
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting trifluoromethyl substituents for tuning orbital character of singlet and triplet states to increase the rate of thermally activated delayed fluorescence†
Jonathan S.
Ward
a,
Andrew
Danos
 b,
Patrycja
Stachelek
b,
Mark A.
Fox
a,
Andrei S.
Batsanov
a,
Andrew P.
Monkman
*b and
Martin R.
Bryce
*a
b,
Patrycja
Stachelek
b,
Mark A.
Fox
a,
Andrei S.
Batsanov
a,
Andrew P.
Monkman
*b and
Martin R.
Bryce
*a
aDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
bDepartment of Physics, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: a.p.monkman@durham.ac.uk
First published on 29th July 2020
Abstract
This work shows that trifluoromethyl (CF3) substituents can be used to increase the rate of thermally activated delayed fluorescence (TADF) in conjugated organic molecules by tuning the excitonic character of the singlet and triplet excited states. The synthesis and detailed photophysical characterization of four new functionalized donor–acceptor–donor (D–A–D) compounds using CF3-substituted-thioxanthene-S,S-dioxide and dimethylacridine units are presented. Several compounds are reported to exhibit rapid blue/green thermally activated delayed fluorescence with major delayed fluorescence lifetime components of ≈1–2 μs. Changes in the orbital character of singlet and triplet states (local vs. charge transfer) result in significant changes in rates of delayed fluorescence, despite similar ΔEST values. The change in orbital character is well supported by TD-DFT calculations. Electrochemical measurements reveal strong shifts in redox potentials that can be induced by σ-electron withdrawing CF3 substituents. Trifluoromethyl substituted compounds with a wider ΔEST also perform more efficiently than might be expected due to the demonstrated changes in excited state character. This study reveals important photophysical and molecular design implications for the future development of TADF emitters.
Introduction
Light emission from organic materials is revolutionizing the color-display and lighting industries.1–3 However, traditional fluorescent molecules have severe drawbacks in these applications, as only 25% of the excited states (singlets) that are generated under electrical excitation are harvested as emission; the remaining 75% of triplet excitons decay non-radiatively.4 Phosphorescent molecules can address this issue and achieve internal quantum efficiencies (IQEs) approaching 100% due to light emission from both singlet and triplet excited states.5 This has led to extensive studies on emitters containing heavy metal atoms, where strong spin–orbit coupling promotes intersystem crossing (ISC) and phosphorescence (PH) emission.6,7 The main disadvantages of organometallic emitters are their high cost due to the necessity of precious and possibly toxic heavy metals (typically iridium and platinum) and the electrochemical and thermal instability of the blue and especially deep-blue emitting complexes.8E-type delayed fluorescence,9 which has been re-named thermally activated delayed fluorescence (TADF) is an alternative mechanism that harvests triplet excited states in all-organic emitters for light emission.10,11 The main application of TADF materials is in organic light-emitting diodes (OLEDs).12–15 TADF materials have also been deployed in fluorescence imaging,16,17 optical temperature sensing18,19 and catalysis.20 Specific molecular design features enable fast reverse intersystem crossing (rISC), which can convert up to 100% of triplet excited states to emissive singlet states, leading to TADF.21 Despite intense global research efforts, full understanding of the specific molecular features that enable rapid rISC remain elusive. Indeed, several different rISC mechanisms with different design rules have been discovered, including spin-vibronic coupling in donor–acceptor–donor (D–A–D) charge transfer (CT) emitters,22 upper triplet state crossings,23 resonant non-overlapping orbital distributions in planar boron compounds,24 exciplexes,25,26 and hybrid D–A compounds.27
Ideal TADF materials possess both high photoluminescence quantum yields (PLQYs) and fast rISC rates, the latter of which are enabled by a small energy difference between the lowest singlet and triplet excited states (typically ΔEST < 0.25 eV). These properties can be found in heteroaromatic donor–acceptor (D–A) molecules with charge-transfer (CT) excited states. In these D–A molecules, a large average dihedral angle between the planes of the D and A units ensures the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO, respectively) are spatially separated. This minimizes electron exchange energy and thus also ΔEST. A detailed analysis of the mechanism of TADF in D–A–D materials has revealed a second-order spin-vibronic coupling process, in which a triplet state of local excitonic character (3LE) mediates rISC between the triplet and singlet charge transfer states (3CT and 1CT, respectively).28–30
The key roles of D–A conjugation and dihedral angles, molecular rigidity, conformation, connectivity, and steric effects are also important factors that influence CT formation and hence TADF efficiency and emission color.31–37 Despite this level of understanding, stable blue emission, which is invaluable for high-quality full-color OLED displays, remains especially challenging to achieve. Only a limited number of blue/deep-blue TADF emitters possessing good color purity have been reported.36,38–44
In this context a systematic series of TADF molecules that are functionalized at specific sites with trifluoromethyl substituents are herein presented. Trifluoromethyl (CF3) is a strongly inductive electron-withdrawing group which makes it a useful substituent for emission colour tuning via donor and acceptor strength manipulation in D–A–D CT emitters. CF3 is not π-conjugative, which makes it distinct from the electron-withdrawing groups that are typically used in TADF emitters, for example: cyano,10,45 sulfone,46,47 ketone,48 triazine49 and oxadiazole.50 Trifluoromethyl groups can also be very stable to chemical, electrochemical, thermal and photochemical degradation, which are valuable properties to impart to TADF materials.51–54
Extensive systematic studies of TADF emitters with CF3 substituents are scarce. Current CF3 functionality in TADF emitters are typically incorporated in the ‘Ar–CF3’ type motif.55 Examples include CF3–phenyl56–59 or CF3–pyridyl52,60 as an acceptor unit, or CF3-carbazole as a donor unit.61,62 This present study further diversifies the library of reported CF3 functionalised TADF molecules and has led to new blue and green TADF emitters by manipulation of the electron-donating and electron-accepting abilities of the D and A subunits with CF3 substituents. In particular, we have investigated the use of the CF3 unit as part of the bridge of the acceptor unit as opposed to the typical ‘Ar–CF3’ motif currently observed in the literature. There are few examples of CF3 ‘bridges’ being utilized in TADF molecules.53,63 Swager and co-workers showed that geminal-CF3 substituents on (non-TADF) polyfluorene derivatives were more photostable compared to geminal-CH3 substituents.53 Adachi and co-workers used a flexible bis(CF3)-substituted methylene bridge directly between donor and acceptor in TADF emitters.63
The synthetic routes, X-ray crystal structures, solution electrochemical data and a thorough photophysical investigation of the materials are presented here. These results show that trifluoromethyl substituents can be used to impart large shifts in redox potentials and emission properties through inductive effects. Molecules 1 and 2 show highly similar ΔEST values, but 2 shows faster delayed fluorescence. Detailed analysis and supporting hybrid-DFT calculations reveal how the tuning of orbital character with trifluoromethyl substituents leads to an increase in TADF emission rates. OLEDs were also fabricated, although these devices showed unexpected instability.
Experimental section
The synthesis and structural characterization of the molecules are given in the ESI.†X-ray diffraction experiments: full data (including structure factors) in CIF format is available as ESI† and have been deposited with the Cambridge Structural Database, CCDC 1887606 (3) and 1887607 (4).†
Optical and optoelectronic methods are given in the ESI.†
Results and discussion
Molecular design and synthesis
Starting from the well-characterized blue TADF emitter 2,7-bis(9,9-dimethylacridin-10-yl)-9,9-dimethylthioxanthene-S,S-dioxide (DDMA-TXO2) 1, (Fig. 1), the motivation was to explore the effect on photophysical properties that arise from tuning the donor and acceptor abilities of the sub-units using systematic CF3 substitution.64,65 Compound 1 was selected as it has efficient TADF properties and can be systematically modified. The DDMA-TXO2 triplet level (T1) is a localized triplet (3LE) state and the energy splitting between 1CT and 3LE is ΔEST = (0.06 ± 0.03) eV in evaporated DPEPO host.65 Two independent studies have shown that DDMA-TXO2 affords blue OLEDs with EQEs > 20%.64,65 | ||
| Fig. 1 Chemical structures of the previously-reported D–A–D molecule 1 and the new trifluoromethylated analogs 2–5. | ||
Fig. 1 presents the chemical structures of 1 and the new analogs 2–5. The methyl groups of the thioxanthene acceptor in 1 are replaced by CF3 groups in 2–4 (Fig. 1), whereas CF3 groups are added directly on to the aryl rings of the D unit in 5 (with both D and A modifications in 4). The acceptor CF3 groups in 2–4 are on the bridging sp3 carbon atom and are not directly attached to the π-electron system. The potential ability of these CF3 groups to alter the singlet and triplet levels (and ΔEST) are therefore of both fundamental and practical interest. The donor within the series 1–4 is systematically changed from the 9,9-dimethylacridine units (in 1 and 2) to the 3,6-dimethoxycarbazole (in 3) and 2,7-bis(trifluoromethyl)-9,9-dimethylacridine (in 4 and 5).
The synthetic routes to 2–5 proceeded via palladium-catalysed Buchwald–Hartwig coupling of suitably functionalized D and A fragments.66 The identity and high purity of 2–5 were confirmed by a combination of NMR spectroscopy, high resolution mass spectrometry, and X-ray crystallography (see the ESI†). The synthesis of molecule 4 is shown in Scheme 1, as a representative example.
