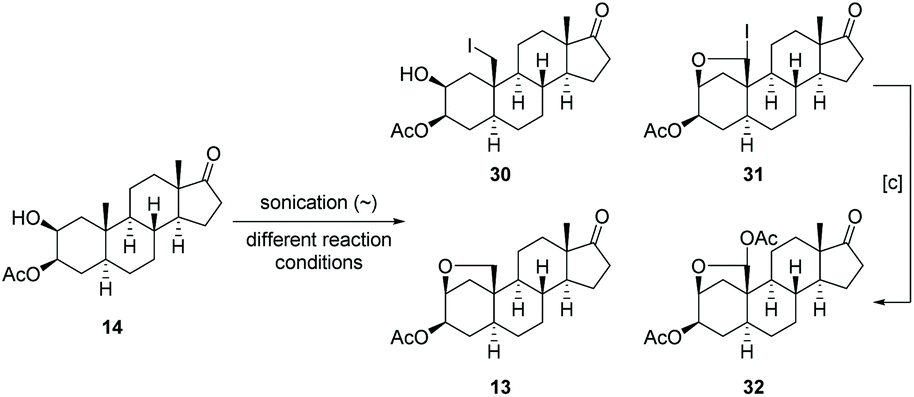
In the beginning, 1.5 equiv. of (diacetoxyiodo)benzene (DIB) and 1.3 equiv. of iodine were used for the hypoiodite reaction and the reaction was conducted at r.t. for 120 min under sonication (entry 1). After column chromatography on silica gel, the desired product 13 was isolated with 34% yield together with 20% reisolated starting material. Therefore, both the amounts of reagents and the reaction time were increased (entries 2 and 3) giving finally the product with synthetically useful 53% yield, although still small amounts of starting material 14 remained. In order to run the reaction at higher concentrations, we added benzene as co-solvent to increase the solubility of 14 and DIB. Keeping the amounts of reagents constant, we screened the reaction times for a higher concentrated solution of 14 (entries 4–6). After 140 min the desired ether 13 was isolated with an improved yield of 69% (entry 6) and no starting material was remained. Instead, the formation of hemiacetal acetate 32 was observed presumably as a result of a second hypoiodite reaction due to an excess of reagents. In order to figure out which parameters promote overoxidation, we applied harsher reaction conditions. At higher temperatures for example the ratio of the desired and double oxidized product shifts towards overoxidation (entry 7). Since the amount of applied reagents should have an effect on the ratio of the two products, we kept reaction time at 140 min, but reduced the amounts of reagent. In this case we observed a dramatic drop in yield of the desired ether 13 (entry 8). In summary, increased amounts of reagent or elevated reaction times led to higher and faster conversion of the starting material, but also to an enhanced formation of the hemiacetal acetate 32 (not shown). Fine tuning of all these parameters finally provides the ether 13 with 72% yield (entry 9). Since the hemiacetal acetate 32 already possess the correct oxidation state at C-19, we were also interested how its formation can be promoted. By the addition of higher equivalents of reagents (entry 10) and prolonged reaction times (entry 11), the ratio of the ether 13 and the hemiacetal acetate 32 could be increased in the direction of the latter.
The molecular structure of the C-17 benzyl protected ether derivative 13-Bn by X-ray crystallography confirms the formation of the bridging ether (Fig. 3) and shows a deformation of the chair geometry on the A-ring induced by the bridging ether. In particular, the angles close to the bridging THF ring differ by up to 10% from the usual angle of 111.4° with values of 106.72(14)° (C5–C10–C1) or 100.00(14)° (C10–C1–C2).
