
Cobalt(III)-catalyzed ketone-directed C–H vinylation using vinyl acetate†
Md Raja
Sk
and
Modhu Sudan
Maji
 *
*
Department of Chemistry, Indian Institute of Technology Kharagpur, Kharagpur 721302, WB, India. E-mail: msm@chem.iitkgp.ac.in
First published on 1st November 2019
Abstract
Weakly coordinating, ketone-directed C–H vinylation using vinyl acetate is reported here for a wide range of aromatic ketones such as acetophenones, diaryl ketones, chromones and biologically relevant chalcones under cost-effective and air-stable cobalt(III)-catalysis. Regioselective, mono-vinylation occurs for challenging vinyl substitution-free styrenes in moderate to good yields, and this moiety has been used to synthesize functionalized indanone, α-naphthol and an advanced intermediate for bruguierol A synthesis. An acrylate-surrogate provided the corresponding alkenylated product under these vinylation conditions. Detailed mechanistic studies are carried out to support the proposed catalytic cycle.
Introduction
Vinylarenes are very important structural motifs for the preparation of a vast array of organic molecules, fine chemicals and polymers.1 The development of various synthetic methods by using vinyl arenes as starting materials, and transforming them into the desired synthetic target molecules through cross-metathesis,2 cross-coupling,3 or olefin functionalizations4 are clear and key motivations behind the development of styrene synthesis methods. Considering a step- and atom-economic5 approach, as well as the availability of the required precursors, styrenes have become an attractive target with the recent progress in the field of C–H bond functionalization. Although various directing groups have been used for the synthesis of styrenes, the use of more challenging and weakly coordinating aromatic ketones is still elusive.6The opportunity of facile functional group transformation with ketones and a vinyl group makes 2-vinyl aromatic ketones a convenient synthetic intermediate for heterocycle, carbocycle, and natural product synthesis.7 In line with this, a ketone-directed C–H vinylation could be an effective strategy as it does not require any prefunctionalization.8 As a coupling partner for the vinylation, vinyl acetate could be a useful electrophile because it is cheap, and non-volatile unlike ethylene, and it could be a perfect alternative for ethylene or acetylene gas.9 Moreover, vinylation using vinyl acetate does not need a stoichiometric amount of oxidant or additive compared to electrophiles such as ethylene,3b,10 vinyltriethoxysilane,6j or vinylstannates.6e Ketone-directed C–H vinylations face a common problem due to the low stability of the cyclometallated species due to the low Lewis basicity of ketones. When vinyl acetate is used as an electrophile, the challenges are further increased due to the enolizability of the α-proton and the high reactivity of styrenes.11,12 Although ketone-directed vinylation with Ru-catalysts using vinyl boronate6b,c has been reported, the scope is limited to tert-butyl aromatic ketones and alkoxyacetophenones, restricting the application of the 2-vinyl aromatic ketone moiety. For a similar transformation under Rh-catalysis using vinyltriethoxysilane, only tert-butyl aromatic ketones were explored with only two examples.6j Hence, we look forward to addressing these challenges associated with ketone-directed C–H vinylation in a way where a broad range of aromatic ketones can be used with a relatively stable, economic catalyst, and using vinyl acetate as a simple and cost-effective vinyl source. To the best of our knowledge, a ketone-directed C–H vinylation has not been reported under any first-row transition metal catalysis, and vinyl acetate has not been studied under ketone directed C–H functionalization with any transition metal catalyst to date.11e,13
Considering the aforementioned obstacles, and inspired by the pioneering work by Kanai and Matsunaga on air-stable and cost-effective Cp*Co(III)-catalysis, as well as other relevant work,14,15 herein, ketone-directed vinylation has been studied for the first time under cobalt-catalysis, and electron-rich alkene vinyl acetate has been used as a coupling partner to get 2-vinyl aromatic ketones. Including acetophenone derivatives, a broad range of ketones such as chalcones, diaryl ketones, chromones, and acetylcarbazoles have been employed for the regioselective, mono-vinylation reaction (Scheme 1). We also realized C–H alkenylation using an acrylate equivalent that contains an oxidizing group or leaving group (carbonate).