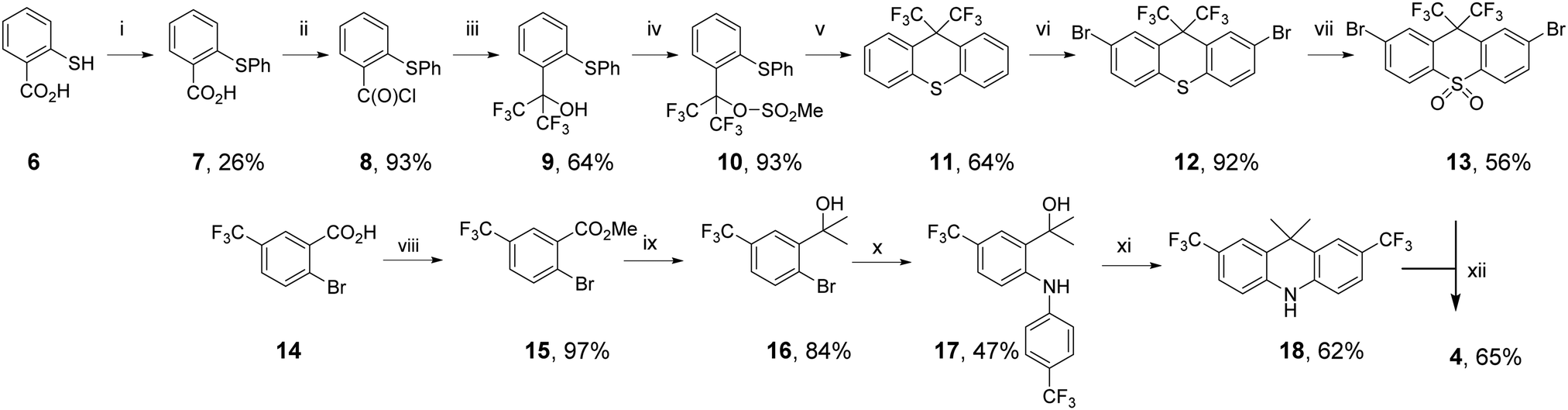 | ||
| Scheme 1 The synthetic route to 4. Reagents and conditions: (i) PhBr, CuI, K2CO3, DMF, reflux. (ii) oxalyl chloride, DMF (cat.), DCM, (iii) TMSCF3, NMe4F, DME −60 °C → rt. (iv) MeSO2Cl, Et3N, DCM, rt. (v) Amberlyst-15, toluene, reflux. (vi) Br2, AcOH, reflux. (vii) H2O2(aq), AcOH, reflux. (viii) K2CO3, MeI, acetone, reflux. (ix) MeMgBr, THF, rt. (x) CuI, 1,10-phenanthroline, Cs2CO3, toluene, reflux. (xi) Amberlyst-15, CHCl3, reflux (xii) Pd2(dba)3·CHCl3, HPtBu3BF4, NaOtBu, toluene, reflux. | ||
To prepare CF3 substituted compound 4, the acceptor precursor 13 with a bis-CF3 methylene bridge was required. Using modified literature procedures, acid chloride-containing 8 was synthesized using copper-mediated thioether synthesis, followed by conversion to acid chloride with oxalyl chloride. Pleasingly, 8 could be converted to bis-CF3-alcohol 9 using NMe4F and TMSCF3,67 avoiding the use of gaseous hexafluoroacetone typically used to synthesize bis-CF3 functionality.53 Attempts to close alcohol 9 to thioxanthene 11 with standard reagents did not yield the desired product.68 Therefore, 9 was converted to the mesylate 10 to introduce a more suitable leaving group. Treatment of 10 with a solid supported sulfonic acid resin (Amberlyst-15) resulted in formation of bis-CF3 substituted thioxanthene 11. Interestingly GCMS analysis of the reaction revealed that 10 was also partly converted back to 9. Upon complete conversion of mesylate 10, no additional thioxanthene 11 was formed. This analysis shows that the mesylate leaving group was essential to form thioxanthene 11. Bromination of 11 to give dibromothioxanthene 12 was achieved with bromine in AcOH, but required a higher temperature to proceed than the corresponding dimethylthioxanthene derivative used to prepare the acceptor precursor of 1.69 Typical oxidation of 12 gives the thioxanthene-S,S-dioxide acceptor precursor 13.
To prepare the 2,7-bis-CF3 substituted acridine donor 18, the rings were constructed using starting materials with CF3 functionality already installed. Ester formation yielded compound 15 from 14. Installation of the dimethylmethylene bridge was achieved by reaction of MeMgBr with 15 to give alcohol 16. Successful utilization of a recently reported protecting-group-free copper-catalyzed C–N coupling gave diarylamine compound 17.70 Compound 17 was cyclized to give new bis-CF3-acridine donor 18 using Amberlyst-15. Compound 4 was synthesized from the coupling of 13 and 18 using typical Buchwald–Hartwig conditions, which generally tolerate a wide range of substituents for donor–acceptor couplings.47,65,71,72 Compounds 2, 3 and 5 were also coupled using Buchwald–Hartwig conditions, with 2–5 prepared with yields ranging from 56 to 73%.
X-Ray crystal structures
The crystal structures of 3 and 4 are shown in Fig. 2. Structure 3 is modulated along the [1 1 1] direction: three symmetrically independent molecules are related roughly by pseudo-translations (a + b + c)/3 and adopt nearly identical conformations (Fig. S6a and b, ESI†). In each, the acceptor moiety is slightly twisted, with an angle of 6.9°, 14.8° and 5.4° between the arene ring planes. The donor (D) moieties are approximately planar (including the methoxy carbon atoms) and are inclined in a propeller-like fashion with respect to the acceptor (A) moiety; the twist angles around the C(A)–N(D) bonds vary from 38.7° to 53.7°, average 46.5(5.2)°. In 4, the acceptor moiety is twisted in the same way as in 3 (by 6.3°); the two donor moieties are folded along the N⋯C(sp3) vectors by 29.9° and 7.9°, and inclined with respect to the acceptor more than in 3, the twists around the C(A)–N(D) bonds being 74.7° and 87.9°, respectively. These C(A)–N(D) twists are typically observed in other TADF molecules.10,48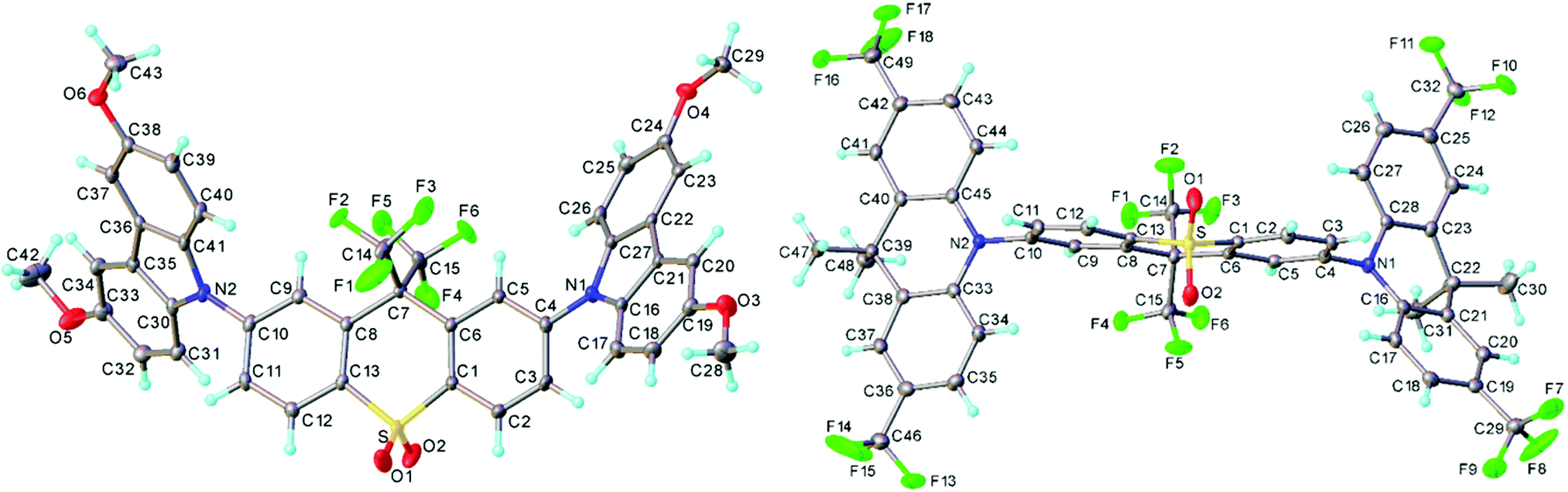 | ||
| Fig. 2 X-Ray molecular structures of 3 (one of the three independent molecules) and 4, showing thermal ellipsoids at 50% probability level. | ||
Solution electrochemistry
Solution state cyclic voltammetry (CV) was performed to assess the effect of the trifluoromethyl substituents on the redox properties of 1–5. All CV measurements were performed with acetonitrile (MeCN) as solvent due to its wide electrochemical window. Trifluoromethyl substituents significantly improve the solubility of 2–5 compared to 1 in MeCN, making it possible to obtain good signal-to-noise CV traces for 2–5. The solubility of 1 is poor in MeCN but the measurements are in agreement with the reported oxidation potential of 1.73Table 1 and Fig. 3 summarize the CV data.| Entry | E OxOnset (V) | E REDOnset (V) | HOMO (eV) | LUMO (eV) | HLG (eV) |
|---|---|---|---|---|---|
| 1 | 0.55 | −2.34 | −5.65 | −2.76 | 2.89 |
| 2 | 0.56 | −2.03 | −5.66 | −3.07 | 2.59 |
| 3 | 0.60 | −2.09 | −5.70 | −3.01 | 2.69 |
| 4 | 0.93 | −2.02 | −6.03 | −3.08 | 2.95 |
| 5 | 0.88 | −2.28 | −5.98 | −2.82 | 3.16 |
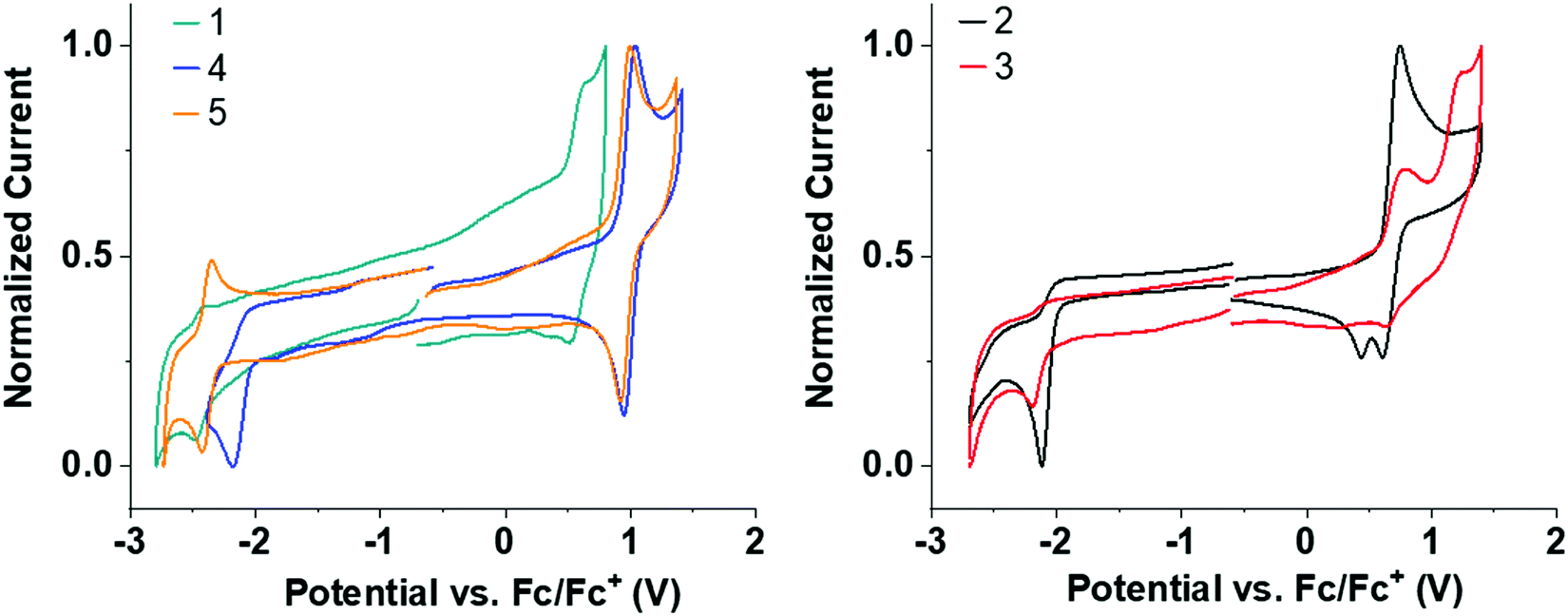 | ||
| Fig. 3 Normalized CV redox curves for 1–5 in deoxygenated MeCN solvent. All curves are referenced to the Fc/Fc+ redox couple. | ||
Several clear trends can be observed by systematic CF3 substitution on 1. Addition of CF3 groups in the para-position on the donor shifts the oxidation to significantly higher potential in 4 and 5 compared to unsubstituted 1 (≈+0.55 → ≈+0.9 V). This shift is in agreement with the strongly σ-withdrawing effect of the CF3 substituent, destabilizing any resulting cation. Addition of CF3 groups on the donor makes the oxidation wave reversible in 4 and 5 compared to 1 (see Fig. S4b and c for multiple CV cycles, ESI†). The stability imparted is similar to that seen by introduction of tBu substituents in other acridine analogs,23 but CF3 groups are electron withdrawing in contrast to the inductive electron donating tBu group. Interestingly, 5 has a highly reversible oxidation and reduction despite a wide electrochemical window of 3.16 V.