The final step for the synthesis focused on the nucleophilic substitution in order to open the previously constructed bridging THF ring which leads concurrently to an integration of the desired oxygen functionality at C-19. Since the ring-opening requires an activation for instance by a Lewis acid, we selected boron trifluoride diethyl etherate being widely used for the opening of cyclic ethers75,76 or the transformation of methyl ethers into acetyl ester.77–80 If applied to the C-17 acetyl protected ether 13-Ac or the C-17 benzyl protected ether 13-Bn, the same ring-opened product was obtained, both in good yields of greater than 70% (see ESI†). Unfortunately, the selective cleavage of just one ether moiety of the C-17 benzyl protected ether derivative 13-Bn did not succeeded. However, same reaction conditions applied to ether 13 (Table 2, entry 1) did not cleave the cyclic ether. Even if the reaction temperature and/or the amount of BF3·OEt2 were increased, mainly starting material was recovered accompanied by small amounts of an unknown byproduct. Hernandez et al.81 reported that a combination of ZnI2 and Ac2O can initiate the opening of cyclic ethers. At elevated reaction temperatures of 40 °C, we did observe the formation of the desired product for all three cyclic ethers (compare entries 2 and 3), along with the iodide 34 – in most cases as a byproduct, but occasionally even as the main product. To overcome this drawback further zinc(II) salts were tested (entries 4–7). When Zn(OAc)2 was added (entry 4), no conversion to the desired product 29 was determined. In case of ZnBr2, a similar reaction outcome was observed as with ZnI2 as Lewis acid (entry 5) yielding the undesired bromide 33 as byproduct. Luckily, a Zn(OTf)2 induced ring-opening reaction gave the desired product 29 in 68% yield (entry 6). Since small amounts of starting material remained, more reagent was applied, but instead of improving the yield, the latter decreased even though full conversion was achieved (entry 7). Noteworthy, the hemiacetal acetate 32 forms under the same reaction conditions the corresponding aldehyde 43 (not shown).
|
|
|||
|---|---|---|---|
| Entry | Lewis acid | Reaction conditions | Isolated yield [%] |
| a Mainly starting material was detected accompanied by an unidentified byproduct. b The formation of 34 was observed. c The formation of 33 was observed. | |||
| 1 | 20 equiv. BF3·OEt2 | Ac2O, −30 °C → 0 °C, 4 h | 0a |
| 2 | 1.4 equiv. ZnI2 | Ac2O, r.t., 16 h | 0a |
| 3 | 1.5 equiv. ZnI2 | Ac2O, 40 °C, 16 h | 20b |
| 4 | 1.5 equiv. Zn(OAc)2 | Ac2O, 40 °C, 16 h → 60 °C, 2 h | 0a |
| 5 | 1.5 equiv. ZnBr2 | Ac2O, 40 °C, 16 h → 60 °C, 2 h | 0c |
| 6 | 1.5 equiv. Zn(OTf)2 | Ac2O, 45 °C, 16 h → 60 °C, 2 h | 68 |
| 7 | 2.0 equiv. Zn(OTf)2 | Ac2O, 45 °C, 16 h → 60 °C, 2 h | 56 |

Experimental section
General procedure for the dihydroxylation (GP-1)
The androst-2-ene derivative (1.0 equiv.) was dissolved in tert-BuOH and the required commercially AD-mix, dissolved in the same amount of H2O, was added. The mixture was stirred for 10 min at r.t. before MeSO2NH2 (0.40 equiv.) was added. The yellow suspension was stirred vigorously for 7 d at r.t. and was then quenched with saturated aqueous Na2S2O3 solution. After extraction with EtOAc (3×) the combined organic phases were washed consecutively with 2 m aqueous solution of KOH, H2O and brine. The organic phases were dried over Na2SO4, filtered and the solvent was removed. The residue was purified by flash column chromatography on silica gel (cHex/EtOAc) to obtain the corresponding cis-diol as a colorless powder.Please note that the reaction times can be reduced when 2.5 equiv. of “self-made” AD-mix were used (please see ESI†).
General procedure for the C–H oxidation via a hypoiodite reaction (GP-2)
A suspension of DIB (1.5 equiv.) and I2 (1.3 equiv.) in a mixture of cyclohexane and benzene (10:1, 25–30 mm) was degassed by bubbling with argon (15 min) which was followed by the addition of the 2β-acetoxy-3β-hydroxyandrostane derivative (1.0 equiv.). The reaction was sonicated at the given temperature and for the given time before it was quenched with saturated aqueous Na2S2O3 solution. After phase separation the aqueous phase was extracted with CH2Cl2 (3×) and the combined organic extracts were washed with brine and dried over Na2SO4. After filtration and evaporation the obtained crude product was purified by flash column chromatography on silica gel (cHex/EtOAc) to afford the 2β,19-epoxy-5α-androstane derivative as colorless solid.
General procedure for the Lewis acid-mediated opening of the THF ring (GP-3)
For the work up, the reaction was added to a solution of saturated aqueous NaHCO3 and stirred until the gas formation stopped. After phase separation, the aqueous phase was extracted with EtOAc (3×). The combined organic phases were washed with H2O and brine, then dried over Na2SO4 and filtered. Solvent removal gave the crude product which was purified by flash column chromatography on silica gel (cHex/EtOAc).
