
Aggregation-induced emission characteristics of o-carborane-functionalized fluorene and its heteroanalogs: the influence of heteroatoms on photoluminescence†
Xueyan
Wu
a,
Jixi
Guo
 *a,
Yan
Lv
a,
Dianzeng
Jia
*a,
Jianzhang
Zhao
ab,
Huici
Shan
a,
Xiaoping
Jin
a and
Yongdong
Ma
a
*a,
Yan
Lv
a,
Dianzeng
Jia
*a,
Jianzhang
Zhao
ab,
Huici
Shan
a,
Xiaoping
Jin
a and
Yongdong
Ma
a
aKey Laboratory of Energy Materials Chemistry, Ministry of Education, Key Laboratory of Advanced Functional Materials, Autonomous Region, Institute of Applied Chemistry, Xinjiang University, Urumqi 830046, P. R. China. E-mail: jxguo1012@163.com; jdz@xju.edu.cn
bState Key Laboratory of Fine Chemicals, Dalian University of Technology, 2 Ling gong Road, Dalian 116024, P. R. China
First published on 19th November 2019
Abstract
o-Carborane was used to alter the electronic states of π-conjugated organic aryls and was demonstrated as an effective optical and electronic control unit to tune the photophysical properties, endowing the molecules with certain luminescence properties and modifying the HOMO and LUMO energies for electron transfer. We introduced fluorene and its heteroanalogs (carbazole, dibenzofuran and dibenzothiophene), four types of substitutes with different electron-rich or electron-deficient heteroatoms, to the two carbon atoms of o-carborane to synthesize CDC, CDN, CDO and CDSvia a modified nickel-catalyzed cross-coupling reaction. The photophysical properties of o-carborane-containing heterocyclic compounds were investigated and compared with each other. The results showed that all compounds were AIE luminogens with tunable fluorescence colors from blue to yellow emissions (λem = 465–570 nm) and possessed excellent emission quantum efficiencies in the solid state (ΦF > 70%). Among the four o-carborane-containing heterocyclic compounds, the ground state structures confirmed by crystallographic measurements provided a close correlation of the structure and electronic properties between the π-conjugated organic aryl groups and o-carborane. Besides, the heteroatoms controlled and altered the electronic interaction between aryls and o-carborane due to the different electron-rich or electron-deficient attributes. The HOMO and LUMO energies were estimated by DFT calculations. The excited states were calculated by TD-DFT and correlated with the corresponding absorption and emission spectra to systematically elucidate the electronic perturbation. This work not only demonstrates an efficient strategy for constructing o-carborane-containing heterocyclic AIE luminogens but also indicates that they are promising as advanced optoelectronic materials.
Introduction
With the tremendous growth in the full-color display technology, it is imperative to meet the increasing demand for high-performance luminescent materials. Solid-state luminescent materials, which possess excellent photoluminescence (PL) quantum yields, high electroluminescence efficiencies, and small efficiency roll-off, are becoming hot topics of current research worldwide.1–3 However, the practical application of luminescent materials has long been a challenge because of their aggregation-caused quenching (ACQ), which has become an obstacle for their high-tech applications.4–7 Thus, a lot of research efforts have been devoted to develop luminescent materials with high PL quantum yields both in the solid and aggregated states. In 2001, Tang and co-workers proposed the concept of aggregation-induced emission (AIE), which emphasized that the AIE materials significantly promote bright luminescence in the aggregated and solid states.4 By realizing light emission in the solid state that is crucial for their practical application, the AIE fluorophores have an impressive impact on the fields of energy storage and conversion, optoelectronics, life science, public security and environment.7–12 Many groups have conducted in-depth studies in this field and developed various types of new AIE luminogens. Among them, organoboron compounds and boron complexes are beginning to draw more and more attention.13 Recent studies have reported that some o-carborane-containing molecules can exhibit multiple emissions, thermally activated delayed fluorescence (TADF) and ultralong organic phosphorescence,14 AIE, crystallization-induced emission (CIE), and mechanochromic and thermochromic luminescence behaviors.o-Carborane (C2B10H12) is known as an inorganic analog of benzene, in which two carbon and ten boron atoms form a 12-vertex icosahedral structure.15 The electron-withdrawing characteristics of the skeletal electrons originate from their delocalization over the 3-center-2-electron bonds and they possess a highly polarizable σ-aromatic character. Due to the electron-deficient nature of the cluster bonding found in o-carborane, it is often regarded as an electron-withdrawing and thermally and light stable functional moiety. Therefore, o-carborane is a versatile component for fabricating multi-functional optical materials. The o-carborane unit can act the part of a strong electron acceptor in the excited state of conjugated systems, stemming from the electron-deficient nature of the σ*–π* conjugation on the Ccage–Ccage bond of the o-carborane unit; when the cage carbon atoms combine with aryls, the intramolecular charge transfer (ICT) process can be readily developed in the excited state.13 Based on these observations, various types of organic or polymeric materials functionalized by o-carborane were prepared, and their advantageous photophysical properties, such as multiple emissions, thermally activated delayed fluorescence (TADF), AIE, crystallization-induced emission (CIE), and mechanochromic and thermochromic luminescence, were obtained. However, the emission color of previously reported o-carborane-containing luminescent materials is mainly located in the yellow to pink region. As we all know, the dyes emitting blue color are indispensable as a host material in a series of energy transfers for conversion into any color.16 The development of full-color emissive organic materials is an area of particular interest with high relevance, especially for improving the quality of organic full-color luminescent devices. Previously, full-color emissive organic materials were established by introducing oligoacenes from benzene to tetracene into o-carborane based on the idea that full-color emissions could be obtained by tuning the degree of the π-conjugated system.17 Thus, developing a new design strategy not only for obtaining full-color emission but also for observing stable optical properties even in the solid state is still of great significance.