 | ||
| Scheme 1 Ketone-directed C–H vinylation. | ||
Results and discussion
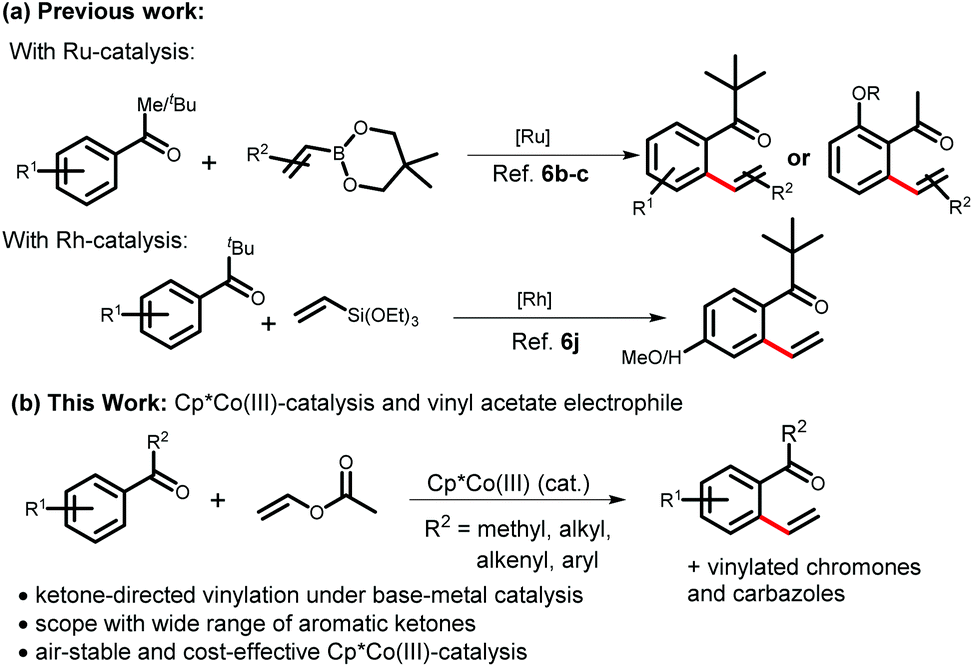
We started the optimization of the reaction conditions by taking 1-(benzo[d][1,3]dioxolo-5-yl)ethan-1-one 1a as the limiting substrate in the presence of a dimeric cobalt catalyst, [Cp*CoI2]2 (Table 1). Combination of 5 mol% of cobalt catalyst along with 20 mol% Ag-salt and 20 mol% of acetate was found to be crucial for the optimization of the vinylation reaction. Initially, the desired product 3a was isolated in 49% yield using 1,2-dichloroethane (1,2-DCE) as a solvent (Table 1, entry 1). The weak-coordination of the 1,3-dioxole oxygen played a vital role in the selective functionalization of the sterically more hindered C–H bond of the substrate 1a by acting as a secondary directing group. Among the various solvents tested (entries 2–4), only fluorinated solvent 2,2,2-trifluoroethanol (TFE) provided the best yield (entry 3). This could be explained by considering the assistance of fluorinated solvents on cobalt-catalyzed C–H bond functionalizations as reported in the recent work by the Pérez-Temprano group14i and other results.15a,b The reaction can be performed in the absence of solvent using an excess amount of vinyl acetate (61%, entry 5). Then, changing either the silver-salt (entries 6–10) or the additive (entries 11–15) while keeping other parameters unchanged did not give better results. A more detailed optimization with 4-methoxyacetophenone 1b revealed that a dimeric cobalt catalyst gives little advantage over the monomeric one and 10 mol% [Cp*CoI2]2 provides the best yield of the corresponding vinylation product 3b.16 It is important to note that no oxidative coupling product to furnish substituted vinyl acetate was observed under the reaction conditions. The product 2-vinyl aromatic ketones can be a ligand for the active catalyst by coordinating through the carbonyl oxygen and alkene group. This could be a possible reason for the high catalyst loading for the vinylation reaction. The mono-vinylation/alkenylation can also be explained in a similar way as the approaching catalyst will not be able to reach the second ortho-position of the monovinylated product to yield a divinylated product.6c|
|
||||
|---|---|---|---|---|
| Entry | Ag-Salt | Additive | Solvent | Yieldb [%] |
| a Reaction conditions: 1a (0.3 mmol, 1.0 equiv.), 2a (1.05 mmol, 3.5 equiv.), [Cp*CoI2]2 (5.0 mol%), Ag-salt (20 mol%), additive (20 mol%), solvent (1.8 mL), 100 °C, 20 h. b Isolated yield of 3a. c 0.5 mL of 2a. TCE = 1,1,2,2-tetrachloroethane. | ||||
| 1 | AgSbF6 | Cu(OAc)2 | DCE | 49 |
| 2 | AgSbF6 | Cu(OAc)2 | TCE | 41 |
| 3 | AgSbF6 | Cu(OAc)2 | TFE | 78 |
| 4 | AgSbF6 | Cu(OAc)2 | EtOH | 0 |
| 5c | AgSbF6 | Cu(OAc)2 | neat | 61 |
| 6 | AgPF6 | Cu(OAc)2 | TFE | 46 |
| 7 | AgOAc | Cu(OAc)2 | TFE | 0 |
| 8 | AgBF4 | Cu(OAc)2 | TFE | 65 |
| 9 | AgOTf | Cu(OAc)2 | TFE | 59 |
| 10 | AgNTf2 | Cu(OAc)2 | TFE | 76 |
| 11 | AgSbF6 | NaOAc | TFE | 42 |
| 12 | AgSbF6 | KOAc | TFE | 37 |
| 13 | AgSbF6 | Cu(OTf)2 | TFE | 0 |
| 14 | AgSbF6 | Cu(OAc)2. H2O | TFE | 77 |
| 15 | AgSbF6 | Cu(OAc) | TFE | 73 |
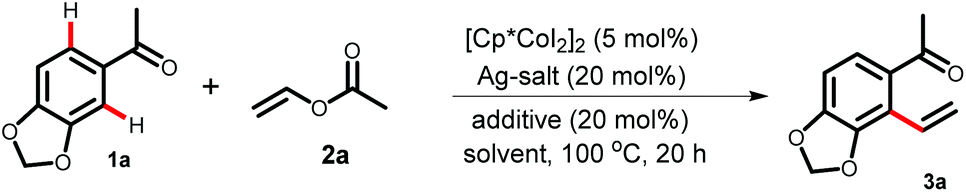
Having optimized the reaction conditions, first, we studied the scope of the vinylation reaction with various acetophenones, diaryl ketones, and chromones (Table 2). Depending on the substrates, the catalyst loading was varied between 5 to 10 mol%. The acetophenone 1b gave 3b in 71% yield with 10 mol% catalyst loading. However, a more electron rich acetophenone bearing a free hydroxyl- or dialkylamine-substituent at the para-position provided 3c–3d in 66–68% yield. Notably, the free hydroxyl functional group present in piceol 1c was compatible under the vinylation conditions. Thiomethyl, phenyl, bromine, and chlorine functional groups tolerated the reaction conditions to provide products 3e–3h in moderate to good yields. The acetophenone 1i produces 3i in moderate yield because of the steric effect arising due to the presence of the ortho-methoxy group. The regioselectivity of the vinylation becomes opposite for the acetophenone 1j bearing a 3,4-dimethoxy group. This could be due to the steric interaction (the buttressing effect)17 between two methoxy-groups, which favours the formation of the cyclometallated species at the sterically less demanding side of the acetophenone 1j. Vinylation on the electron rich carbazole ring was also achieved as products 3k–3l were isolated in 41–48% yields. Upon changing the methyl group attached to the ketone with other alkyl groups, products 3m–3n were isolated in 63–64% yields. However, in the case of diaryl ketones 1o–1p and chromone 1q, the monomeric-catalyst Cp*Co(CO)I2 was found to be relatively better than dimeric [Cp*CoI2]2 and provided the mono vinylated product. For substrate 1p, 3p is the major product and the vinylation occurs selectively at the more electron-rich aromatic ring. The biologically relevant chromone 1q was also a suitable substrate (3q, 37%). A thiophene-derived ketone was also a suitable substrate for the vinylation reaction (3s, 51%).