Crystal structure determinations
The single-crystal X-ray diffraction study was carried out on a Bruker D8 Venture diffractometer with Photon 100 detector PhotonII CPAD (for 27) at 123(2) K using Cu-Kα radiation (λ = 1.54178 Å. Direct Methods (SHELXS-97)82 or dual space methods (SHELXT for 13-Bn and 27)83 were used for structure solution and refinement was carried out using SHELXL-2014 (full-matrix least-squares on F2).83 Hydrogen atoms were localized by difference electron density determination and refined using a riding model (H(O) free, except 21). Semi-empirical absorption corrections were applied. For 15, 14-Ac, 27 and 20-Ac an extinction correction were applied. The absolute configuration was determined by refinement of Parsons’ x-parameter84 or the enantiomer has been assigned by reference to an unchanging chiral center in the synthetic procedure (see cif-files for details). In 21 there seems to be a possible disorder of the OH-groups at C3 and C4 (see cif-files for details).656 reflections, of which 3011 were independent (Rint = 0.021), 182 parameters, R1 = 0.031 (for 2981 I > 2σ(I)), wR2 = 0.079 (all data), S = 1.08, largest diff. peak/hole = 0.287/−0.172 e Å−3. x = −0.05(5).
480 reflections, of which 6149 were independent (Rint = 0.032), 399 parameters, 7 restraints, R1 = 0.042 (for 5585 I > 2σ(I)), wR2 = 0.104 (all data), S = 1.03, largest diff. peak/hole = 0.239/−0.179 e Å−3. x = 0.08(17).
546 reflections, of which 3541 were independent (Rint = 0.044), 232 parameters, 3 restraints, R1 = 0.045 (for 3243 I > 2σ(I)), wR2 = 0.109 (all data), S = 1.04, largest diff. peak/hole = 0.317/−0.191 e Å−3. x = 0.14(18).
27 colourless crystals, C19H30O3·H2O, Mr = 324.44, crystal size 0.28 × 0.14 × 0.06 mm, orthorhombic, space group P212121 (No. 19), a = 6.3125(4) Å, b = 13.3434(8) Å, c = 10.8575(12) Å, V = 1756.83(18) Å3, Z = 4, ρ = 1.227 Mg m−3, μ(Cu-Kα) = 0.671 mm−1, F(000) = 712, 2θmax = 144.4°, 17174 reflections, of which 3469 were independent (Rint = 0.025), 220 parameters, 5 restraints, R1 = 0.032 (for 3439 I > 2σ(I)), wR2 = 0.087 (all data), S = 1.05, largest diff. peak/hole = 0.302/−0.267 e Å−3. x = −0.09(4).
308 reflections, of which 1455 were independent (Rint = 0.030), 230 parameters, 2 restraints, R1 = 0.029 (for 3588 I > 2σ(I)), wR2 = 0.076 (all data), S = 1.04, largest diff. peak/hole = 0.187/−0.160 e Å−3. x = 0.01(6).
473 reflections, of which 4591 were independent (Rint = 0.024), 290 parameters, 1 restraint, R1 = 0.031 (for 4482 I > 2σ(I)), wR2 = 0.086 (all data), S = 1.05, largest diff. peak/hole = 0.260/−0.161 e Å−3. x = 0.13(6).
103 reflections, of which 4139 were independent (Rint = 0.025), 260 parameters, R1 = 0.028 (for 4106 I > 2σ(I)), wR2 = 0.075 (all data), S = 1.07, largest diff. peak/hole = 0.214/−0.167 e Å−3. x = −0.02(4).
893 reflections, of which 7180 were independent (Rint = 0.047), 40 parameters, 1 restraint, R1 = 0.064 (for 5010 I > 2σ(I)), wR2 = 0.147 (all data), S = 1.02, largest diff. peak/hole = 0.178/−0.173 e Å−3. x = 0.4(3).