Currently, in most of the reported organic substances with strong luminescence, there are always pure polycyclic aromatic hydrocarbon compounds, but sometimes there are some heteroatoms in the molecules, such as N, O, S, and P.18 These heteroatoms are typically electron-deficient or electron-rich, which can provide some special bonding modes or stabilization effects, such as intra/inter-molecular hydrogen bonding or super-conjugation. Since the lone pair electrons of the heteroatoms can increase the molecular electron density, they can increase the intermolecular dipole–dipole interactions or decrease the energy gap for electron transitions. Therefore, it seems that heteroatoms always have a great influence on the photophysical properties of compounds. Herein, we elaborated on the unique photophysical properties of o-carborane when it was functionalized by an opto-electronically active substituent fluorene and its heteroanalogs, namely, carbazole (Cz), dibenzofuran (DBF) and dibenzothiophene (DBT). Four types of substitutes with different electron-deficient or electron-rich heteroatoms were introduced into the cage carbon atoms of o-carborane. Four color tunable AIE luminogens were obtained and showed blue to yellow emissions (λem = 465–570 nm) with excellent emission quantum efficiencies (ΦF > 70%) in the solid state. The photophysical properties of the heteroatom compounds were studied with steady-state UV-vis absorption and fluorescence spectroscopy and DFT calculations. To the best of our current knowledge, it is the first time where heteroaromatic compounds and o-carborane are combined using a modified nickel-catalyzed cross-coupling reaction to study the effect of o-carborane on the photophysical properties of heteroaromatic compounds. Furthermore, the luminescent color was tuned without a substantial loss of emission efficiencies by utilizing heteroatoms.
Experimental
Materials and methods
All reactions were carried out under a dry N2 atmosphere. Solvents of tetrahydrofuran (THF) and toluene were distilled from sodium/benzophenone and stored over molecular sieves. Other chemicals were purchased from Adamas and Aladdin chemical reagent company and used as received. All reactions were monitored by thin layer chromatography (TLC). Column chromatography was conducted using a silica gel (300–400 mesh).Analytical measurements
NMR spectra were recorded on a Bruker AVANCE III 400 MHz spectrometer (1H NMR: 400 MHz, 13C NMR: 100 MHz, 11B NMR: 128 MHz). All chemical shifts were reported in δ units with references to the residual solvent resonances of the deuterated solvents for proton and carbon chemical shifts and to external BF3·OEt2 (0.00 ppm) for boron chemical shifts. Mass spectra were obtained on an Ultimate 3000/Q-Exactive spectrometer. Melting points were measured using a Nikon Polarizing Microscope ECLIPSE 50i POL equipped with an INTEC HCS302 heating stage. UV-vis absorption spectra were recorded on HITACHI U-3900H. Fluorescence spectra were collected on a Horiba FluoroLog-3 spectrofluorometer (Horiba-Jobin-Yvon, Edison, NJ, USA). Luminescence lifetime measurements were carried out by using a FluoroLog-3 spectrofluorometer; absolute PL quantum yield (ΦF) was determined using a Horiba FL-3018 Integrating Sphere. The temperature-dependent luminescence spectra were measured with a Janis VPF-100 liquid nitrogen low temperature thermostat (Janis, USA).Synthetic procedures
Results and discussion
Design and synthesis of the compounds