Addition of CF3 groups on the acceptor in 2–4 reduces the reduction potential compared to unsubstituted 1 (≈–2.3 V → ≈–2.1 V). This shift in reduction is quite substantial considering that the CF3 groups on the acceptor in 2–4 are on an sp3 hybridized bridge which is not in direct conjugation with the π-system, and highlights the strongly σ-withdrawing effect of the CF3 substituent. Unexpectedly, 2–4 all have irreversible reduction waves in contrast to 5. While the exact origin of this irreversibility is unclear, it is suggested that the σ-bridge of the acceptor is likely to be highly δ+ due to the CF3 functionality and the sulfone group. These multiple electron withdrawing groups in combination could reduce the strength of neighbouring C–C alkyl–aryl bonds on the σ-bridge resulting in bond cleavage upon reduction.
Optical and photophysical properties
All the molecules in this study have been characterized using UV/Vis absorption and a range of photoluminescence techniques.Solution-state absorption
The UV/Vis absorption spectra of molecules 1–5 were obtained in DCM solution and are shown in Fig. 4.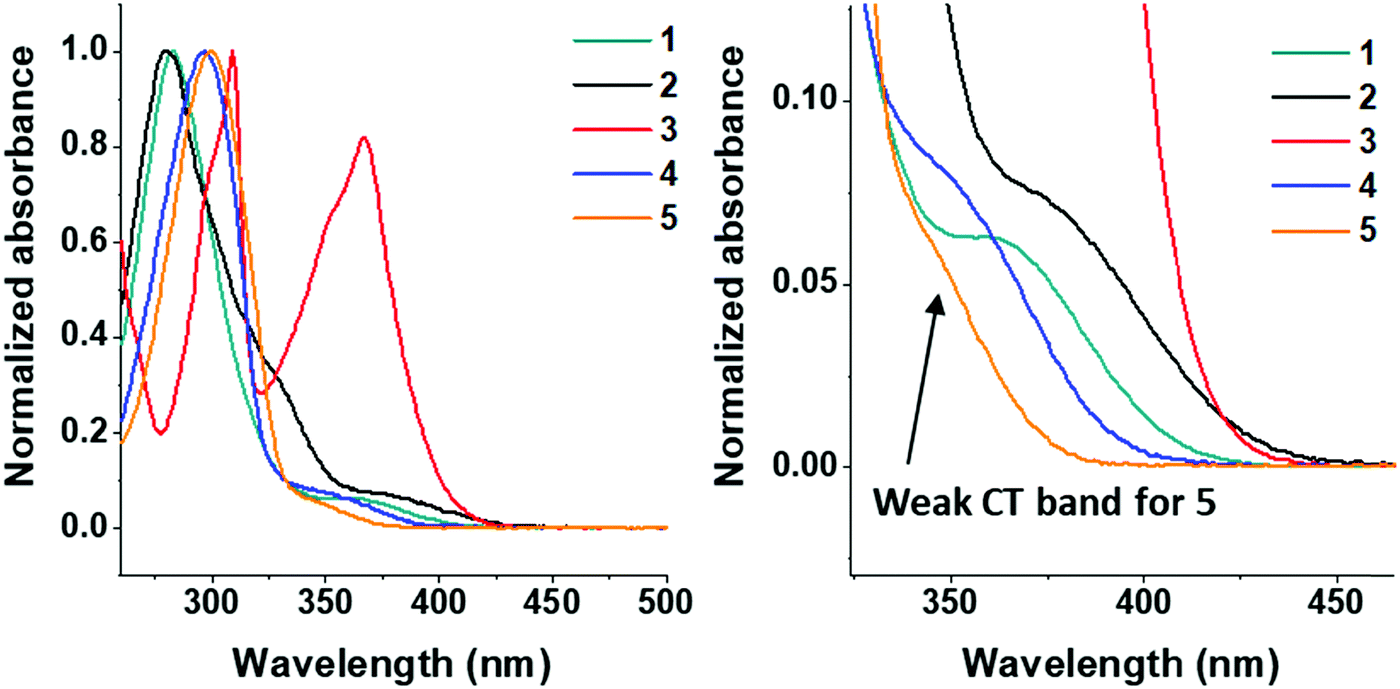 | ||
| Fig. 4 Normalized absorption spectra for molecules 1–5 in DCM solution at 25 μM (left) and 75 μM (right). The weak shoulder attributed to the direct CT absorbance band in 5 is indicated. Higher concentration was used to obtain accurate measurement for onset of CT absorption band. Negligible red-shift in this band between the two concentrations was observed. | ||
All molecules in the series have π–π* transitions in the 275–320 nm region. Molecule 2 has an additional shoulder at 325 nm. Compounds 1–5 have charge transfer (n–π*) transitions at ≈350–375 nm. The dimethoxycarbazole moiety in 3 induces intense absorption bands in the 360–375 nm region,74 which is due to a smaller torsion angle between the D–A units in carbazole functionalized systems (see Fig. 2: <60° dihedral angle for molecule 3 and >70° for molecule 4). The 360–375 nm band shape in 3 is not pure CT in character, i.e. not classically Gaussian; it appears as a normal ππ* transition with some vibronic structure. However, this is very similar to what was previously observed in other carbazole systems,75 where the carbazole units are partially conjugated giving a highly mixed LE/CT ground state and had CT character as shown by the electroabsorption. The previous observations are in agreement with 3 as the D and A are more conjugated in the ground state in 3 with a smaller D–A torsion angle than typically observed in carbazole units (Fig. 2). The highly mixed LE/CT nature of the state here gives rise to the far stronger absorbance on excitation in a polar solvent, the CT character is fully stabilized and emission from 3 falls in sequence with the other compounds.
Steady-state solution photoluminescence
The emission spectra of all compounds were obtained in solution to probe for solvatochromic behaviour.All molecules 1–5 show solvatochromic behavior in solution, with a red-shift in onset of emission ranging from 17–33 nm upon switching from toluene to DCM as solvent (Table 2). Solvatochromism in combination with the broad Gaussian band shapes establishes predominately CT character in the emissive states of molecules 1–5. 1CT emission is an important type of transition character typically required for efficient vibronically coupled TADF. The CF3 functionality is electron withdrawing via inductive effects, yet it has a profound effect of the emission properties of 2–5 when compared with unsubstituted 1. Molecule 5 shows a large blue-shift (36 nm in toluene; 49 nm in DCM) in the emission onset compared to molecule 1 due to the reduced donor strength of 5. In contrast, molecules 2–3 are substituted with CF3 groups on the thioxanthene-S,S-dioxide methylene bridge, and are not directly attached to the aromatic rings. Even in this bridge position, the σ-withdrawing effects of the CF3 group significantly increase the acceptor strength leading to a substantial red-shift (>50 nm at λmax) in emission in 2–3 compared to 1. As a result, CF3 substituents can be strategically positioned to tune the emission onset over the 373–446 nm range (toluene λmax range 410–515 nm). The utilization of CF3 substituents on both donor and acceptor units in molecule 4 results in emission of similar color to unsubstituted 1. The red-shift in the emission of 4 with the stronger CF3 substituted acceptor (as in 2) is offset by the blue-shift associated with weaker donor (as in 5), leading to a similar emission profile as in 1. Unsubstituted 1 has slightly bluer emission in toluene and the emission of 4 is slightly bluer in dichloromethane, suggesting weaker CT emission character in 4 compared to 1. This difference is reflected in the solvatochromic shifts from toluene to dichloromethane in molecules 1 and 4.
| Onset abs. (25 μM)/nma | Onset abs. (75 μM)/nma | Onset em./nmb | Onset em./nma | λ max em./nmb | λ max em./nma | Onset em. solvent shift/nmc | λ max em. solvent shift/nmc | |
|---|---|---|---|---|---|---|---|---|
| a Measured in DCM solvent. b Measured in toluene solvent. c Shift in emission onset or λmax from toluene to DCM solvent. | ||||||||
| 1 | 406 | 407 | 409 | 441 | 452 | 503 | 32 | 51 |
| 2 | 428 | 427 | 446 | 479 | 515 | 572 | 33 | 57 |
| 3 | 413 | 416 | 427 | 457 | 502 | 554 | 30 | 52 |
| 4 | 391 | 391 | 406 | 423 | 462 | 492 | 17 | 30 |
| 5 | 374 | 374 | 373 | 392 | 410 | 441 | 19 | 31 |
Steady-state solid state photoluminescence
Compounds 1–5 were also characterized in three solid state host materials of differing polarities: zeonex, 1,3-bis(triphenylsilyl)benzene (UGH3), and (bis[2-(diphenylphosphino)phenyl]ether oxide) (DPEPO). UGH3 and DPEPO were chosen due to their high triplet energies.All molecules 1–5 show some degree of red-shift in emission from apolar zeonex to DPEPO and UGH3 hosts. This red-shift is mainly due to packing effects in the solid state, as can be seen through the consistent similarity between UGH3 and DPEPO film spectra. Molecule 4 shows a significantly smaller shift in emission color between hosts. It is suggested that the weak CT strength makes 4 less sensitive to polarity of the surrounding medium, as found in solution state emission measurements (Fig. 5). Interestingly, the high degree of hydrophobicity imparted by the more extensive trifluoromethylation on molecule 4 may limit spectral shifts associated with modifications in molecular packing, considering that 5 (with a weaker acceptor than 4) and subsequently weaker charge transfer still significantly shifts from zeonex to DPEPO or UGH3 hosts.
