
Structural evolution of BCN systems from graphene oxide towards electrocatalytically active atomic layers†
 *a
and
*a
and
Abstract
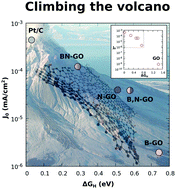
The boron–carbon–nitrogen (B–C–N) ternary phase diagram is rich with several stable phases. But experimentally exploring the vast array of plausible phases in atomically thin layers is quite challenging. BCN structures vary in nature with the method of preparation, which contributes to differences in their physical properties, particularly while studying photo/electrocatalytic applications. Here, we present a molecular dynamics (MD) approach to study the structural evolution of BCN systems, using the reactive force field (ReaxFF). ReaxFF-based MD simulations allow observation of bond breaking/making in BCN systems, in real time. This helps unravel physically stable structures for B,N-singly and co-doped GO. These predicted structures are subsequently realized in experiments, and characterised via various spectroscopic techniques. We further study the electrochemical activity of these structures for the hydrogen evolution reaction. Specifically, in B,N co-doped systems, we find that the electrocatalytic activity changes considerably if we consider separated doping or domain doping. Together, by combining simulation and experiments, this work shows a structure–property relationship of BCN structures, which ultimately governs their electrocatalytic activity towards the hydrogen evolution reaction.
 Please wait while we load your content...
Please wait while we load your content...