Fluorene and its heteroanalogs, namely, Cz, DBF and DBT for the use of their photochemical features have been described in recent works of literature.19–21 The outstanding electronic properties of these compounds, characterized primarily by their extensive π conjugation, make them promising candidates in organic light-emitting diodes (OLEDs), organic thin-film transistors (OTFTs), chemical sensors, photovoltaics, electronic data storage, and electroluminescent devices.22 DBT is a chalcogenophene compound, one of the most abundant substances in diesel oil, and it is cheap and commercially available. It has a low HOMO level and a large band gap (no absorption in the visible light region) compared with anthracene, fluorene, and carbazole, which are generally used in organic semiconductors to improve the stability of the device. Furthermore, it has a planar structure that favors intermolecular interactions. Additionally, the sulfur atom in DBT gives rise to a strong intermolecular S⋯S and S⋯π overlap, contributing to high charge migration.23,24 All of these advantages indicate that DBT should be an ideal conjugated unit for organic luminogens. However, recent studies have demonstrated that DBF shows better charge-transporting properties and solubility than DBT.24,25 The small oxygen atom size endows DBF with smaller torsional angles and higher planarity, which is beneficial for orderly stacking and charge transport in the solid state.24,26 Even more striking is that furan derivatives can avoid the heavy atom effect that occurs in thiophene-based luminogens.25 Thus, they tend to exhibit stronger and short-wavelength luminescence and are promising candidates for fabricating advanced full-color display optoelectronic devices. o-Carborane was selected to tune the excited state of organic chromophores. We introduced fluorene and its heteroanalogs (Cz, DBF and DBT), four types of substitutes with different electron-rich or electron-deficient heteroatoms, to the two carbon atoms of o-carborane to synthesize CDC, CDN, CDO, and CDSvia a modified nickel-catalyzed cross-coupling reaction (Scheme 1).27–30 Four compounds were obtained with satisfactory yields and high purity. Furthermore, the four compounds exhibited good stability even when stored in THF solutions at room temperature for 15 days, which was enough for spectroscopic measurements. The molecular structures were fully verified with 1H NMR, 13C NMR and HRMS spectroscopies. The single crystals of CDO and CDS were obtained by the slow evaporation of dichloromethane and hexane mixtures at ambient conditions and analyzed crystallographically. | ||
| Scheme 1 (a) THF, 2.5 eq. i-PrMgCl, 0 °C for 1 h, r.t. 12 h; (b) toluene, 2.4 eq. 2-bromo-9,9-dimethylfluorene, NiCl2, 115 °C, 10 h, yield: 83%. (c) Toluene, 2.4 eq. 3-bromo-9-propyl-9H-carbazole, NiCl2, 115 °C, 10 h, yield: 75%. (d) Toluene, 2.4 eq. 2-bromodibenzofuran, NiCl2, 115 °C, 10 h, yield: 93%. (e) THF, 2.4 eq. 2-bromodibenzothiophene, NiCl2, 100 °C, 8 h, yield: 33%. | ||
The optical properties of CDC, CDN, CDO and CDS were characterized by UV-vis spectroscopy (Fig. 1) and the corresponding photochemical data are summarized in Table 1. The UV-vis spectra of CDC, CDN, CDO and CDS showed a strong bathochromic shift as well as enhanced transition probability, arising from the effective electronic alteration by o-carborane. The shorter absorption bands correspond to the lowest π–π* transition and the weak absorption peaks at longer wavelengths could be attributed to the n–π* transitions of fluorene and its heteroanalogs.31–33 The absorption intensities of CDC, CDN, CDO and CDS were observed to be approximately the same as that of corresponding organic chromophores (fluorene and its heteroanalogs). This result implies that all compounds have a significant electronic coupling between the two organic chromophores in the ground state.34,35 The solvent dependence of the UV-vis absorption spectra for CDC, CDN, CDO and CDS was hardly observed (Fig. S1, ESI†), further indicating that the ground state was not affected by the solvent polarity.