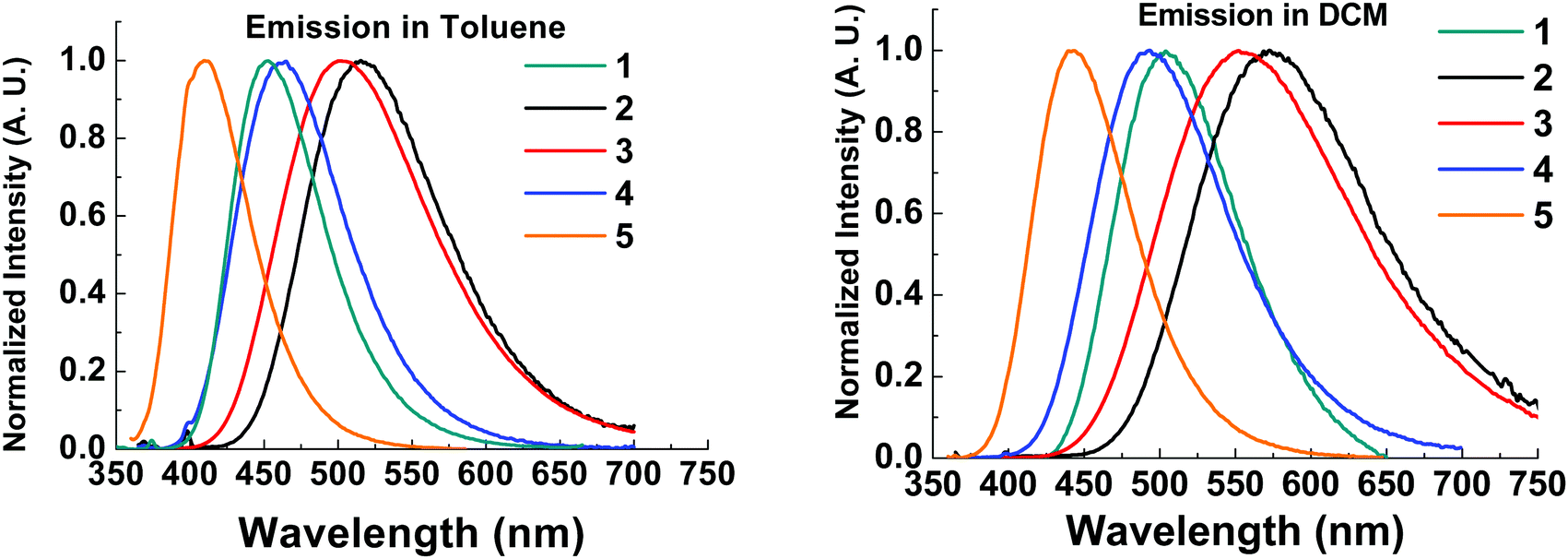 | ||
| Fig. 5 Solution steady state emission spectra in toluene (left) and dichloromethane (right) of compounds 1–5. λexc = 355 nm. | ||
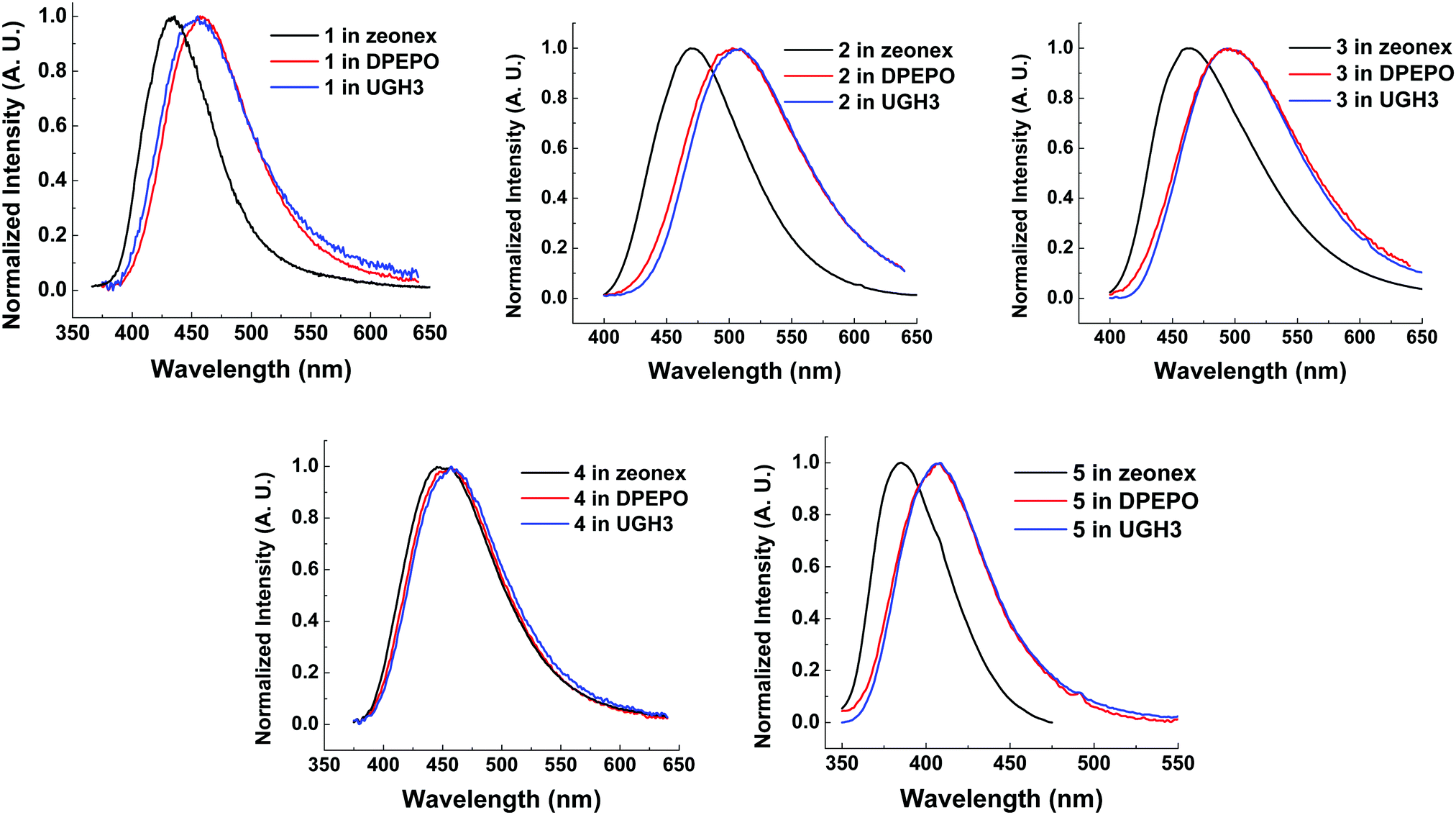 | ||
| Fig. 6 Normalized steady state emission spectra from drop cast films of molecules 1–5 at 10 wt% in solid state hosts at room temperature. λexc = 355 nm. | ||
Time-resolved photoluminescence
Time-resolved emission spectroscopy was performed on films of 1–5 in DPEPO host to determine the effect of trifluoromethyl substituents on the TADF properties of 2–5 in comparison to 1. Fig. 7 shows the time resolved emission decays and Fig. 8 shows snapshots of the emission at various time delays during the time resolved decay.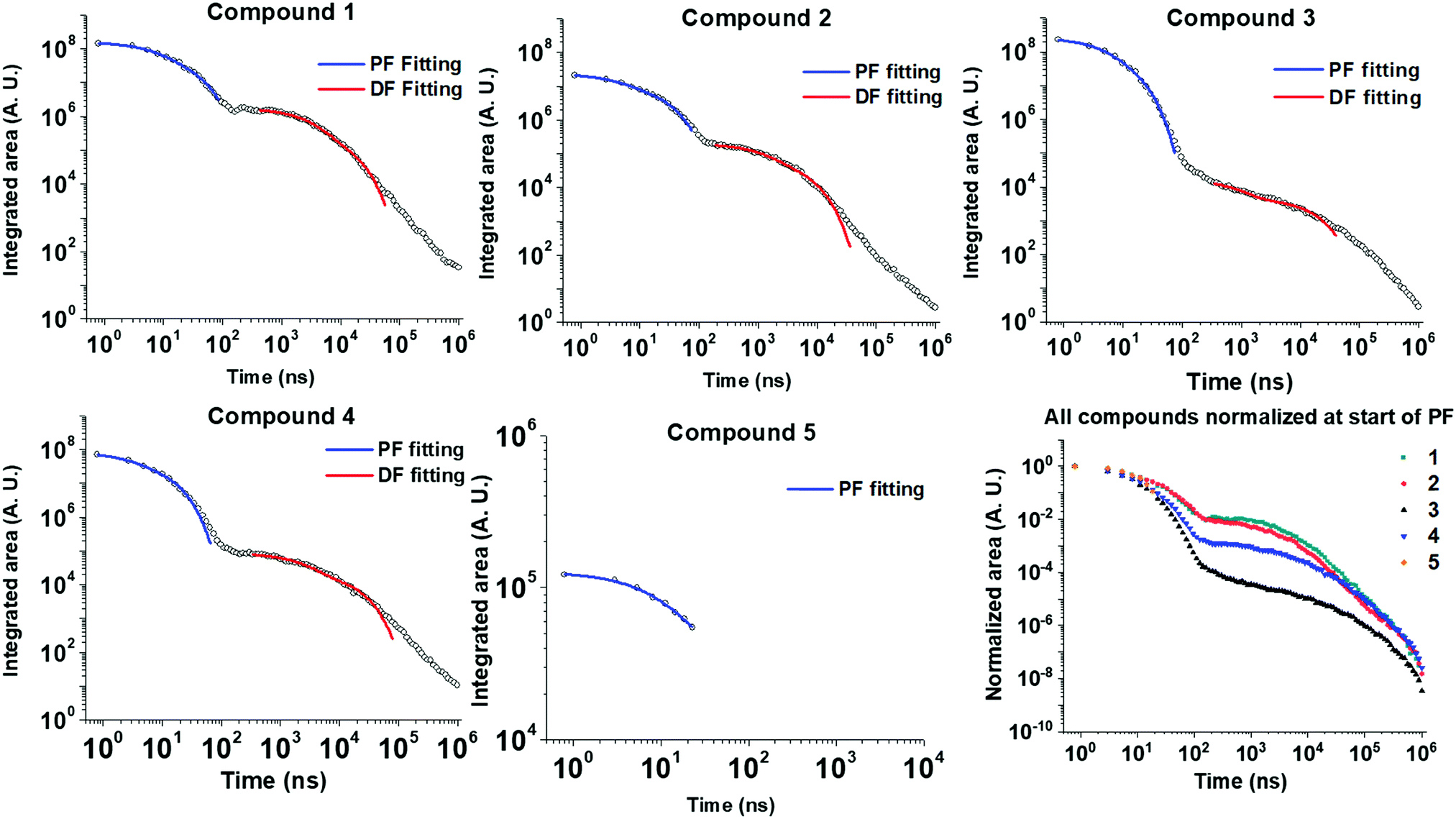 | ||
| Fig. 7 Time-resolved photoluminescence decays of drop-cast films of 1–5 at 10 wt% in DPEPO host matrix. Dual exponential fittings performed for the PF (prompt fluorescence) in the early ns region and for the DF (delayed fluorescence) in the late ns/early μs region. Fitting parameters are shown in Table S1 (ESI†). Lifetimes and corresponding weightings are shown in Table 3. | ||
 | ||
| Fig. 8 Time-resolved photoluminescence spectra of 1–5 in drop cast films at 10 wt% in DPEPO. Spectra are snapshots in time from the data in Fig. 7 at the specific times given each figure panel. A separate measurement was performed at 80 K to obtain the phosphorescence spectrum at 80 ms delay times to give an unambiguous phosphorescence spectrum. | ||
Compounds 1–4 all show charge transfer (CT) emission in the early ns region with a slight red-shift of the emission during in the first 80 ns of decay (Fig. 8 and Table 4). The CT emission at 80 ns representing the prompt fluorescence (PF) closely resembles the delayed fluorescence (DF) emission at 2 μs. This is typical of emission originating from a TADF process, where intersystem crossing to the triplet excited state, and reverse intersystem crossing back to the 1CT state has occurred. Molecules 1–4 have ΔEST gaps ≤ 0.22 eV with varying DF emission lifetimes. Temperature dependence measurements on 2–4 show increased emission intensity in the μs delayed fluorescence region (Fig. S5a and b, ESI†). Power dependence measurements (Fig. S5c, ESI†) rule out multi-exciton emission processes with a slope close to one for 2–4, confirming that vibronically coupled TADF processes are occurring.