| a Reaction conditions: 1 (0.3 mmol, 1.0 equiv.), 2a (1.05 mmol, 3.5 equiv.), and 1.8 mL of TFE were used. b Isolated yields. c 5.0 mol% of [Cp*CoI2]2, 20 mol% AgSbF6 and 20 mol% of Cu(OAc)2 were used. d 7.5 mol% of [Cp*CoI2]2, 30 mol% AgSbF6 and 30 mol% of Cu(OAc)2 were used. e 20 mol% of Cp*Co(CO)I2, 40 mol% AgSbF6 and 40 mol% of Cu(OAc)2 were used. |
|---|
|
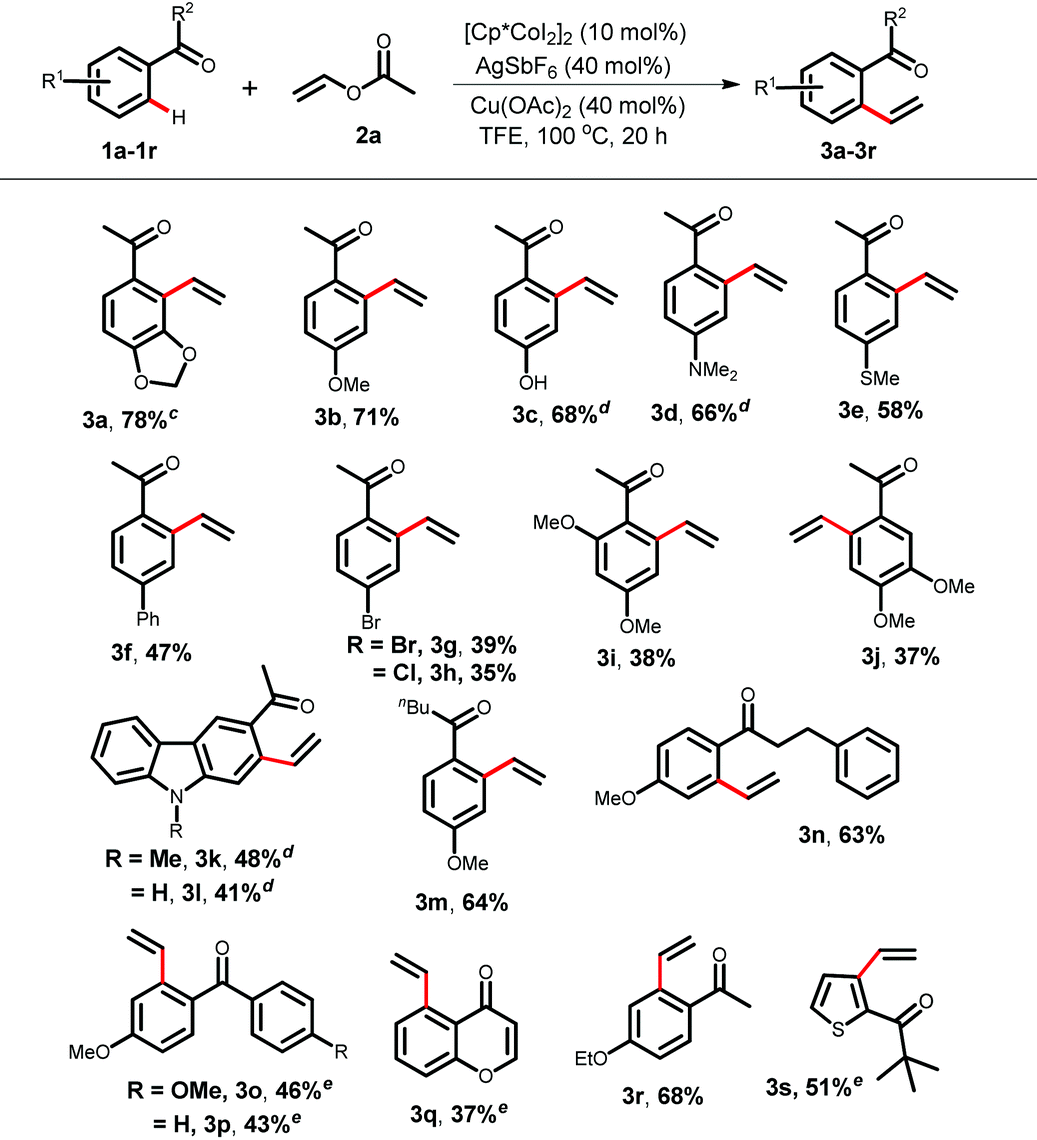
After successfully investigating the vinylation reaction with acetophenones and benzophenones, we next studied the scope with chalcone 4 (Table 3). Moreover, chalcones are widespread in medicinally important molecules as they exhibit interesting biological activities.18 To our delight, only the aromatic C–H bond underwent selective vinylation over the olefinic one. The chalcone containing a 1,3-dioxole group took part in the vinylation at the sterically more crowded side to produce the desired dialkenylated product 5a in 68% yield. Chalcones bearing a methoxy-, free hydroxyl-, dimethyl amine- or thiomethyl-group at the para-position of the ketone yielded vinylation products in moderate to good yields (5b–5e, 44–61%). A heteroaryl chalcone was also suitable under the reaction conditions (5f, 49%). Moreover, a conjugated dienone 4g furnished the corresponding styrene product 5g.