CCDC 1998175 (27), CCDC 1822521 (15), CCDC 1822522 (21), CCDC 1822523 (22), CCDC 1822524 (23), CCDC 1822525 (33), CCDC 1822526 (14), CCDC 1822527 (13-Bn), CCDC 1822528 (14-Ac), and 1822529 (20-Ac) contain the supplementary crystallographic data for this paper.†
Conclusions
Due to the interesting biological properties of calotropin (4) and related cardenolides, we herein presented a modular synthetic route for these cardenolide aglyocones. The presented work enables the regioselective C–H oxidation of the methyl group at C-19 followed by the subsequent introduction of the 2α,3β-trans-diol moiety in order to enable the connection to the sugar moiety. We faced two synthetic routes for the preparation of the precursor 14 for the remote C–H oxidation starting from commercially available 3-epiandrosterone (12). The first generation route based on traditional steroid chemistry could be replaced by a more convenient and efficient sequence of an asymmetric Sharpless dihydroxylation and a regioselective acetylation. With androst-2-ene as intermediate not only the synthesis of the required 2β,3β-cis-diol was accomplished, but also two more diastereomeric 2,3-diols became accessible. The corresponding 2α,3β-trans-diol is not accessible from androst-2-ene, but can be accessed from 3-epiandrosterone via a Rubottom oxidation and a consecutive reduction of the 3-keto group. Due to the close proximity of the axial C-2 hydroxyl and C-19 methyl group in the steroidal framework, a remote intramolecular radical C–H oxidation of C-19 with DIB and I2 (hypoiodite reaction) led to the formation of the A-ring bridging ether which was then opened by a Lewis acid-mediated ether cleavage to give the 2,3,19-triacetoxy derivative 29. The overoxidation product 32, resulting from a double hypoiodite reaction under harsher reaction conditions, can be directly converted to the desired aldehyde functionality at C-19 after acidic treatment.In summary, we have devised a novel strategy towards all 2,3-diol diastereomers and the introduction of an oxygen functionality at C-19 via a remote C–H oxidation without using any light source or transition metal. The herein presented method should not only allow the synthesis of calotropin (4) and related cardenolides, but should also inspire upcoming synthesis of steroids and cyclic terpenes.
Acknowledgements
We thank S. Al Muthafer, H. Kühner, C. Schissler for their help during the synthesis of the precursor. V. K. gratefully acknowledges the Studienstiftung des Deutschen Volkes for not exclusively financial support.Notes and references
- A. A. Agrawal, G. Petschenka, R. A. Bingham, M. G. Weber and S. Rasmann, Toxic cardenolides: chemical ecology and coevolution of specialized plant–herbivore interactions, New Phytol., 2012, 194, 28 CrossRef CAS PubMed.
- O. L. Wade and I. Two, Hundred Years of Digitalis, J. Clin. Pharm. Ther., 1986, 11, 3 CrossRef CAS PubMed.
- H. A. Bessen, Therapeutic and toxic effects of digitalis: William Withering, 1785, Int. J. Emerging Technol., 1986, 4, 243 CAS.
- A. J. Levi, M. R. Boyett and C. O. Lee, The cellular actions of digitalis glycosides on the heart, Prog. Biophys. Mol. Biol., 1994, 62, 1 CrossRef CAS PubMed.
- H. P. Albrecht and K. H. Geiss, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2005 Search PubMed.
- M. Michalak, K. Michalak and J. Wicha, The synthesis of cardenolide and bufadienolide aglycones, and related steroids bearing a heterocyclic subunit, Nat. Prod. Rep., 2017, 34, 361 RSC.
- H. R. El-Seedi, S. A. M. Khalifa, E. A. Taher, M. A. Farag, A. Saeed, M. Gamal, M.-E. F. Hegazy, D. Youssef, S. G. Musharraf, M. M. Alajlani, J. Xiao and T. Efferth, Cardenolides: Insights from chemical structure and pharmacological utility, Pharmacol. Res., 2019, 141, 123 CrossRef CAS PubMed.
- G. Hesse and F. Reicheneder, Über das afrikanische Pfeilgift Calotropin. I, Justus Liebigs Ann. Chem., 1936, 526, 252 CrossRef CAS.
- G. Hesse, F. Reicheneder and H. Eysenbach, Die Herzgifte im Calotropis-Milchsaft. II. Mitteilung über afrikanische Pfeilgifte, Justus Liebigs Ann. Chem., 1939, 537, 67 CrossRef.
- G. Hesse, L. J. Heuser, E. Hütz and F. Reicheneder, Zusammenhänge zwischen den wichtigsten Giftstoffen der Calotropis procera V. Mitteilung über afrikanische Pfeilgifte, Justus Liebigs Ann. Chem., 1950, 566, 130 CrossRef CAS.
- G. Hesse and H. W. Gampp, Der heterocyclische Bezirk des Uscharins; VI. Mitteil. über afrikanische Pfeilgifte, Chem. Ber., 1952, 85, 933 CrossRef CAS.