 | ||
| Fig. 1 UV-vis absorption spectra of CDC, CDN, CDO and CDS in THF, c = 1.0 × 10−5 M, 20 °C. | ||
| Sample | λ abs (nm) | λ em , (nm) | τ F (ns) | Φ F (%) | k rad (107 s−1) | k nr (107 s−1) |
|---|---|---|---|---|---|---|
| a Measured in THF solution (1 × 10−3 M) at room temperature. b Taken by excitation at 300 nm for CDC and CDO, 330 nm for CDN and CDS, respectively. c For lifetimes excited at 340 nm. d f w = 99%. e In the solid state. f Values of krad and knr were calculated according to the equations, krad = ΦF/τF and knr = (1/τF) − krad, respectively. g Not detected or not available. | ||||||
| CDC | 285, 310 | 336, 351, 526, 500e | 2.9,d 4.1e | —,g 57,d 78e | 19.8,d 19.1e | 14.7,d 5.3e |
| CDN | 278, 334, 349 | 377, 431, 598, 570e | 9.4,d 5.9e | 0.5,a 34,d 80e | 3.6,d 13.6e | 7.0,d 3.4e |
| CDO | 289, 299 | 344, 530, 465e | 14.1,d 8.8e | —,g 49,d 71e | 3.5,d 8.0e | 3.6,d 3.3e |
| CDS | 317, 330 | 380, 560, 526e | 5.0,a 4.5,d 3.2e | 0.6,a 10,d 97e | 2.3,d 30.3e | 19.9,d 0.9e |
Fig. S2 (ESI†) shows the photoluminescence (PL) spectra of CDC, CDN, CDO and CDS in various solvents. All compounds exhibited dual emissions consisting of the locally excited (LE) and ICT emission bands in the solution state; the former emission band was attributed to the LE (localized at the aryl backbone) state emission, and the latter belonged to the ICT state emission because the intensity of the ICT emission is sensitive to solvent polarity.36 Moreover, we measured the emission spectra at the low temperature of 77 K in a 2-methyltetrahydrofuran (2-MeTHF) solution, where the molecular motions are completely frozen (Fig. S3, ESI†). Significantly, in contrast to these features from the LE and ICT emissions at room temperature, all compounds showed strong broad emissions in the longer-wavelength region but the sharp bands with vibrational structures were similar to those at room temperature, indicating that the emission bands should be from the LE and ICT states in solutions.36
Owing to the introduction of fluorene and its heteroanalogs into the two carbon atoms of optically functional “element-block” o-carborane,37–39CDC, CDN, CDO and CDS showed a typical AIE characteristic: the weak emission in a dilute THF solution was enhanced upon aggregate formation in solution and also in the solid state. The AIE characteristics of CDC, CDN, CDO and CDS were further proved by studying their PL behaviors in THF and THF/water mixtures with different water fractions (fw) (Fig. 2). In a dilute THF solution, CDC, CDN, CDO and CDS were weakly emissive because the ICT from the π-conjugated moieties to the Ccage–Ccage bond of o-carborane quenched the emission in solution,40 whereas the freezing of the Ccage–Ccage bond vibration caused enhanced emission in the aggregates (AIE).41,42
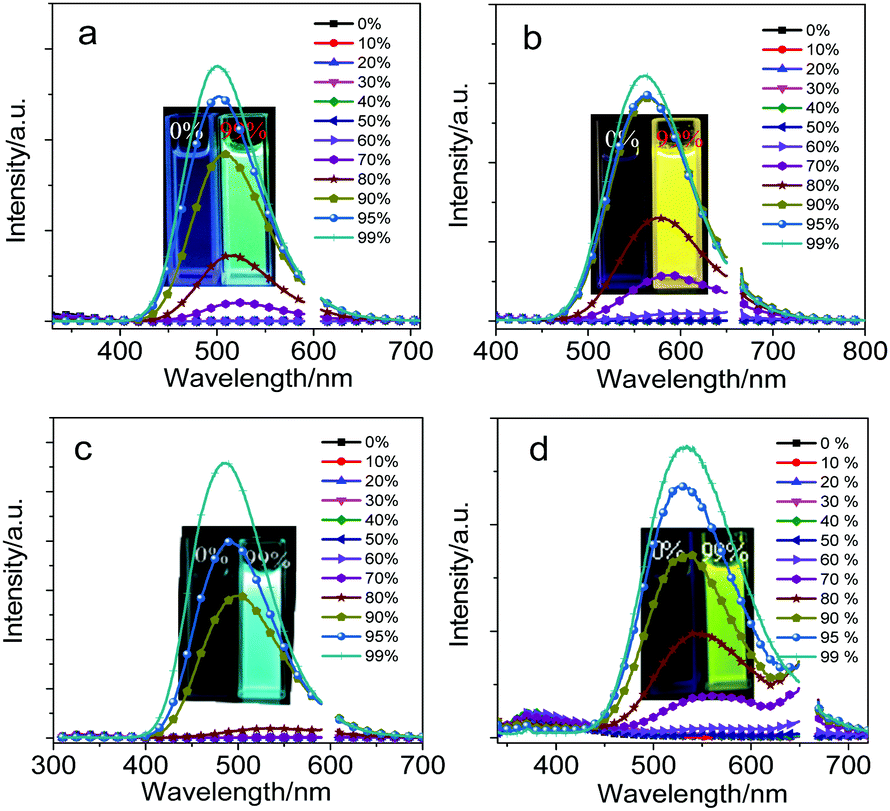 | ||
| Fig. 2 Fluorescence spectra of (a) CDC, (b) CDC, (c) CDF and (d) CDS in THF/water mixtures with different water volume fractions; λex = 300 nm for CDC and CDO, λex = 330 nm for CDN and CDS, c = 5.0 × 10−5 M, 20 °C (light scattering from the incident light was omitted in the spectra). | ||