The lifetimes for PF and DF emission for 1–4 were determined by fitting with dual exponential decay curves (Fig. 7). Dual exponential curves gave good R2 values (>0.98, ESI†) for all fittings with <10% standard error in all cases (Table S1, ESI†). The emission for 3 and 5 was very weak and consequently the error in fitting the emission is larger. The start of the fitting for the DF was carefully considered to minimize interference from the overlapping PF signal as much as possible at approx. 100 ns. For 1, 2 and 4 both components of the dual exponential decays contribute significantly to the total PF and DF emission (Table 3). This kinetic analysis for 1, 2 and 4 suggests that there are two ‘groups’ of varying conformation/packing effects in the films that result in either a faster or a slower emission lifetime. In most cases the faster component of the DF or PF is the major emission component (Table 3). Unexpectedly, CF3-substituted compound 2 has a faster DF emission lifetime than 1 for both exponential components. This is a highly thought-provoking finding considering the ΔEST values for 1 and 2 are comparable (0.07 vs. 0.08 eV). Interestingly, 4 shows a similar DF lifetime to 1 despite a much larger ΔEST (0.22 eV vs. 0.07 eV). The above discussion brings about the general observation that the introduction of CF3 substituents on the acceptor methylene bridge in 2 induces faster rISC rates than in 1. There are longer components in the spectra of 3 and 4 (Fig. 7) due to the wider ΔEST, but 4 has a rather rapid delayed fluorescence component considering the ΔEST magnitude. The relaxation of the 1CT energy over time (Fig. 8 + relaxed S1 energies Table 4) lowers the 1CT energy closer to the T1 energy, and this may explain why 3 and 4 still have TADF with respectable rates despite the 0.22 eV ΔEST gap.
| # | τ 1 PF/ns | A 1 PF(% cont.)a | τ 2 PF/ns | A 2 PF (% cont.)a | τ 1 DF/μs | B 1 DF (% cont.)b | τ 2 DF/μs | B 2 DF (% cont.)b |
|---|---|---|---|---|---|---|---|---|
| a % contribution of each PF exponential to the total PF contribution based on A1 and A2 values. b % contribution of each DF exponential to the total DF contribution based on B1 and B2 values. c The signal for the delayed fluorescence in 3 is very weak in comparison to molecules 1, 2 and 4 (see intensity in Fig. 7 and 8), and consequently only one lifetime from the fitting of 3 is meaningful due to strong overlapping PF signal at earlier times. | ||||||||
| 1 | 6.5 | 9.1 × 107 (62) | 28.8 | 5.6 × 107 (38) | 2.5 | 1.2 × 106 (81) | 11.6 | 2.9 × 105 (19) |
| 2 | 4.0 | 1.1 × 107 (52) | 24.5 | 1.0 × 107 (48) | 1.1 | 1.2 × 105 (66) | 6.0 | 6.1 × 104 (34) |
| 3 | 3.5 | 1.4 × 108 (62) | 10.9 | 8.7 × 107 (38) | 15.6 | 4.5 × 103 (100)c | — | — |
| 4 | 2.4 | 3.2 × 107 (46) | 11.8 | 3.8 × 107 (54) | 2.2 | 5.1 × 104 (71) | 17.8 | 2.1 × 104 (29) |
| 5 | 12.9 | 7.5 × 104 (100) | — | — | — | — | — | — |
| # | E S1/eV (nm)a | E S1/eV (nm)b | E T1/eV (nm)c | ΔEST/eVd | ΔEST/eVe | λ MAX all Em./nmf | Φ em ± 5/% |
|---|---|---|---|---|---|---|---|
| a Onset of steady state emission in DPEPO host (Fig. 6). b Onset of relaxed CT emission at approx. 75 ns as indicated in Fig. 8 for 1–4. For 5 the relaxed onset was taken at 11 ns due to little signal beyond this time. c Onset of phosphorescence emission at 80 K with a time delay of 80 ms (Fig. 8). d Calculated from steady state emission in DPEPO as in a. T1 energy taken from c e Calculated from relaxed CT emission as in b. T1 energy taken from c. f Taken from steady state emission in DPEPO host (Fig. 6). g Taken from evaporated DPEPO film data from literature.73 h Measured at 25 wt% in DPEPO evaporated film. Evaporated film at higher emitter concentration required in the blue region to obtain strong signal due to scattering and lower detector sensitivity in the blue region of the spectrum. i 1 wt% in zeonex matrix. Signal was too low to be measured in DPEPO film as shown by the signal to noise ratio in Fig. 7. | |||||||
| 1 | 3.08 (403) | 3.00 (413) | 3.01 (412) | 0.07 | −0.01 | 460 | 80g |
| 2 | 2.86 (433) | 2.77 (448) | 2.78 (446) | 0.08 | −0.02 | 504 | 74 |
| 3 | 2.93 (423) | 2.81 (442) | 2.71 (458) | 0.22 | 0.10 | 495 | 46 |
| 4 | 3.14 (395) | 3.05 (406) | 2.92 (425) | 0.22 | 0.13 | 454 | 50h |
| 5 | 3.42 (363) | 3.30 (376) | 3.07 (404) | 0.35 | 0.22 | 407 | 5i |
This raises the question: why is 2 emitting delayed fluorescence faster than 1 when the ΔEST values and structures are so similar? The emission spectrum of 2 is red-shifted compared to 1 (Fig. 5 and 6), highlighting a more stabilized CT state. The triplet (T1) energies of 2–4 are lower compared to 1, showing that the CF3 groups on the acceptor are responsible for the reduction in T1 energy. The discussed analysis suggests that the lower T1 energy is 3CT in character and less localized due to the significantly increased acceptor strength. This change in triplet state character in 2 and 4 is likely to adjust the vibrational modes to the system, and it is proposed that in this case there is efficient mixing of 3LE and 3CT to allow rISC to occur. It is suggested that the differences in triplet state character improve the vibronic mixing of states, resulting in faster delayed fluorescence in 2. This may also explain why 4 exhibits DF with a similar lifetime to 1 despite a wider ΔEST in 4 (Table 4).
Computations
Geometry optimizations on the ground state (S0) geometries of 1–5 at the hybrid DFT functional CAM-B3LYP/6-31G(d) revealed D–A orientations to be orthogonal in all molecules except for 3 where the averaged D–A dihedral angle of approximately 50° is obtained (Table 5). Good agreements between computed and experimental geometries were found for 3 and 4 (Fig. S7a, ESI†) so the optimized geometries for 1, 2 and 5 are likely to be present as major conformers in solutions. Simulated absorption spectra from time-dependent DFT (TD-DFT) data (Fig. S7b, ESI†) on 1–5 are in accord with experimental absorption spectra with the intense mixed 1CT/1LE band for 3 present in the 370 nm region (oscillator strength, f = 0.7). The much greater intensity and strong CT/LE mixing in 3 compared to CT bands in 1, 2, 4 and 5 is due to the different D–A orientations. Constraining the D–A orientations to 90° in 3 results in a CT band with near zero intensity (f = 0.0003) (Fig. S7c and Table S5, ESI†). The perpendicular D–A energy barrier in 3 is estimated to be 2 kcal mol−1 thus a range of conformers of 3 in solution would be present.| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| a Two averaged dihedral angles are reported because of the D–A–D architecture. When 1–5 are excited and charge transfer occurs, only one electron is transferred from a single donor to the central acceptor. In this excited state the other donor is acting as a ‘pendant’ substituent that will play a role in the electronics, but in a more passive fashion compared to the donor that transfers an electron. The ‘pendant’ substituents in the excited state geometries (S1, T1) contain similar dihedral angles to those in the ground state geometries (S0). | |||||
| S0 | 89.6, 89.6 | 84.1, 87.1 | 47.9, 49.1 (46.5) | 84.4, 88.5 (81.3) | 89.6, 89.6 |
| S1 | 85.8, 90.0 | 87.3, 87.8 | 52.2, 87.6 | 88.2, 89.0 | 82.3, 89.5 |
| T1 | 57.7, 89.4 | 55.8, 87.1 | 45.2, 49.3 | 54.7, 85.8 | 54.4, 89.3 |
Excited state geometries of 1–5 were also optimized here by TD-DFT for S1 states and by unrestricted DFT (UDFT) for T1 states with D–A dihedral angles of the optimized geometries listed in Table 5. For all molecules except 3, the D–A orientations remain orthogonal on going from S0 to S1 so a relatively small geometrical rearrangement would take place after excitation and geometry relaxation to the most stable S1 state for each geometry. To gain insight into the observed different emission data for 1–5, results from TD-DFT data carried out on the optimized S1 geometries are analyzed here with the assumption that these S1 geometries are responsible for the charge-transfer emissions and are summarized (Table S8, ESI†).
