
Porous organic polymers: a promising platform for efficient photocatalysis
 *a
*a
Abstract
Porous organic polymers (POPs) are a class of multi-dimensional porous network materials, which are built via strong covalent linkages between various organic building blocks with different geometries and topologies, and have recently become a rising research field in porous materials. POPs can be generally divided into two categories based on their degree of long-range order, including amorphous (e.g., CMPs, HCPs, PIMs, and PAFs) and crystalline (e.g., COFs). Owning to their advantages of light-weight, superior inherent porosity, excellent stability, pre-designable and tunable structures and functions, POPs have received increasing attention and research interest for their tremendous potential applications in gas storage/separation, heterogeneous catalysis, photoelectric conversion, chemical- and bio-sensing, energy storage and conversion, etc. Their porous structure, pore size, specific surface areas and functions can be directly designed and facilely tuned by introducing specific functional building blocks. In this review article, we summarize the latest representative advances in the field of POPs, focusing on their design and synthetic strategies, with emphasis on their specific applications in photocatalysis, including photocatalytic chemical transformation, photodetoxification of contaminants from water, and water splitting.
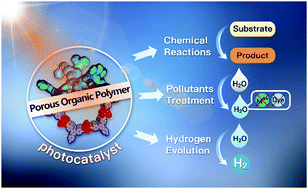
- This article is part of the themed collection: Materials Chemistry in Tianjin University
 Please wait while we load your content...
Please wait while we load your content...