The addition of a large amount of water into their THF solutions aggregated their molecules and enhanced their PL. Besides, with the increase in the water content in the THF solutions, blue-shifted emission bands were observed, which may be attributed to the micro-environmental polarity that is lowered with the formation of aggregates. Accompanying the increase in the water content, the solvating power of the mixture decreased. At a certain point with a high water content, the aqueous mixture could not dissolve all the molecules anymore. Some of the molecules thus began to aggregate into nanoparticles, whose polarity inside was lower than that on the outside. The emission bands blue-shifted similar to the typical ICT emission.4,43,44 The highest emission was achieved at fw = 99%. The quantum yields of CDC, CDN, CDO and CDS in dilute THF solutions were determined to be less than 1% by a calibrated integrating sphere. In the aggregated state, their quantum yields were measured to be 57%, 34%, 49% and 10%, and the emission colors ranged from blue to yellow. These results indicate that CDC, CDN, CDO and CDS are all AIE luminophores and the photophysical properties of CDC, CDN, CDO and CDS are summarized in Table 1.
The emission properties in the crystal state were examined (Fig. 3, Table 1 and Fig. S4, ESI†). From the PL spectra in the crystalline state, CDO showed an emission band in the shortest wavelength region. CDN, however, showed emission bands in the longest wavelength region. Blue and bright yellow emissions were observed from CDO and CDN (Fig. 3c), respectively. All compounds showed stronger emission intensity (ΦF > 70%) in the crystalline state than that in the solution and aggregated states, indicating that the four compounds are AIE-active compounds. As shown in Fig. 3, the emission color of the compounds can be tuned from blue to yellow (λ > 100 nm) simply via changing the heteroatom in the organic aryl group. As mentioned above, the emission bands for the aryl–carborane dyads are attributed to the ICT state emission, and there are mainly two factors for the wide wavelength range of ICT state emissions. One is the strength of the electron-donating property of the aryl moiety. The electron-donating ability of the aryl moieties can be tuned with different electron-deficient or electron-rich heteroatoms. The extent of electron-rich or electron-deficient heteroatoms can tune the degree of ICT between π-conjugated organic aryls and o-carborane, which results in an excited state with varied emission colors. The other factor is the dihedral angle between the Ccage–Ccage bond in o-carborane and the aryl π-plane. As depicted in Fig. S3 (ESI†), four aryl–carborane dyads show a large ICT emission band at longer wavelengths. In addition, the values of the dihedral angle between the aryl substituent and the Ccage–Ccage bond in o-carborane are −89.9(3)° for CDO, −79.6(2)° for CDC, and −75.9(3)° for CDS (see below). These results suggest that the most stable molecular structure of the four aryl–carborane dyads should be the twisted conformation in the ground state, and this twisted conformation favors the ICT process in the excited state.45,46 Therefore, the degree of the ICT process was regulated by the strength of the electron-donating property of the aryl moiety and the dihedral angle between the aryl substituent and the Ccage–Ccage bond in o-carborane. The solids CDC, CDN, CDO and CDS were observed to be white or light pale-yellow under daylight, as shown in Fig. 3b. However, strong cyan emissions for CDC, bright yellow emission for CDN, blue emission for CDO and green emission for CDS were observed from the crystal state under 365 nm UV light irradiation (Fig. 3c). This fact strongly suggests that the emission band in the solid-state is induced by the ICT state and the heteroatoms play an important role in manipulating the degree of the ICT process.