Table S8 (ESI†) and Fig. 9 show the energies of the S1, T1 and T2 states and their orbital/excitonic characters for 1 and 2. The 3LE state is necessary to allow ISC/rISC processes to occur since the 1CT and 3CT exchange is formally forbidden.28,29 Large spin–orbit couplings (SOC) between 1CT and 3LE (Table S9, ESI†) with vibronic coupling (VC) between 3CT and 3LE do take place if the 3LE state, 3CT and 1CT are close together. Compound 2 has a fast DF rate and T1 with 3CT character, whereas compound 4 has a slower observed DF rate and T1 with 3LE character like 1 (Table 3, and Fig. S7d, ESI†). Comparing compounds 1, 2 and 4, where very small systematic substitutions are made to the structure, it appears that the different T1 character predicted is related to the DF rate where a faster rate occurs when T1 has 3CT character.
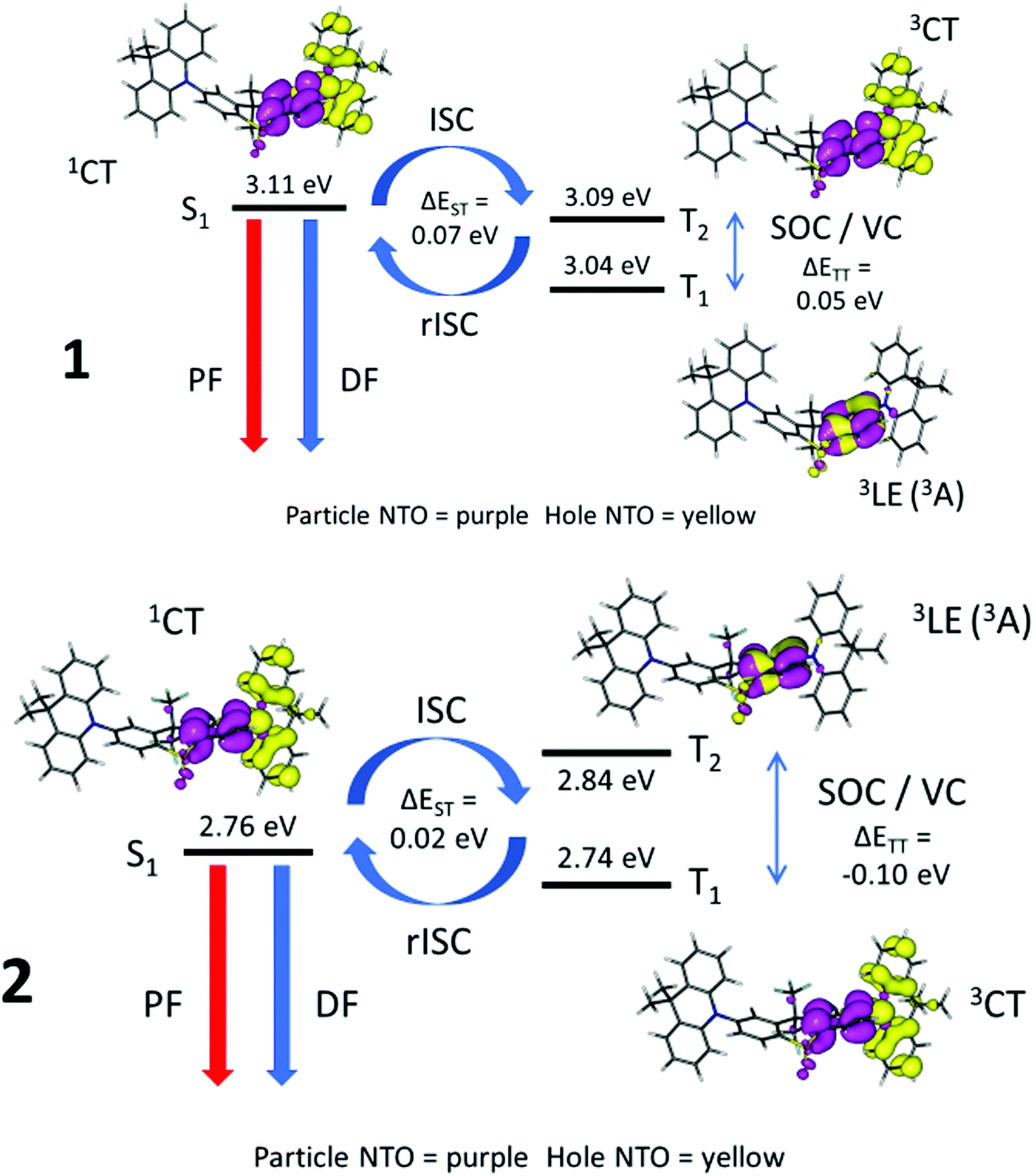 | ||
| Fig. 9 Energy diagrams illustrating TADF in 1 and 2 with natural transition orbitals for each state on optimized S1 excited state geometries from TD-DFT computations. PF = prompt fluorescence, DF = (thermally activated) delayed fluorescence, ISC = intersystem crossing, rISC = reverse intersystem crossing, SOC = spin orbit coupling, VC = vibronic coupling, ΔEST = S1 energy – T1 energy, ΔETT = 3CT energy – 3LE energy, 1CT = singlet charge transfer state, 3CT = triplet charge transfer state, 3LE = local triplet excitation state, 3A = local triplet excitation at the acceptor unit. | ||
Fig. 9 highlights the differences between 1 where the 1CT–3LE gap dominates whereas in 2 the 3CT–3LE gap dominates. Thus in 1 rIC to populate T2 is the rate limiting step, whereas rIC is not rate limiting in 2, and 2 has faster rISC than 1.76
In the case of 3, there is considerable early prompt fluorescence 1CT (Fig. 8) and relatively low DF intensity, making it difficult to make quantitative comparisons in rates between 3 and the other compounds (Fig. 7). If the geometry in 3 contains D–A dihedral angles constrained at 50° in the S1 state, the TD-DFT data provide an entirely different picture where the S1, T1 and T2 states are up to 0.59 eV apart and no rISC process would be expected (Fig. S7e and Table S4, ESI†). For 4, the S1, T1 and T2 states are all within 0.17 eV and the larger energy gap suggests a less efficient rISC process in 4. The nature of the 1CT character in 4 with only 74% CT (and thus 26% local) is much less than the 1CT characters in the other molecules (1–3 and 5) with 93–95% CT. This is in accord with the much weaker solvatochromic effect on the emissions of 4 compared to those of 1–3 and 5. Compound 5 has no observable TADF as expected with the large S1–T1 gap of 0.31 eV from TD-DFT (Fig. S7f, ESI†) and 0.35 eV from experimental measurements. In general, the TD-DFT data is in good agreement with the experimental results and supports the proposed differences in TADF rates.
Device fabrication
OLEDs using compounds 2 and 4 in various hosts (Section S5, ESI†) were found to be unstable, with brightness dropping below half its initial value within seconds at constant voltage driving. Simultaneous drops in device current were observed at constant voltage, and despite the reasonable PLQY values, device EQEs were below 1%. This emitter instability is in accordance with the irreversibility of the reduction in the electrochemical measurements (Fig. 3). Considering that 1 produces more stable devices,65 it is likely that the acceptor is responsible for the instability in the OLED devices of 2 and 4. The instability is likely to be due to the very high acceptor strength which results in cleavage of a C–C or C–S bond of the acceptor on reduction, rather than instability of the CF3 functionality itself.Conclusions and outlook
The CF3-substituted acceptor in 2–4 has been an invaluable structural tool for probing the effects of orbital/exciton character on the rates of TADF emission to further understand the TADF mechanism and improve future molecular design. A key finding is that changing the character of the TADF-active singlet and triplet states through tuning with trifluoromethyl substituents improves the rate of TADF with rapid delayed emission components in the early microsecond regime. This work highlights how changes in the orbital character of singlet and triplet states (local vs. charge transfer) lead to significant changes in rates of delayed fluorescence, despite the molecules possessing similar ΔEST values. The change in orbital character is supported by TD-DFT calculations. These results open up new important considerations for the design of TADF emitters where the ordering of the excited states has an impact on the rates of delayed fluorescence. To further enhance the rate of delayed fluorescence, and thereby reduce the probability of triplet interactions which cause detrimental annihilation effects, it is necessary to gain a deeper understanding of how singlet and triplet state character and orbital distribution affect emission rates. This will be a focus of future studies.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. J. S. W. wrote the main body of the manuscript with assistance from all authors. J. S. W. designed the molecules and performed all the chemical synthesis and characterization under the supervision of M. R. B. A. D. performed the majority of the photophysical measurements with assistance from P. S. under the supervision of A. P. M. Processing of the photophysical data was performed by J. S. W., A. D. and P. S. M. A. F. performed and reported the DFT calculations. A. S. B. performed X-ray diffraction experiments and analysis on crystals grown by J. S. W. OLED devices were fabricated by A. D. Solution electrochemistry measurements were performed by J. S. W. and P. S. The funding for this work was obtained jointly by M. R. B. and A. P. M.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank EPSRC grant EP/L02621X/1 and EU Horizon 2020 grant agreement number 732013 (HyperOLED) for funding. Dr Paloma Lays dos Santos is thanked for technical assistance. Dr Luke O’ Driscoll is thanked for useful synthetic discussions. Dr Kleitos Stavrou and Dr Nils Haase are thanked for useful photophysical discussions.References
- R. Mertens, The OLED handbook. A Guide to OLED Technology, Industry and Market, http://www.oled-info.com/handbook, 2020, (accessed 4-6-20) Search PubMed.
- S. Reineke, M. Thomschke, B. Lussem and K. Leo, White organic light-emitting diodes: Status and perspective, Rev. Mod. Phys., 2013, 85, 1245–1293 CrossRef CAS.
- S. Reineke, Complementary LED technologies, Nat. Mater., 2015, 14, 459–462 CrossRef CAS PubMed.
- M. Segal, M. A. Baldo, R. J. Holmes, S. R. Forrest and Z. G. Soos, Excitonic singlet-triplet ratios in molecular and polymeric organic materials, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 075211 CrossRef.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Highly efficient phosphorescent emission from organic electroluminescent devices, Nature, 1998, 395, 151–154 CrossRef CAS.
- X. L. Yang, G. J. Zhou and W. Y. Wong, Functionalization of phosphorescent emitters and their host materials by main-group elements for phosphorescent organic light-emitting devices, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC.