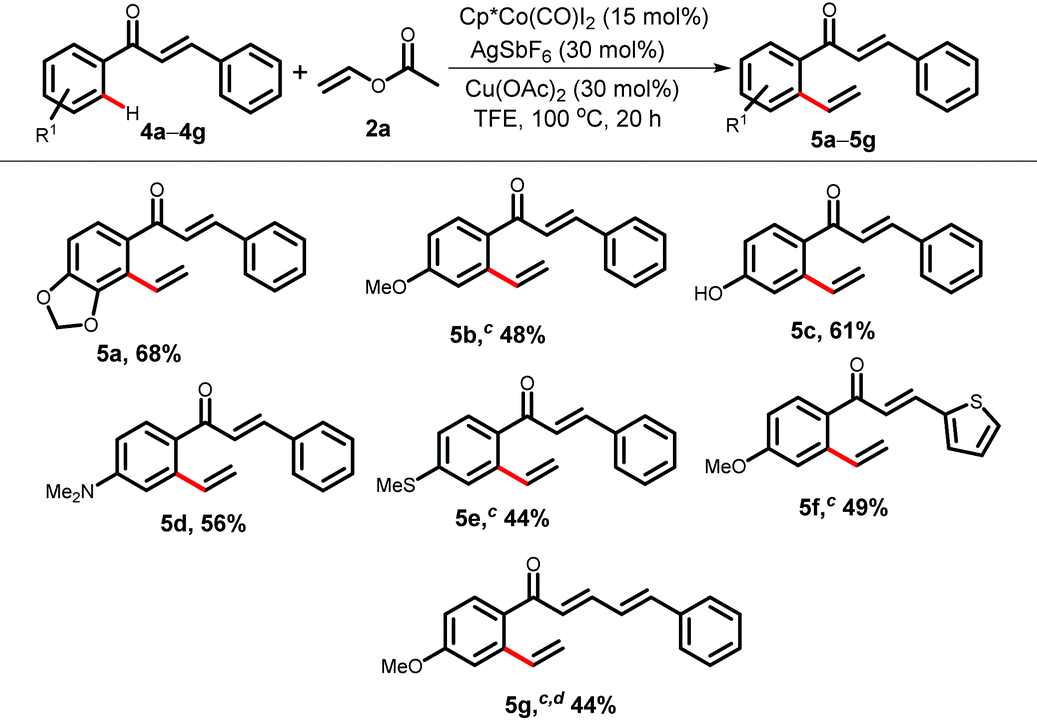
We envisioned that an electrophile 2b can be employed as an acrylate equivalent to achieve a formal Fujiwara–Moritani type coupling reaction without using a stoichiometric amount of oxidant. Interestingly, the carbonate acts as a leaving group and such types of acrylate equivalent are under used in C–H alkenylation (Table 4).9d,i Variously substituted acetophenones, and aromatic ketones were alkenylated using 2b under redox neutral conditions to furnish alkenylated products in moderate to good yields (6a–6h, 34–67%). A γ-PPAR antagonist could be synthesized from compound 6h following the literature procedure.15b A heteroaryl ketone also took part in the reaction and provided 6i in 43% yield.