 | ||
| Fig. 3 (a) The normalized emission spectra of CDC, CDN, CDO and CDS in the solid state, λex = 300 nm for CDC and CDO, λex = 330 nm for CDN and CDS. (b) Photos of compounds CDC, CDN, CDO and CDS as solid powders under room light and (c) fluorescence images under UV illumination (365 nm), 20 °C. | ||
From the emission spectra at 77 K (Fig. 4), CDC, CDN, CDO and CDS in the solid state presented narrow and sharp bands attributable to the ICT emissions at 500, 570, 465 and 526 nm, respectively. The intramolecular rotation was likely prohibited because of the steric hindrance of the substituents in the o-carborane units.43 This is why the o-carborane-containing heterocyclic compounds demonstrated ICT emissions rather than LE emissions in the solid state. Moreover, the compounds were insensitive to temperature due to their rigid structure, and only enhanced emission intensity was observed for all compounds rather than a change in the emission wavelength during cooling from 333 to 77 K (Fig. 4). From the above results, it can be concluded that o-carborane acts as a versatile “element-block” not only for enabling ICT emissions but also for designing AIE luminogens with strong fluorescence in the solid state. Besides, the species and the difference in the electronegativities of the heteroatoms played a key role in fixing the molecular configuration and adjusting the emission colors, respectively.19
 | ||
| Fig. 4 The PL spectra of (a) CDC, (b) CDN, (c) CDO and (d) CDS in the solid state during cooling from 333 to 77 K, λex = 300 nm for CDC and CDO, λex = 330 nm for CDN and CDS. | ||
To better understand the AIE characteristics and stronger fluorescence of CDC, CDN, CDO and CDS, the fluorescence lifetime was measured in the aggregated and solid states, and the radiative and non-radiative decay processes were also investigated (Fig. 5, Table 1 and Fig. S5, ESI†). From the aggregated to the solid state, the radiative decay rate (krad) of CDS increased by around 13 times (krad,agg = 2.3 × 107 s−1 and krad,solid = 33.1 × 107 s−1), while the non-radiative decay rate (knr) decreased almost by 22 times (knr,agg = 19.9 × 107 s−1 and knr,solid = 0.9 × 107 s−1). This suggests that the inhibition of the non-radiative decay of the excited state in the solid state results in its high fluorescence. In sharp contrast, krad of CDO increased by around 2.3 times upon conversion to the solid state, while its knr decreased by only about 1.2 times, whose extent was much smaller than that in CDS. The incorporation of DBT restricted the non-radiative decay of the excited state to a greater extent than DBF, generating AIE luminogens with stronger fluorescence. In other words, the heteroatoms have a noticeable impact on the photophysical properties of o-carborane-containing heterocyclic compounds.
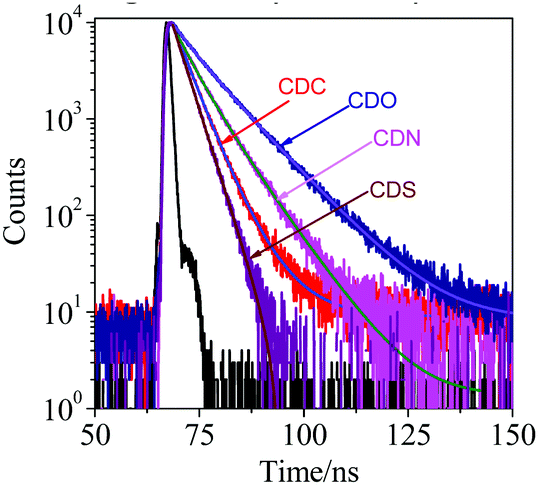 | ||
| Fig. 5 Emission decay profiles of CDC, CDN, CDO and CDS in the solid state, λex = 340 nm, 20 °C. | ||
Crystal packing from X-ray crystallography
Single-crystals of CDO and CDS were obtained from CH2Cl2 and n-hexane solutions via slow evaporation at ambient temperatures by the solvent layering method. The data were obtained at 296 and 173 K for CDO and CDS, respectively, on a Bruker APEX-II CCD diffractometer using Mo-Kα radiation. The structure was solved with the ShelXT structure solution program using intrinsic phasing and refined with the ShelXL refinement package using least squares minimization.47,48 All hydrogen atoms were geometrically fixed using the riding model. The X-ray crystal structure analysis crystallographic data have been deposited with the Cambridge Crystallographic Data Center as supplementary publication No. CCDC 1838965 (CDO) and 1843555 (CDS).†The specified molecular arrangements of CDO and CDS in the solid state were investigated by X-ray crystallographic analysis. The single-crystal structures of CDO and CDS are depicted in Fig. 6a and c, and the associated crystallographic data, such as the distances of the Ccage–Ccage carbons and the dihedral angles between a cage carbon vector and the planes of the aryl substituents, are given in Fig. 6 and Fig. S6 as well as Tables S1 and S2 (ESI†). The molecular packing patterns of compounds CDO and CDS are shown in Fig. 6b, d and Fig. S6 (ESI†). With the twisted conformation, the molecules piled up loosely in the crystal lattice without π–π stacking among the CDO and CDS fragments. The adjacent O⋯O distances in DBF and S⋯S in the DBT substitutes were 3.897 and 8.783 Å, respectively. Also, no closely overlapping carbon atoms were observed. The shortest O⋯H distance was 3.028 Å, which was too long to form an effective intermolecular hydrogen bonding interaction. We also note that for a reasonable estimation of cage carbon deformation, a distance factor needs to be considered, which is related to the distance between the aryl substituents and the midpoint of the cage carbons.