- G. Hesse and G. Lettenbauer, Ein zweiter Schwefel-haltiger Stoff aus dem Milchsaft von Calotropis procera, Angew. Chem., 1957, 69, 392 CrossRef CAS.
- G. Hesse and K. Mix, Über afrikanische Pfeilgifte, IX Konstitution und Teilsynthese des Uscharins, Justus Liebigs Ann. Chem., 1959, 625, 146 CrossRef CAS.
- G. Hesse and G. Ludwig, Über afrikanische Pfeilgifte, XIV. Voruscharin, ein zweites schwefelhaltiges Herzgift aus Calotropis procera L, Justus Liebigs Ann. Chem., 1960, 632, 158 CrossRef CAS.
- F. Brüschweiler, W. Stöcklin, K. Stöckel and T. Reichstein, Die Glykoside von Calotropis procera, R. BR. Glykoside und Aglykone, 320. Mitteilung, Helv. Chim. Acta, 1969, 52, 2086 CrossRef PubMed.
- R. Coombe and T. Watson, The cardiac glycosides of Gomphocarpus fruticosus R.Br. III. Gomphoside, Aust. J. Chem., 1964, 17, 92 CrossRef CAS.
- H. T. A. Cheung, F. C. K. Chiu, T. R. Watson and R. J. Wells, Reduction of hemiacetal ring-opened forms of Asclepiadaceae cardenolide glycosides, J. Chem. Soc., Perkin Trans. 1, 1986, 55 RSC.
- R. Watson and S. Wright, The Cardiac Glycosides of Gomphocarpus fruticosus, R.Br. II. Gomphoside, Aust. J. Chem., 1957, 10, 79 CrossRef.
- H. T. A. Cheung and T. R. Watson, Stereochemistry of the hexosulose in cardenolide glycosides of the asclepiadaceae, J. Chem. Soc., Perkin Trans. 1, 1980, 2162 RSC.
- H. T. A. Cheung, F. C. K. Chiu, T. R. Watson and R. J. Wells, Cardenolide glycosides of the asclepiadaceae. New glycosides from asclepias fruticosa and the stereochemistry of uscharin, voruscharin and calotoxin, J. Chem. Soc., Perkin Trans. 1, 1983, 2827 RSC.
- C. Theurer, H.-J. Treumann, T. Faust, U. May and W. Kreis, Glycosylation in cardenolide biosynthesis, Plant Cell, Tissue Organ Cult., 1994, 38, 327 CrossRef CAS.
- B. Singh and R. P. Rastogi, Structure of asclepin and some observations on the NMR spectra of Calotropis glycosides, Phytochemistry, 1972, 11, 757 CrossRef CAS.
- P. Brown, J. von Euw, T. Reichstein, K. Stöckel and T. R. Watson, Cardenolides of Asclepias syriacaL., Probable Structure of Syrioside and Syriobioside. Glycosides and aglycones, 334th communication, Helv. Chim. Acta, 1979, 62, 412 CrossRef CAS.
- J.-d.-D. Tamokou and V. Kuete, in Toxicological Survey of African Medicinal Plants, ed. V. Kuete, Elsevier, 5 edn, 2014, ch. 7, pp. 135 Search PubMed.
- R. F. Keeler and A. T. Tu, Handbook of Natural Toxins, Taylor & Francis, 1983 Search PubMed.
- S. M. Kupchan, J. R. Knox, J. E. Kelsey and J. A. S. Renauld, Calotropin, a Cytotoxic Principle Isolated from Asclepias curassavica L, Science, 1964, 146, 1685 CrossRef CAS PubMed.
- N. Mascolo, R. Sharma, S. C. Jain and F. Capasso, Ethnopharmacology of Calotropis procera flowers, J. Ethnopharmacol., 1988, 22, 211 CrossRef CAS PubMed.
- V. L. Kumar and N. Basu, Anti-inflammatory activity of the latex of Calotropis procera, J. Ethnopharmacol., 1994, 44, 123 CrossRef CAS PubMed.
- A. Basu and A. K. N. Chaudhuri, Preliminary studies on the antiinflammatory and analgesic activities of Calotropis procera root extract, J. Ethnopharmacol., 1991, 31, 319 CrossRef CAS PubMed.
- H. Singh, S. Kumar, S. Dewan and V. L. Kumar, Inflammation induced by latex of Calotropis procera—a new model to evaluate anti-inflammatory drugs, J. Pharmacol. Toxicol. Methods, 2000, 43, 219 CrossRef CAS.