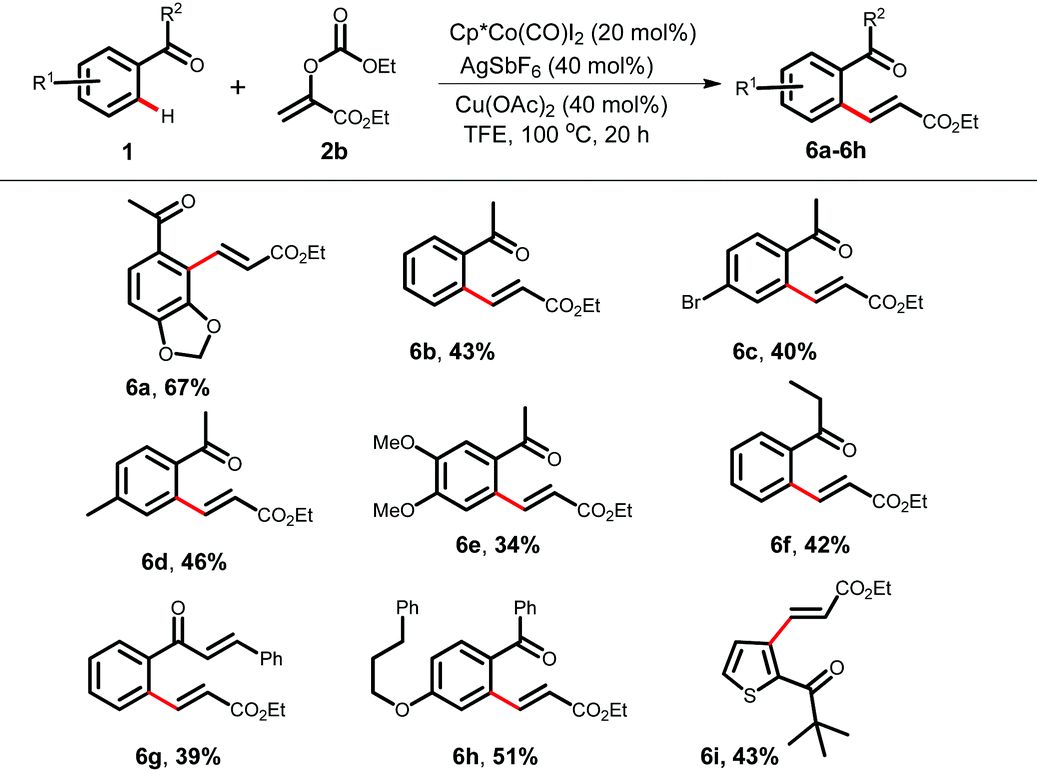
After completing the substrate scope, we focused on several transformations using the 2-vinyl ketones (Scheme 2). The dihydroxylation of alkenes with a catalytic amount of OsO4 followed by diol cleavage with NaIO4 yielded the aldehyde 7, which cyclized to functionalized indanone 8 in 56% yield via an intramolecular Aldol reaction in the presence of LiOH. Phenyl acetaldehyde derivative 9, a key intermediate for the synthesis of the natural product bruguierol A, was also prepared from 2-vinyl ketone 3r.19a Furthermore, vinylated product 11 can be synthesized through a late-stage vinylation of the HBV inhibitor 10.19b The terminal selective Wacker oxidation of 3a provided phenyl acetaldehyde derivative 12, which was transformed to densely functionalized α-naphthol 13.
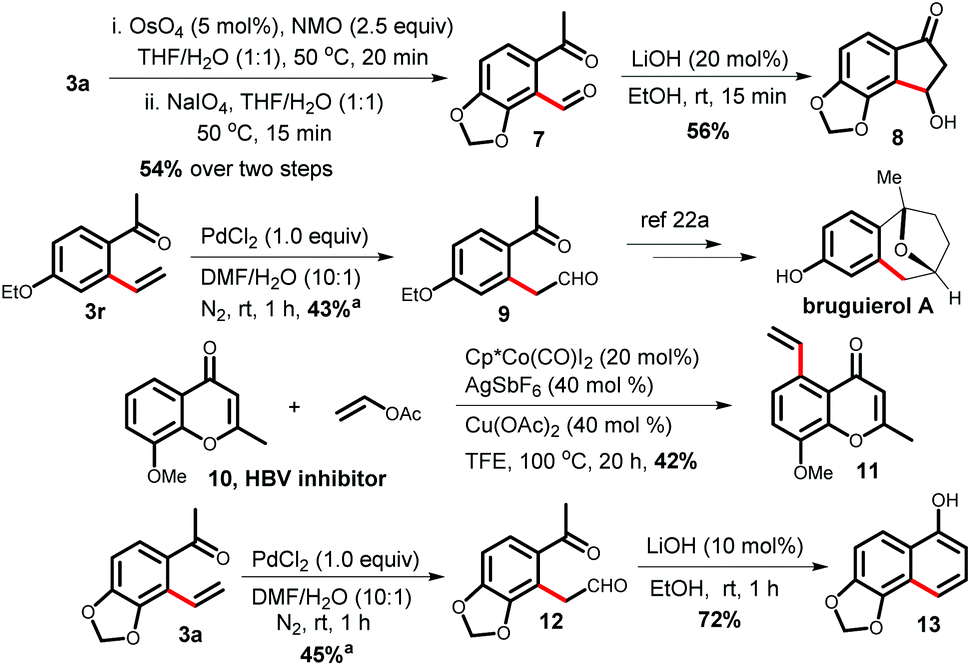 | ||
| Scheme 2 Application of the vinylated products. aYield based on recovered starting material. | ||
To gain insight into the nature of the present cobalt catalyzed C–H bond activation, several experiments have been carried out (Scheme 3). A competition experiment revealed that electron rich acetophenone 1b reacted preferentially over the electron deficient 1h, indicating that metallation occurs via a base-assisted intermolecular electrophilic substitution (BIES) pathway (Scheme 3a).20a The deuterium exchange experiments implied a reversible metalation step (Scheme 3b and c). Furthermore, the kinetic isotope effect determined from competition (KIE value of approximately 2.1) and the kH/kD value of approximately 2.0 from the parallel experiments indicated that a C–H activation step might not be involved in the rate determining step (Scheme 3d).20b,c The LCMS analysis of the reaction mixture for the acetophenone 1a indicated that the presence of molecular species [C19H22CoO3]+ and [C23H28CoO5]+ might be due to the involvement of the cyclometallated species 14 and the intermediate 15 or 17 during the course of the reaction (Scheme 3e–f).16
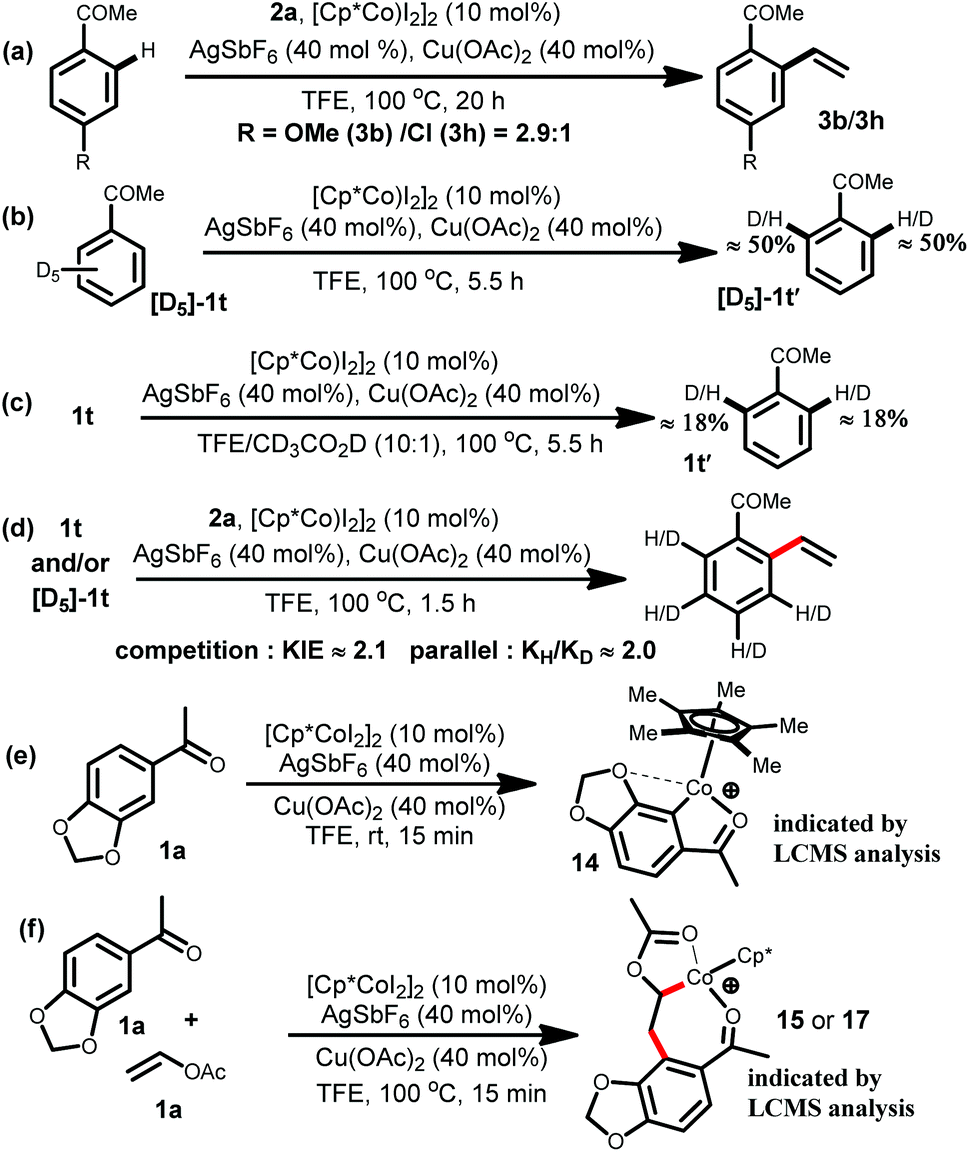 | ||
| Scheme 3 Mechanistic studies. | ||