 | ||
| Fig. 6 The ORTEP drawing (30% probability for thermal ellipsoids) of (a) CDO and (c) CDS; molecular packing pattern of (b) CDO and (d) CDS; Carbon (gray), boron (orange) and oxygen (red), and chlorine (green). Hydrogen atoms are omitted for clarity. | ||
The value of the dihedral angle determines the location of the aryl substituent with respect to the cage carbon vector, and the higher deviation of its angle from 90° minimizes the interaction with the Ccage–Ccage bond. Based on this standard, it is clear that CDO shows a dihedral angle of −89.9(3)°, which is closer to 90° than that of CDC (−79.6(2)°) or CDS (−75.9(3)°). This indicates that there is a large orbital overlap between DBF and o-carborane.33 The dihedral angle of the two aryl groups at o-carborane contributed greatly to the orbital interaction between the aryl π-bond and cage carbon σ*-bond.49 Additional deformation was correlated with the parallel orientation of DBF. One of the obvious features found in each structure is that the Ccage–Ccage distances in the o-carborane cage are significantly elongated and CDS (1.743(3) Å) has the longest distance of the three compounds of CDC (1.727(4) Å), CDO (1.727(2) Å), and CDS, indicating that CDS significantly generates cage deformation.50,51 Based on these serial comparisons, it is now clear that not only the distance but also the steric factor influences the cage carbon deformation significantly and is directly related to the interaction between the aryl substitutes. Thus, the cage carbon distance represents the degree of the aryl substituent interaction. Among the four o-carborane-containing heterocyclic compounds, CDO showed the most o-carborane–aryl interaction and such a ground state interaction further contributed to the electronic state of the resulting excited state with the lowest quantum yields and shortest emission wavelength.
Electrochemical study (cyclic voltammetry)
To understand the effect of the heteroatoms in π-conjugated organic aryls on their donating strength, the redox properties of the compounds were studied with cyclic voltammetry (Fig. 7 and Fig. S7, ESI†) and the data are summarized in Table 2. The electron-donating strength of the π-conjugated organic aryls containing heteroatoms can be evaluated based on their oxidation potential.52 However, no reversible oxidation waves were observed in CDC, CDN, CDO and CDS, and the onset oxidation (Eox) potentials were in the order CDN (+0.90 V) < CDC (+1.22 V) < CDS (+1.30 V) < CDO (+1.40 V). This is probably ascribed to the highest electron-donating strength of 9-propyl-9H-Cz in the four π-conjugated organic aryls, and the lesser electronegativity of the sulphur atom in DBT compared with that of the oxygen atom in DBF. | ||
| Fig. 7 Cyclic voltammogram of CDC, CDN, CDO and CDS, c = 1.0 × 10−3 M. The deaerated acetonitrile solution contained the compounds, 0.10 M Bu4NPF6 as supporting electrode, Ag/AgNO3 as a reference electrode, scan rates: 100 mV s−1. For all compounds, ferrocene (Fc) (c = 1.0 × 10−3 M) was used as an internal reference, 20 °C. | ||
| Samples | Reduction (V vs. Fc/Fc+) | Oxidation (V vs. Fc/Fc+)b | ||||
|---|---|---|---|---|---|---|
| E pc1 | E pc2 | E pa1 | E pa2 | E red1/2 | E oxonset | |
| a CVs were recorded at room temperature in N2 purged acetonitrile/0.1 M Bu4NPF6 (scan rate = 100 mV s−1). b Onset potentials (V vs. Fc/Fc+) from oxidation (Eox). | ||||||
| CDC | −1.45 | −1.57 | −1.62 | −1.70 | −1.51, −1.66 | +1.22 |
| CDN | −1.58 | −1.70 | −1.72 | −1.80 | −1.64, −1.75 | +0.9 |
| CDO | −1.44 | −1.56 | −1.60 | −1.70 | −1.50, −1.65 | +1.40 |
| CDS | −1.42 | −1.54 | −1.58 | −1.66 | −1.48, −1.62 | +1.30 |
