
Modified diterpenoids from the tuber of Icacina oliviformis as protein tyrosine phosphatase 1B inhibitors†
 *a
Guangmin
Yao,
d
*a
Guangmin
Yao,
d
Abstract
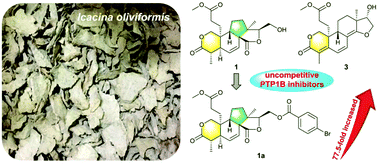
Seven new diterpenoids (1–7) and five known analogs (8–12) were isolated from the tuber of Icacina oliviformis. Their chemical structures were elucidated by spectroscopic analyses, with the spatial configurations defined based on calculated 13C NMR-DP4 analysis and electronic circular dichroism methods. Oliviformislactones A (1) and B (2) are the first examples of rearranged 3,4-seco-pimarane possessing a 6/6/5/5 tetracyclic ring system featuring an unprecedented 4,12-dioxatetracyclo[8.6.0.02,7.010,14]hexadecane core; secopimaranlactone A (3) and secocleistanthone A (4) are the first examples of 3,4-seco-pimarane and 3,4-seco-cleistanthane type diterpenoids, respectively, obtained from the Icacinaceae family. The plausible biosynthetic routes for 1–3 are proposed. Compounds 1–4 and 12 exhibited PTP1B inhibitory activity with IC50 values ranging from 3.24 to 58.05 μM. Among them, 1 (IC50 = 6.78 μM, Ki = 1.51 μM) and 3 (IC50 = 3.24 μM, Ki = 1.25 μM) displayed uncompetitive inhibition responses. In addition, the 16-p-bromobenzoyl derivative of oliviformislactone A (1a) displayed higher potency (IC50 = 87.51 nM, Ki = 0.44 μM) than its natural precursor.
 Please wait while we load your content...
Please wait while we load your content...