- A. K. Pathak and A. Argal, Analgesic activity of Calotropis gigantea flower, Fitoterapia, 2007, 78, 40 CrossRef CAS PubMed.
- S. Dewan, H. Sangraula and V. L. Kumar, Preliminary studies on the analgesic activity of latex of Calotropris procera, J. Ethnopharmacol., 2000, 73, 307 CrossRef CAS.
- G. Nenaah, Antimicrobial activity of Calotropis procera Ait. (Asclepiadaceae) and isolation of four flavonoid glycosides as the active constituents, World J. Microbiol. Biotechnol., 2013, 29, 1255 CrossRef CAS PubMed.
- P. T. Deshmukh, J. Fernandes, A. Atul and E. Toppo, Wound healing activity of Calotropis gigantea root bark in rats, J. Ethnopharmacol., 2009, 125, 178 CrossRef PubMed.
- V. Saratha, S. Subramanian and S. Sivakumar, Evaluation of wound healing potential of Calotropis gigantea latex studied on excision wounds in experimental rats, Med. Chem. Res., 2010, 19, 936 CrossRef CAS.
- M. C. Roy, F.-R. Chang, H.-C. Huang, M. Y. N. Chiang and Y.-C. Wu, Cytotoxic Principles from the Formosan Milkweed, Asclepias curassavica, J. Nat. Prod., 2005, 68, 1494 CrossRef CAS PubMed.
- T. Lhinhatrakool and S. Sutthivaiyakit, 19-Nor- and 18,20-Epoxy-cardenolides from the Leaves of Calotropis gigantea, J. Nat. Prod., 2006, 69, 1249 CrossRef CAS PubMed.
- J.-Z. Li, C. Qing, C.-X. Chen, X.-J. Hao and H.-Y. Liu, Cytotoxicity of cardenolides and cardenolide glycosides from Asclepias curassavica, Bioorg. Med. Chem. Lett., 2009, 19, 1956 CrossRef CAS PubMed.
- H. You, M. Lei, W. Song, H. Chen, Y. Meng, D. Guo, X. Liu and L. Hu, Cytotoxic cardenolides from the root bark of Calotropis gigantea, Steroids, 2013, 78, 1029 CrossRef CAS PubMed.
- R.-R. Zhang, H.-Y. Tian, Y.-F. Tan, T.-Y. Chung, X.-H. Sun, X. Xia, W.-C. Ye, D. A. Middleton, N. Fedosova, M. Esmann, J. T. C. Tzen and R.-W. Jiang, Structures, chemotaxonomic significance, cytotoxic and Na+,K+-ATPase inhibitory activities of new cardenolides from Asclepias curassavica, Org. Biomol. Chem., 2014, 12, 8919 RSC.
- E.-P. Mo, R.-R. Zhang, J. Xu, H. Zhang, X.-X. Wang, Q.-T. Tan, F.-L. Liu, R.-W. Jiang and S.-H. Cai, Calotropin from Asclepias curasavica induces cell cycle arrest and apoptosis in cisplatin-resistant lung cancer cells, Biochem. Biophys. Res. Commun., 2016, 478, 710 CrossRef CAS PubMed.
- I. Prassas and E. P. Diamandis, Novel therapeutic applications of cardiac glycosides, Nat. Rev. Drug Discovery, 2008, 7, 926 CrossRef CAS PubMed.
- H. Y. Park, K. Toume, M. A. Arai, S. K. Sadhu, F. Ahmed and M. Ishibashi, Calotropin: A Cardenolide from Calotropis gigantea that Inhibits Wnt Signaling by Increasing Casein Kinase 1α in Colon Cancer Cells, ChemBioChem, 2014, 15, 872 CrossRef CAS PubMed.
- K. K. Chen, R. C. Anderson and E. B. Robbins, The potency of five additional cardiac glucosides, calotropin, α-antiarin, emicymarin, folinerin and sarmentocymarin, J. Am. Pharm. Assoc., 1937, 26, 214 CAS.
- F. W. Lichtenthaler, E. Cuny and O. Sakanaka, A Concise and General Method for Doubly Attaching 2-Ketosugars to Aglycon Diols: Synthesis of the Gomphosides and Spectinomycin, Angew. Chem., Int. Ed., 2005, 44, 4944 CrossRef CAS PubMed.