Based on the literature precedence6,9 and the aforementioned studies, a plausible mechanism for the present cobalt-catalyzed C–H vinylation is depicted in Scheme 4. The active catalyst A, generated from the catalyst [Cp*CoI2]2 in the presence of Ag-salt and the acetate, forms the cyclometallated species 14. The formation of the intermediate 14 is supported by the LCMS analysis. The 1,2-migratory insertion of a Co–C bond into the alkene 2a generates the intermediate 15. The presence of the intermediate 15 was again supported by LCMS analysis, which is in agreement with the previous report.6g A β-hydride elimination in the intermediate 15 followed by the reinsertion produces 17. Finally, the facile β-acetate elimination yielded the desired vinylated product 3a.
 | ||
| Scheme 4 Proposed catalytic cycle. | ||
Conclusions
In conclusion, we successfully developed ketone-directed C–H vinylation of aromatic ketones and more challenging acetophenones using first-row transition metal cobalt-catalysis. This eliminated the multi-step approach for the synthesis of 2-vinyl aromatic ketones. A broad range of aromatic ketones were reacted under the reaction conditions. Several ethyl cinnamates were also successfully prepared by using this protocol employing an appropriate acrylate equivalent. The importance of 2-vinyl ketones was demonstrated by the synthesis of indanone and α-naphthol, formal synthesis of the natural product bruguierol A, and the late-stage vinylation of an HBV-inhibitor. We believe that this method could be useful to access several challenging vinyl ketones via late stage C–H vinylation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. S. M. gratefully acknowledges SERB, Department of Science and Technology, New Delhi, India (Sanction No. CRG/2018/000317) for funding. M. R. S. sincerely thanks IIT Kharagpur for the fellowship. We thank the DST (Sanction No. SR/FST/CSII-026/2013) for the 500 MHz NMR facility at IIT Kharagpur.Notes and references
- (a) F. Agbossou, J.-F. Carpentier and A. Mortreux, Chem. Rev., 1995, 95, 2485 CrossRef CAS; (b) A. Zapf and M. Beller, Top. Catal., 2002, 19, 101 CrossRef CAS; (c) X. Wang and H. Gao, Polymers, 2017, 9, 188 CrossRef.