- Iridium(III) in Optoelectronic and Photonics Applications, ed. E. Zysman-Colman, John Wiley & Sons, 2017 Search PubMed.
- M. J. Jurow, A. Bossi, P. I. Djurovich and M. E. Thompson, In Situ Observation of Degradation by Ligand Substitution in Small-Molecule Phosphorescent Organic Light-Emitting Diodes, Chem. Mater., 2014, 26, 6578–6584 CrossRef CAS.
- C. A. Parker and C. G. Hatchard, Triplet-Singlet Emission in Fluid Solutions - Phosphorescence of Eosin, Trans. Faraday Soc., 1961, 57, 1894–1904 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly efficient organic light-emitting diodes from delayed fluorescence, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- C. Adachi, Third-generation organic electroluminescence materials, Jpn. J. Appl. Phys., 2014, 53, 060101 CrossRef.
- Y. Im, M. Kim, Y. J. Cho, J. A. Seo, K. S. Yook and J. Y. Lee, Molecular Design Strategy of Organic Thermally Activated Delayed Fluorescence Emitters, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Thermally Activated Delayed Fluorescence Materials Towards the Breakthrough of Organoelectronics, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- M. Y. Wong and E. Zysman-Colman, Purely Organic Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Y. C. Liu, C. S. Li, Z. J. Ren, S. K. Yan and M. R. Bryce, All-organic thermally activated delayed fluorescence materials for organic light-emitting diodes, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- X. Xiong, F. Song, J. Wang, Y. Zhang, Y. Xue, L. Sun, N. Jiang, P. Gao, L. Tian and X. Peng, Thermally activated delayed fluorescence of fluorescein derivative for time-resolved and confocal fluorescence imaging, J. Am. Chem. Soc., 2014, 136, 9590–9597 CrossRef CAS PubMed.
- N. R. Paisley, C. M. Tonge and Z. M. Hudson, Stimuli-Responsive Thermally Activated Delayed Fluorescence in Polymer Nanoparticles and Thin Films: Applications in Chemical Sensing and Imaging, Front. Chem., 2020, 8, 229 CrossRef PubMed.
- A. Steinegger, I. Klimant and S. M. Borisov, Purely Organic Dyes with Thermally Activated Delayed Fluorescence—A Versatile Class of Indicators for Optical Temperature Sensing, Adv. Opt. Mater., 2017, 5, 1700372 CrossRef.
- C. J. Christopherson, D. M. Mayder, J. Poisson, N. R. Paisley, C. M. Tonge and Z. M. Hudson, 1,8-Naphthalimide-Based Polymers Exhibiting Deep-Red Thermally Activated Delayed Fluorescence and Their Application in Ratiometric Temperature Sensing, ACS Appl. Mater. Interfaces, 2020, 12, 6525–6535 CrossRef PubMed.
- Z. J. Yu, W. Y. Lou, H. Junge, A. Papcke, H. Chen, L. M. Xia, B. Xu, M. M. Wang, X. J. Wang, Q. A. Wu, B. Y. Lou, S. Lochbrunner, M. Beller and S. P. Luo, Thermally activated delayed fluorescence (TADF) dyes as efficient organic photosensitizers for photocatalytic water reduction, Catal. Commun., 2019, 119, 11–15 CrossRef CAS.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Triplet Harvesting with 100% Efficiency by Way of Thermally Activated Delayed Fluorescence in Charge Transfer OLED Emitters, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- R. J. Huang, J. Avo, T. Northey, E. Chaning-Pearce, P. L. dos Santos, J. S. Ward, P. Data, M. K. Etherington, M. A. Fox, T. J. Penfold, M. N. Berberan-Santos, J. C. Lima, M. R. Bryce and F. B. Dias, The contributions of molecular vibrations and higher triplet levels to the intersystem crossing mechanism in metal-free organic emitters, J. Mater. Chem. C, 2017, 5, 6269–6280 RSC.
- R. J. Huang, N. Kukhta, J. S. Ward, A. Danos, A. S. Batsanov, M. R. Bryce and F. B. Dias, Balancing charge-transfer strength and triplet states for deep-blue thermally activated delayed fluorescence with an unconventional electron rich dibenzothiophene acceptor, J. Mater. Chem. C, 2019, 7, 13224–13234 RSC.
- Y. W. Zhang, D. D. Zhang, J. B. Wei, Z. Y. Liu, Y. Lu and L. Duan, Multi-Resonance Induced Thermally Activated Delayed Fluorophores for Narrowband Green OLEDs, Angew. Chem., Int. Ed., 2019, 58, 16912–16917 CrossRef CAS PubMed.
- M. Sarma and K. T. Wong, Exciplex: An Intermolecular Charge-Transfer Approach for TADF, ACS Appl. Mater. Interfaces, 2018, 10, 19279–19304 CrossRef CAS PubMed.
- B. Zhao, T. Y. Zhang, B. Chu, W. L. Li, Z. S. Su, Y. S. Luo, R. G. Li, X. W. Yan, F. M. Jin, Y. Gao and H. R. Wu, Highly efficient tandem full exciplex orange and warm white OLEDs based on thermally activated delayed fluorescence mechanism, Org. Electron., 2015, 17, 15–21 CrossRef CAS.
- D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. Rivett, S. Jones, T. H. Thomas, M. Abdi Jalebi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, High-performance light-emitting diodes based on carbene-metal-amides, Science, 2017, 356, 159–163 CrossRef CAS PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, The Importance of Vibronic Coupling for Efficient Reverse Intersystem Crossing in Thermally Activated Delayed Fluorescence Molecules, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Revealing the spin–vibronic coupling mechanism of thermally activated delayed fluorescence, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- P. K. Samanta, D. Kim, V. Coropceanu and J. L. Bredas, Up-Conversion Intersystem Crossing Rates in Organic Emitters for Thermally Activated Delayed Fluorescence: Impact of the Nature of Singlet vs Triplet Excited States, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS PubMed.
- P. L. dos Santos, M. K. Etherington and A. P. Monkman, Chemical and conformational control of the energy gaps involved in the thermally activated delayed fluorescence mechanism, J. Mater. Chem. C, 2018, 6, 4842–4853 RSC.
- N. A. Kukhta, A. S. Batsanov, M. R. Bryce and A. P. Monkman, Importance of Chromophore Rigidity on the Efficiency of Blue Thermally Activated Delayed Fluorescence Emitters, J. Phys. Chem. C, 2018, 122, 28564–28575 CrossRef CAS.
- G. Valchanov, A. Ivanova, A. Tadjer, D. Chercka and M. Baumgarten, Understanding the Fluorescence of TADF Light-Emitting Dyes, J. Phys. Chem. A, 2016, 120, 6944–6955 CrossRef CAS PubMed.
- R. Pashazadeh, P. Pander, A. Lazauskas, F. B. Dias and J. V. Grazulevicius, Multicolor Luminescence Switching and Controllable Thermally Activated Delayed Fluorescence Turn on/Turn off in Carbazole-Quinoxaline-Carbazole Triads, J. Phys. Chem. Lett., 2018, 9, 1172–1177 CrossRef CAS PubMed.
- N. A. Kukhta, H. F. Higginbotham, T. Matulaitis, A. Danos, A. N. Bismillah, N. Haase, M. K. Etherington, D. S. Yufit, P. R. McGonigal, J. V. Gražulevičius and A. P. Monkman, Revealing resonance effects and intramolecular dipole interactions in the positional isomers of benzonitrile-core thermally activated delayed fluorescence materials, J. Mater. Chem. C, 2019, 7, 9184–9194 RSC.
- L. S. Cui, H. Nomura, Y. Geng, J. U. Kim, H. Nakanotani and C. Adachi, Controlling Singlet–Triplet Energy Splitting for Deep-Blue Thermally Activated Delayed Fluorescence Emitters, Angew. Chem., Int. Ed., 2017, 56, 1571–1575 CrossRef CAS PubMed.
- R. S. Nobuyasu, J. S. Ward, J. Gibson, B. A. Laidlaw, Z. J. Ren, P. Data, A. S. Batsanov, T. J. Penfold, M. R. Bryce and F. B. Dias, The influence of molecular geometry on the efficiency of thermally activated delayed fluorescence, J. Mater. Chem. C, 2019, 7, 6672–6684 RSC.
- T. T. Bui, F. Goubard, M. Ibrahim-Ouali, D. Gigmes and F. Dumur, Recent advances on organic blue thermally activated delayed fluorescence (TADF) emitters for organic light-emitting diodes (OLEDs), Beilstein J. Org. Chem., 2018, 14, 282–308 CrossRef CAS PubMed.
- J. A. Seo, Y. R. Im, S. H. Han, C. W. Lee and J. Y. Lee, Unconventional Molecular Design Approach of High-Efficiency Deep Blue Thermally Activated Delayed Fluorescent Emitters Using lndolocarbazole as an Acceptor, ACS Appl. Mater. Interfaces, 2017, 9, 37864–37872 CrossRef CAS PubMed.
- I. S. Park, H. Komiyama and T. Yasuda, Pyrimidine-based twisted donor-acceptor delayed fluorescence molecules: a new universal platform for highly efficient blue electroluminescence, Chem. Sci., 2017, 8, 953–960 RSC.
- Y. J. Cho, S. K. Jeon, S. S. Lee, E. Yu and J. Y. Lee, Donor Interlocked Molecular Design for Fluorescence-like Narrow Emission in Deep Blue Thermally Activated Delayed Fluorescent Emitters, Chem. Mater., 2016, 28, 5400–5405 CrossRef CAS.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. P. Ni, Y. Ono and T. Ikuta, Ultrapure Blue Thermally Activated Delayed Fluorescence Molecules: Efficient HOMO-LUMO Separation by the Multiple Resonance Effect, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- R. P. Gounder, D. R. Martir and E. Zysman-Colman, Pyridine-functionalized carbazole donor and benzophenone acceptor design for thermally activated delayed fluorescence emitters in blue organic light-emitting diodes, J. Photonics Energy, 2018, 8, 032106 Search PubMed.
- J. W. Sun, J. Y. Baek, K. H. Kim, C. K. Moon, J. H. Lee, S. K. Kwon, Y. H. Kim and J. J. Kim, Thermally Activated Delayed Fluorescence from Azasiline Based Intramolecular Charge-Transfer Emitter (DTPDDA) and a Highly Efficient Blue Light Emitting Diode, Chem. Mater., 2015, 27, 6675–6681 CrossRef CAS.