- L. F. Fieser and R. Stevenson, Cholesterol and Companions. IX. Oxidation of Δ5-Cholestene-3-one with Lead Tetraacetate, J. Am. Chem. Soc., 1954, 76, 1728 CrossRef CAS.
- H. B. Henbest, D. N. Jones and G. P. Slater, 881. Reactions of ketones with oxidising agents. Part I. Catalysis of the ketone-lead tetra-acetate reaction with boron trifluoride, J. Chem. Soc., 1961, 4472 RSC.
- K. Yamakawa, S. Kidokoro, N. Umino, R. Sakaguchi, T. Takakuwa and M. Suzuki, Studies on the Terpenoids and Related Alicyclic Compounds. I. Synthesis of 5α-and 5β-2-Oxosantan-6: 13-olide from Santonin, Chem. Pharm. Bull., 1973, 21, 296 CrossRef CAS.
- V. Koch, M. Nieger and S. Bräse, Stille and Suzuki Cross-Coupling Reactions as Versatile Tools for Modifications at C-17 of Steroidal Skeletons – A Comprehensive Study, Adv. Synth. Catal., 2017, 359, 832 CrossRef CAS.
- F. D'Onofrio and A. Scettri, p-Toluenesulfonic Acid Adsorbed on Silica Gel: An Efficient Dehydrating Agent of Alcohols, Synthesis, 1985, 1159 CrossRef.
- C. W. Davey, E. L. McGinnis, J. M. McKeown, G. D. Meakins, M. W. Pemberton and R. N. Young, Hydroxy-steroids. Part XI. The preparation and infrared spectra of vicinal cholestanediols, J. Chem. Soc. C, 1968, 2674 RSC.
- R. B. Woodward and F. V. Brutcher, cis-Hydroxylation of a Synthetic Steroid Intermediate with Iodine, Silver Acetate and Wet Acetic Acid, J. Am. Chem. Soc., 1958, 80, 209 CrossRef CAS.
- P. S. Ellington, D. G. Hey and G. D. Meakins, Hydroxy-steroids. Part VI. Reactions of olefins with silver salts and iodine, J. Chem. Soc. C, 1966, 1327 RSC.
- R. C. Cambie, R. C. Hayward, J. L. Roberts and P. S. Rutledge, Reactions of thallium(I) carboxylates and iodine with alkenes, J. Chem. Soc., Perkin Trans. 1, 1974, 1858 RSC.
- C. A. Horiuchi and J. Y. Satoh, A New Synthesis of cis-Diol from Olefin Using Iodine-Copper(II) Acetate, Chem. Lett., 1988, 17, 1209 CrossRef.
- H. Becker, M. A. Soler and K. B. Sharpless, Selective asymmetric dihydroxylation of polyenes, Tetrahedron, 1995, 51, 1345 CrossRef CAS.
- V. Richmond, A. P. Murray and M. S. Maier, Synthesis and acetylcholinesterase inhibitory activity of polyhydroxylated sulfated steroids: Structure/activity studies, Steroids, 2013, 78, 1141 CrossRef CAS PubMed.
- M. H. Junttila and O. O. E. Hormi, Methanesulfonamide: a Cosolvent and a General Acid Catalyst in Sharpless Asymmetric Dihydroxylations, J. Org. Chem., 2009, 74, 3038 CrossRef CAS PubMed.
- S. Riva, R. Bovara, G. Ottolina, F. Secundo and G. Carrea, Regioselective acylation of bile acid derivatives with Candida cylindracea lipase in anhydrous benzene, J. Org. Chem., 1989, 54, 3161 CrossRef CAS.
- A. Bertinotti, G. Carrea, G. Ottolina and S. Riva, Regioselective esterification of polyhydroxylated steroids by Candida antarctica lipase B, Tetrahedron, 1994, 50, 13165 CrossRef CAS.
- B. Danieli, G. Lesma, M. Luisetti and S. Riva, Candida antarctica lipase B catalyzes the regioselective esterification of ecdysteroids at the C-2 OH, Tetrahedron, 1997, 53, 5855 CrossRef CAS.
- M. M. Cruz Silva, S. Riva and M. L. Sá e Melo, Regioselective enzymatic acylation of vicinal diols of steroids, Tetrahedron, 2005, 61, 3065 CrossRef CAS.
- J. Kalvoda and K. Heusler, Die Hypojodit-Reaktion (Verfahren zur intramolekularen Substitution an nicht-aktivierten C-Atomen), Synthesis, 1971, 501 CrossRef CAS.