- (a) O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510 RSC.
- (a) M. Shibasaki, E. M. Vogl and T. Ohshima, Adv. Synth. Catal., 2004, 346, 1533 CrossRef CAS; (b) L. Zhou and W. Lu, Chem. – Eur. J., 2014, 20, 634 CrossRef CAS.
- (a) T. V. RajanBabu, Chem. Rev., 2003, 103, 2845 CrossRef CAS; (b) M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368 CrossRef CAS; (c) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333 CrossRef CAS; (d) J. Chen and Z. Lu, Org. Chem. Front., 2018, 5, 260 RSC.
- (a) B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS PubMed; (b) P. A. Wender and B. L. Miller, Nature, 2009, 460, 197 CrossRef CAS.
- For C–H vinylation: (a) Y. Matsuura, M. Tamura, T. Kochi, M. Sato, N. Chatani and F. Kakiuchi, J. Am. Chem. Soc., 2007, 129, 9858 CrossRef CAS; (b) S. Ueno, N. Chatani and F. Kakiuchi, J. Org. Chem., 2007, 72, 3600 CrossRef CAS; (c) S. Ueno, T. Kochi, N. Chatani and F. Kakiuchi, Org. Lett., 2009, 11, 855 CrossRef CAS; (d) F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9982 CrossRef CAS PubMed; (e) S. Prakash, K. Muralirajan and C.-H. Cheng, Chem. Commun., 2015, 51, 13362 RSC; (f) S.-T. Mei, K. Jiang, N.-J. Wang, L. Shuai, Y. Yuan and Y. Wei, Eur. J. Org. Chem., 2015, 6135 CrossRef CAS; (g) K. D. Otley and J. A. Ellman, Org. Lett., 2015, 17, 1332 CrossRef CAS; (h) S.-S. Li, C.-Q. Wang, H. Lin, X.-M. Zhang and L. Dong, Org. Biomol. Chem., 2016, 14, 229 RSC; (i) J. Xu, C. Chen, H. Zhao, C. Xu, Y. Pan, X. Xu, H. Li, L. Xu and F. Fan, Org. Chem. Front., 2018, 5, 734 RSC; (j) J. Zhou, X. Li, G. Liao and B.-F. Shi, Chin. J. Chem., 2018, 36, 1143 CrossRef CAS; (k) N. Muniraj and K. R. Prabhu, Adv. Synth. Catal., 2018, 360, 3579 CrossRef CAS.
- (a) X. Chen, H.-J. Zhang, X. Yang, H. Lv, X. Shao, C. Tao, H. Wang, B. Cheng, Y. Li, J. Guo, J. Zhang and H. Zhai, Angew. Chem., Int. Ed., 2018, 57, 947 CrossRef CAS; (b) B. Xu, B. Wang, W. Xun and F. G. Qiu, Angew. Chem., 2019, 58, 5754 CrossRef CAS; (c) S. Chakraborty and D. Mal, J. Org. Chem., 2018, 83, 1328 CrossRef CAS; (d) Y. Hu, Y. Xie, Z. Shen and H. Huang, Angew. Chem., Int. Ed., 2017, 56, 2473 CrossRef CAS PubMed; (e) D. Arican, S. Braukmüller and R. Brückner, Chem. – Eur. J., 2017, 23, 4537 CrossRef CAS; (f) J. Zhao, J. L. Brosmer, Q. Tang, Z. Yang, K. N. Houk, P. L. Diaconescu and O. Kwon, J. Am. Chem. Soc., 2017, 139, 9807 CrossRef CAS.
- (a) S. E. Denmark and C. R. Butler, Chem. Commun., 2009, 20 RSC; (b) J.-Y. Yu and R. Kuwano, Angew. Chem., Int. Ed., 2009, 48, 7217 CrossRef CAS; (c) J. Lindh, J. Sävmarker, P. Nilsson, P. J. R. Sjöberg and M. Larhed, Chem. – Eur. J., 2009, 15, 4630 CrossRef CAS; (d) Y. Gao, Y. Ou and L. J. Gooßen, Chem. – Eur. J., 2019, 25, 8709 CAS.