- X. D. Cao, D. Zhang, S. M. Zhang, Y. T. Tao and W. Huang, CN-Containing donor-acceptor-type small-molecule materials for thermally activated delayed fluorescence OLEDs, J. Mater. Chem. C, 2017, 5, 7699–7714 RSC.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, The Role of Local Triplet Excited States and D-A Relative Orientation in Thermally Activated Delayed Fluorescence: Photophysics and Devices, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
- J. S. Ward, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, J. Santos, F. B. Dias and M. R. Bryce, Bond rotations and heteroatom effects in donor-acceptor-donor molecules: Implications for thermally activated delayed fluorescence and room temperature phosphorescence, J. Org. Chem., 2018, 83, 14431–14442 CrossRef CAS PubMed.
- Y. Yuan, X. Tang, X.-Y. Du, Y. Hu, Y.-J. Yu, Z.-Q. Jiang, L.-S. Liao and S.-T. Lee, The Design of Fused Amine/Carbonyl System for Efficient Thermally Activated Delayed Fluorescence: Novel Multiple Resonance Core and Electron Acceptor, Adv. Opt. Mater., 2019, 7, 1801536 CrossRef.
- Y. P. Xiang, S. L. Gong, Y. B. Zhao, X. J. Yin, J. J. Luo, K. L. Wu, Z. H. Lu and C. L. Yang, Asymmetric-triazine-cored triads as thermally activated delayed fluorescence emitters for high-efficiency yellow OLEDs with slow efficiency roll-off, J. Mater. Chem. C, 2016, 4, 9998–10004 RSC.
- M. W. Cooper, X. Q. Zhang, Y. D. Zhang, C. Fuentes-Hernandez, S. Barlow, B. Kippelen and S. R. Marder, Control of Singlet Emission Energy in a Diphenyloxadiazole Containing Fluorophore Leading To Thermally Activated Delayed Fluorescence, ACS Omega, 2018, 3, 14918–14923 CrossRef CAS PubMed.
- S. Lee, S. O. Kim, H. Shin, H. J. Yun, K. Yang, S. K. Kwon, J. J. Kim and Y. H. Kim, Deep-Blue Phosphorescence from Perfluoro Carbonyl-Substituted Iridium Complexes, J. Am. Chem. Soc., 2013, 135, 14321–14328 CrossRef CAS PubMed.
- G. Grybauskaite-Kaminskiene, K. Ivaniuk, G. Bagdziunas, P. Turyk, P. Stakhira, G. Baryshnikov, D. Volyniuk, V. Cherpak, B. Minaev, Z. Hotra, H. Agren and J. V. Grazulevicius, Contribution of TADF and exciplex emission for efficient “warm-white” OLEDs, J. Mater. Chem. C, 2018, 6, 1543–1550 RSC.
- J. P. Amara and T. M. Swager, Conjugated polymers with geminal trifluoromethyl substituents derived from hexafluoroacetone, Macromolecules, 2006, 39, 5753–5759 CrossRef CAS.
- M. Tavasli, T. N. Moore, Y. H. Zheng, M. R. Bryce, M. A. Fox, G. C. Griffiths, V. Jankus, H. A. Al-Attar and A. P. Monkman, Colour tuning from green to red by substituent effects in phosphorescent tris-cyclometalated iridium(III) complexes of carbazole-based ligands: synthetic, photophysical, computational and high efficiency OLED studies, J. Mater. Chem., 2012, 22, 6419–6428 RSC.
- H. Li, J. Li, D. Liu, T. Huang and D. Li, Effects of Electron Affinity and Steric Hindrance of the Trifluoromethyl Group on the π-Bridge in Designing Blue Thermally Activated Delayed Fluorescence Emitters, Chem. – Eur. J., 2020, 26, 6899–6909 CrossRef CAS PubMed.
- X. Liang, H. B. Han, Z. P. Yan, L. Liu, Y. X. Zheng, H. Meng and W. Huang, Versatile functionalization of trifluoromethyl based deep blue thermally activated delayed fluorescence materials for organic light emitting diodes, New J. Chem., 2018, 42, 4317–4323 RSC.
- Z. L. Lu, D. D. Fang, Y. Y. Zheng, Y. Q. Jin and B. X. Wang, Preparations and photophysical properties of thermally activated delayed fluorescence materials based on N-phenyl-phenothiazine-S,S-dioxide, Tetrahedron, 2017, 73, 21–29 CrossRef CAS.
- L. Mei, J. Hu, X. D. Cao, F. F. Wang, C. Zheng, Y. T. Tao, X. W. Zhang and W. Huang, The inductive-effect of electron withdrawing trifluoromethyl for thermally activated delayed fluorescence: tunable emission from tetra- to penta-carbazole in solution processed blue OLEDs, Chem. Commun., 2015, 51, 13024–13027 RSC.
- W. B. Yuan, M. C. Zhang, X. P. Zhang, X. D. Cao, N. Sun, S. G. Wan and Y. T. Tao, The electron inductive effect of CF3 on penta-carbazole containing blue emitters: Trade-off between color purity and luminescent efficiency in TADF OLEDs, Dyes Pigm., 2018, 159, 151–157 CrossRef CAS.
- X. Cao, J. Hu, Y. Tao, W. Yuan, J. Jin, X. Ma, X. Zhang and W. Huang, Alkyl effects on the optoelectronic properties of bicarbazole/cyanobenzene hybrid host materials: Double delayed fluorescent host/dopant systems in solution-processed OLEDs, Dyes Pigm., 2017, 136, 543–552 CrossRef CAS.
- M. Yokoyama, K. Inada, Y. Tsuchiya, H. Nakanotani and C. Adachi, Trifluoromethane modification of thermally activated delayed fluorescence molecules for high-efficiency blue organic light-emitting diodes, Chem. Commun., 2018, 54, 8261–8264 RSC.
- R. Keruckiene, M. Guzauskas, E. Narbutaitis, U. Tsiko, D. Volyniuk, P.-H. Lee, C.-H. Chen, T.-L. Chiu, C.-F. Lin, J.-H. Lee and J. V. Grazulevicius, Exciplex-forming derivatives of 2,7-di-tert-butyl-9,9-dimethylacridan and benzotrifluoride for efficient OLEDs, Org. Electron., 2020, 78, 105576 CrossRef CAS.
- Y. Geng, A. D'Aleo, K. Inada, L. S. Cui, J. U. Kim, H. Nakanotani and C. Adachi, Donor-σ-Acceptor Motifs: Thermally Activated Delayed Fluorescence Emitters with Dual Upconversion, Angew. Chem. Int. Ed., 2017, 56, 16536–16540 CrossRef CAS PubMed.
- I. Lee and J. Y. Lee, Molecular design of deep blue fluorescent emitters with 20% external quantum efficiency and narrow emission spectrum, Org. Electron., 2016, 29, 160–164 CrossRef CAS.
- P. L. dos Santos, J. S. Ward, M. R. Bryce and A. P. Monkman, Using Guest-Host Interactions To Optimize the Efficiency of TADF OLEDs, J. Phys. Chem. Lett., 2016, 7, 3341–3346 CrossRef CAS PubMed.
- M. M. Heravi, Z. Kheilkordi, V. Zadsirjan, M. Heydari and M. Malmir, Buchwald-Hartwig reaction: An overview, J. Organomet. Chem., 2018, 861, 17–104 CrossRef CAS.
- G. Yzambart, L. Rincon-Garcia, A. A. Al-Jobory, A. K. Ismael, G. Rubio-Bollinger, C. J. Lambert, N. Agrait and M. R. Bryce, Thermoelectric Properties of 2,7-Dipyridylfluorene Derivatives in Single-Molecule Junctions, J. Phys. Chem. C, 2018, 122, 27198–27204 CrossRef CAS PubMed.
- HCl in AcOH did not yield any conversion to product, presumably due to the highly unstable bis-CF3 cation that would be required to close the ring. Treatment of 9 with pyridine in SOCl2 used to close literature fluorene derivatives53 was not suitable here due to the electron rich nature of the arylthioether rings, resulting in aryl chlorination (observed by GCMS-EI) instead of the desired ring closure.
- P. L. Santos, J. S. Ward, P. Data, A. S. Batsanov, M. R. Bryce, F. B. Dias and A. P. Monkman, Engineering the singlet-triplet energy splitting in a TADF molecule, J. Mater. Chem. C, 2016, 4, 3815–3824 RSC.
- L. Mahendar and G. Satyanarayana, Copper catalyzed coupling of protecting group free and sterically hindered 2-bromobenzyl tertiary alcohols with phenols and anilines: facile synthesis of xanthenes and dihydroacridines, RSC Adv., 2016, 6, 20588–20597 RSC.
- P. L. dos Santos, J. S. Ward, D. G. Congrave, A. S. Batsanov, J. Eng, J. E. Stacey, T. J. Penfold, A. P. Monkman and M. R. Bryce, Triazatruxene: A Rigid Central Donor Unit for a D–A3 Thermally Activated Delayed Fluorescence Material Exhibiting Sub-Microsecond Reverse Intersystem Crossing and Unity Quantum Yield via Multiple Singlet–Triplet State Pairs, Adv. Sci., 2018, 5, 1700989 CrossRef PubMed.
- P. L. dos Santos, J. S. Ward, A. S. Batsanov, M. R. Bryce and A. P. Monkman, Optical and Polarity Control of Donor-Acceptor Conformation and Their Charge-Transfer States in Thermally Activated Delayed-Fluorescence Molecules, J. Phys. Chem. C, 2017, 121, 16462–16469 CrossRef CAS.
- P. Stachelek, J. S. Ward, P. L. dos Santos, A. Danos, M. Colella, N. Haase, S. J. Raynes, A. S. Batsanov, M. R. Bryce and A. P. Monkman, Molecular Design Strategies for Color Tuning of Blue TADF Emitters, ACS Appl. Mater. Interfaces, 2019, 11, 27125–27133 CrossRef CAS PubMed.
- S. H. Wu, M. Aonuma, Q. S. Zhang, S. P. Huang, T. Nakagawa, K. Kuwabara and C. Adachi, High-efficiency deep-blue organic light-emitting diodes based on a thermally activated delayed fluorescence emitter, J. Mater. Chem. C, 2014, 2, 421–424 RSC.
- D. de Sa Pereira, C. Menelaou, A. Danos, C. Marian and A. P. Monkman, Electroabsorption Spectroscopy as a Tool for Probing Charge Transfer and State Mixing in Thermally Activated Delayed Fluorescence Emitters, J. Phys. Chem. Lett., 2019, 10, 3205–3211 CrossRef CAS PubMed.
- J. Gibson and T. J. Penfold, Nonadiabatic coupling reduces the activation energy in thermally activated delayed fluorescence, Phys. Chem. Chem. Phys., 2017, 19, 8428–8434 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization details of the new molecules; methods and results for the photophysics, OLED devices and calculations (PDF). X-ray crystallographic data. CCDC 1887606 (3) and 1887607 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qm00429d |
| This journal is © the Partner Organisations 2020 |