- K. Heusler and J. Kalvoda, Intramolecular Free-Radical Reactions, Angew. Chem., Int. Ed. Engl., 1964, 3, 525 CrossRef.
- Ž. Čeković, Reactions of δ-carbon radicals generated by 1,5-hydrogen transfer to alkoxyl radicals, Tetrahedron, 2003, 59, 8073 CrossRef.
- G. Majetich and K. Wheless, Remote intramolecular free radical functionalizations: An update, Tetrahedron, 1995, 51, 7095 CrossRef CAS.
- R. E. Counsell, G. W. Adelstein, P. D. Klimstra and B. Smith, Anabolic Agents. 19-Nor- and 19-Substituted 5α-Androst-2-ene Derivatives, J. Med. Chem., 1966, 9, 685 CrossRef CAS PubMed.
- F. Kohen, G. Adelstein and R. E. Counsell, Neighbouring-group participation by ether oxygen in displacement reactions of 3α-substituted 2β,19-oxide-androstanes, J. Chem. Soc. D, 1970, 770 RSC.
- G. Habermehl and A. Haaf, Synthese von 2-Aza-, 3-Aza- und 4-Aza-A-homo-5α,10α-androstan, Justus Liebigs Ann. Chem., 1969, 723, 181 CrossRef CAS.
- C. W. Shoppee, T. E. Bellas, J. C. Coll and R. E. Lack, Steroids. Part XXXIII. Attempted preparation of 19-nor-5α-cholestanes via 2β-hydroxy-5α-cholestan-19-oic acid, J. Chem. Soc. C, 1969, 2734 RSC.
- C. W. Shoppee, J. C. Coll and R. E. Lack, Steroids. Part XXXV. Preparation of the epimeric 2-hydroxy-19-nor-5α-cholestanes, J. Chem. Soc. C, 1970, 1893 RSC.
- M. E. Wolff and T. Morioka, C-19 Functional Steroids. X.1a 17β-Hydroxy-1β,19-cyclo-5α-androstan-2-one and Related Compounds1b, J. Org. Chem., 1965, 30, 2553 CrossRef CAS PubMed.
- P. de Armas, J. I. Concepcion, C. G. Francisco, R. Hernandez, J. A. Salazar and E. Suarez, Intramolecular hydrogen abstraction. Hypervalent organoiodine compounds, convenient reagents for alkoxyl radical generation, J. Chem. Soc., Perkin Trans. 1, 1989, 405 RSC.
- J. I. Concepción, C. G. Francisco, R. Hernández, J. A. Salazar and E. Suárez, Intramolecular hydrogen abstraction. Iodosobenzene diacetate, an efficient and convenient reagent for alkoxy radical generation, Tetrahedron Lett., 1984, 25, 1953 CrossRef.
- C. W. Shoppee, J. C. Coll and R. E. Lack, Steroids. Part XXXV. Preparation of the epimeric 2-hydroxy-19-nor-5[small alpha]-cholestanes, J. Chem. Soc. C, 1970, 1893 RSC.
- R. E. Lack and A. B. Ridley, 19-Nor and aromatic steroids. Part 1. The cleavage of 3-oxygenated-2β,19-ethers in the cholestane series, J. Chem. Soc. C, 1970, 1437 RSC.
- C. R. Narayanan and K. N. Iyer, Mode of cleavage of steroid methyl ethers with BF3-etherate, Tetrahedron Lett., 1965, 6, 1369 CrossRef.
- C. R. Narayanan and K. N. Iyer, Opening of steroid methyl ethers with BF3-etherate, Tetrahedron Lett., 1964, 5, 759 CrossRef.
- C. R. Narayanan and K. N. Iyer, Regeneration of Steroid Alcohols from Their Methyl Ethers, J. Org. Chem., 1965, 30, 1734 CrossRef CAS.
- R. D. Youssefyeh and Y. Mazur, A novel method of ether cleavage, Tetrahedron Lett., 1962, 3, 1287 CrossRef.
- R. Hernandez, S. M. Velazquez, E. Suarez and M. S. Rodriguez, Synthesis of (+)-8-Deoxyvernolepin, J. Org. Chem., 1994, 59, 6395 CrossRef CAS.
- G. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- S. Parsons, H. D. Flack and T. Wagner, Use of intensity quotients and differences in absolute structure refinement, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 249 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1822521–1822529. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qo00269k |
| This journal is © the Partner Organisations 2020 |