- (a) H. Horino and N. Inoue, J. Org. Chem., 1981, 46, 4416 CrossRef CAS; (b) N. J. Webb, S. P. Marsden and S. A. Raw, Org. Lett., 2014, 16, 4718 CrossRef CAS; (c) H. Chu, S. Sun, J.-T. Yu and J. Cheng, Chem. Commun., 2015, 51, 13327 RSC; (d) M. Zhang, H.-J. Zhang, T. Han, W. Ruan and T.-B. Wen, J. Org. Chem., 2015, 80, 620 CrossRef CAS; (e) S.-S. Li, C.-F. Liu, Y.-Q. Xia, W.-H. Li, G.-T. Zhang, X.-M. Zhang and L. Dong, Org. Biomol. Chem., 2016, 14, 5214 RSC; (f) H.-J. Zhang, W. Lin, F. Su and T.-B. Wen, Org. Lett., 2016, 18, 6356 CrossRef CAS; (g) G. Liang, J. Rong, W. Sun, G. Chen, Y. Jiang and T.-P. Loh, Org. Lett., 2018, 20, 7326 CrossRef CAS; (h) W.-B. Qin, W.-W. Li, P.-F. Zhu, X.-G. Mo, H.-J. Zhang and T.-B. Wen, Org. Chem. Front., 2018, 5, 1096 RSC; (i) W. Lin, W. Li, D. Lu, F. Su, T.-B. Wen and H.-J. Zhang, ACS Catal., 2018, 8, 8070 CrossRef CAS.
- (a) T. Matsumoto, R. A. Periana, D. J. Taube and H. Yoshida, J. Catal., 2002, 206, 272 CrossRef CAS; (b) T. Yamada, A. Sakakura, S. Sakaguchi, Y. Obora and Y. Ishii, New J. Chem., 2008, 32, 738 RSC.
- (a) R. Mahrwald, Chem. Rev., 1999, 99, 1095 CrossRef CAS; (b) C. C. C. Johansson and T. J. Colacot, Angew. Chem., Int. Ed., 2010, 49, 676 CrossRef CAS; (c) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS; (d) S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461 CrossRef CAS; (e) Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764 RSC.
- (a) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529 CrossRef CAS; (b) P. Gandeepan, K. Parthasarathy and C.-H. Cheng, J. Am. Chem. Soc., 2010, 132, 8569 CrossRef CAS; (c) F. W. Patureau, T. Besset, N. Kuhl and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2154 CrossRef CAS; (d) F. Mo, L. J. Trzepkowski and G. Dong, Angew. Chem., Int. Ed., 2012, 51, 13075 CrossRef CAS; (e) M. Bhanuchandra, M. R. Yadav, R. K. Rit, M. R. Kuram and A. K. Sahoo, Chem. Commun., 2013, 49, 5225 RSC; (f) G. Li, L. Wan, G. Zhang, D. Leow, J. Spangler and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 4391 CrossRef CAS; (g) K. R. Bettadapur, V. Lanke and K. R. Prabhu, Org. Lett., 2015, 17, 4658 CrossRef.
- (a) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192 CrossRef CAS; (b) M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843 RSC; (c) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC; (d) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC.
- (a) T. Yoshino, H. Ikemoto, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 2207 CrossRef CAS; (b) D.-G. Yu, T. Gensch, F. de Azambuja, S. Vásquez-Céspedes and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722 CrossRef CAS; (c) J. R. Hummel and J. A. Ellman, J. Am. Chem. Soc., 2015, 137, 490 CrossRef CAS; (d) J. Park and S. Chang, Angew. Chem., Int. Ed., 2015, 54, 14103 CrossRef CAS; (e) N. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498 CrossRef; (f) S. S. Bera, S. Debbarma, A. K. Ghosh, S. Chand and M. S. Maji, J. Org. Chem., 2017, 82, 420 CrossRef CAS; (g) T. Yoshino and S. Matsunaga, Adv. Synth. Catal., 2017, 359, 1245 CrossRef CAS; (h) S. S. Bera, S. Debbarma, S. Jana and M. S. Maji, Adv. Synth. Catal., 2018, 360, 2204 CrossRef CAS; (i) J. Sanjosé-Orduna, J. M. S. Toro and M. H. Pérez-Temprano, Angew. Chem., Int. Ed., 2018, 57, 11369 CrossRef; (j) D. A. Loginov, L. S. Shul'pina, D. V. Muratov and G. B. Shul'pin, Coord. Chem. Rev., 2019, 387, 1 CrossRef CAS; (k) B. Sun, T. Yoshino, S. Matsunaga and M. Kanai, Adv. Synth. Catal., 2014, 356, 1491 CrossRef CAS; (l) P. G. Chirila and C. J. Whiteoak, Dalton Trans., 2017, 46, 9721 RSC.
- (a) M. R. Sk, S. S. Bera and M. S. Maji, Org. Lett., 2018, 20, 134 CrossRef PubMed; (b) M. R. Sk, S. S. Bera and M. S. Maji, Adv. Synth. Catal., 2019, 361, 585 CrossRef CAS; (c) S. S. Bera, M. R. Sk and M. S. Maji, Chem. – Eur. J., 2019, 25, 1806 CrossRef CAS; (d) R. Mandal, B. Emayavaramban and B. Sundararaju, Org. Lett., 2018, 20, 2835 CrossRef CAS; (e) Y. Yu, Q. Wu, D. Liu, L. Hu, L. Yu, Z. Tan and G. Zhu, J. Org. Chem., 2019, 84, 7449 CrossRef CAS PubMed.
- See ESI for details.†.
- L. Li, W. W. Brennessel and W. D. Jones, Organometallics, 2009, 28, 3492 CrossRef CAS.
- C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chem. Rev., 2017, 117, 7762 CrossRef CAS.
- (a) D. Sarkar and R. V. Venkateswaran, Tetrahedron Lett., 2011, 52, 3232 CrossRef CAS; (b) S. Debbarma, M. R. Sk, B. Modak and M. S. Maji, J. Org. Chem., 2019, 84, 6207 CrossRef CAS.
- (a) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS; (b) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef; (c) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qo01164a |
| This journal is © the Partner Organisations 2020 |