Besides, unpredictable results occurred: the electrochemical behaviour of the four compounds exhibited two one-electron reversible distinctive reduction waves, which were attributed to the two sequential electron reduction processes.51 It is worth noting that the reduction waves appeared in the order CDN (−1.64 V) < CDC (−1.51 V) < CDO (−1.50 V) < CDS (−1.48 V), and the different maximum ICT emissions bands in CDC, CDN, CDO and CDS were correlated with the reduction potentials observed in the CV experiments, showing the lowest reduction potential of CDN. The reductive potential of the derivatives will negatively shift provided they contain an electron donor.53 The extent of electron-rich or electron-deficient heteroatoms can tune the degree of ICT between the π-conjugated organic aryls and “element-block” o-carborane, which results in excited states with varied emission colors.
DFT calculation
To ensure the electronic alteration of the functional aryl groups driven by o-carborane, geometry optimization as well as HOMO and LUMO energies was studied with the B3LYP functional and 6-31G(d,p) basis sets using the Gaussian 09 package;54 the results are shown in Fig. 8 and Fig. S8–S11 (ESI†). For all compounds, the energy gap between the HOMO and LUMO energies changed depending on the interaction of the aryl groups with o-carborane. As expected, HOMOs mainly lie in the aryl groups and LUMOs are on the Ccage–Ccage bond irrespective of the types of aryl functional groups. Furthermore, the LUMOs can mix with the aryl and cage carbon orbitals depending on the types of aryl groups. However, the o-carborane contributions vary markedly with the types of the aryl groups. This finding is another clear evidence for a facile electronic communication between the heterocyclic compounds and the o-carborane unit. | ||
| Fig. 8 The electronic density contours and energy levels of the HOMOs and LUMOs of CDC, CDN, CDO and CDS were calculated by DFT at the B3LYP/6-31G(d,p) level with Gaussian 09W. | ||
Furthermore, to determine the orbital contributions as well as the lowered LUMO energy due to the o-carborane unit, time-dependent DFT (TD-DFT) calculations were performed according to the optimized geometries to locate the origin of the low-energy absorptions of each type of aryl. The low-energy absorptions were attributed to the transitions from HOMO to LUMO, shifting from π (aryl) to σ* (Ccage–Ccage bond), and the transitions observed at λmax as well as their relative energies and oscillator strength values were consistent with the UV-Vis absorption spectra. For CDC, 293 nm, 4.23 eV, f = 0.032; CDN 315 nm, 3.94 eV, f = 0.028; CDO 286 nm, 4.33 eV, f = 0.078; CDS 303 nm, 4.09 eV, f = 0.014, as shown in Fig. S12–S15 and Table S3 (ESI†). In all cases, the transitions attributed to the lowest absorptions are in the following order: HOMO → LUMO, HOMO → LUMO+1, and HOMO−1 → LUMO; the oscillator strength of the lowest energy is greater than the next lowest one (Table S3, ESI†). This is conclusive evidence of the alteration of the excited state energies owing to the interaction between π (aryl) and σ* (Ccage–Ccage bond).
However, the above data tend to support the inference that the ICT process occurs between the π-conjugated organic aryls and “element-block” o-carborane. First, to identify the electronic communication between the π-conjugated organic aryls and “element-block” o-carborane, we optimized the excited-state structures of the four compounds through TD-DFT calculations in the gas phase (Fig. 9). It was revealed that the S0–S1 electronic transition was mainly derived from HOMO to LUMO. Next, the electronic structures were investigated. The HOMOs in the four compounds were localized on the π-conjugated organic aryl moieties. In contrast, the LUMOs were significantly delocalized to the o-carborane moiety. This result also strongly supports that the emission should be derived from the ICT states. The calculated emission wavelengths for the CDC, CDN, CDO and CDS states were 504, 572, 485 and 532 nm, respectively. These values showed good agreement with the experimental results.
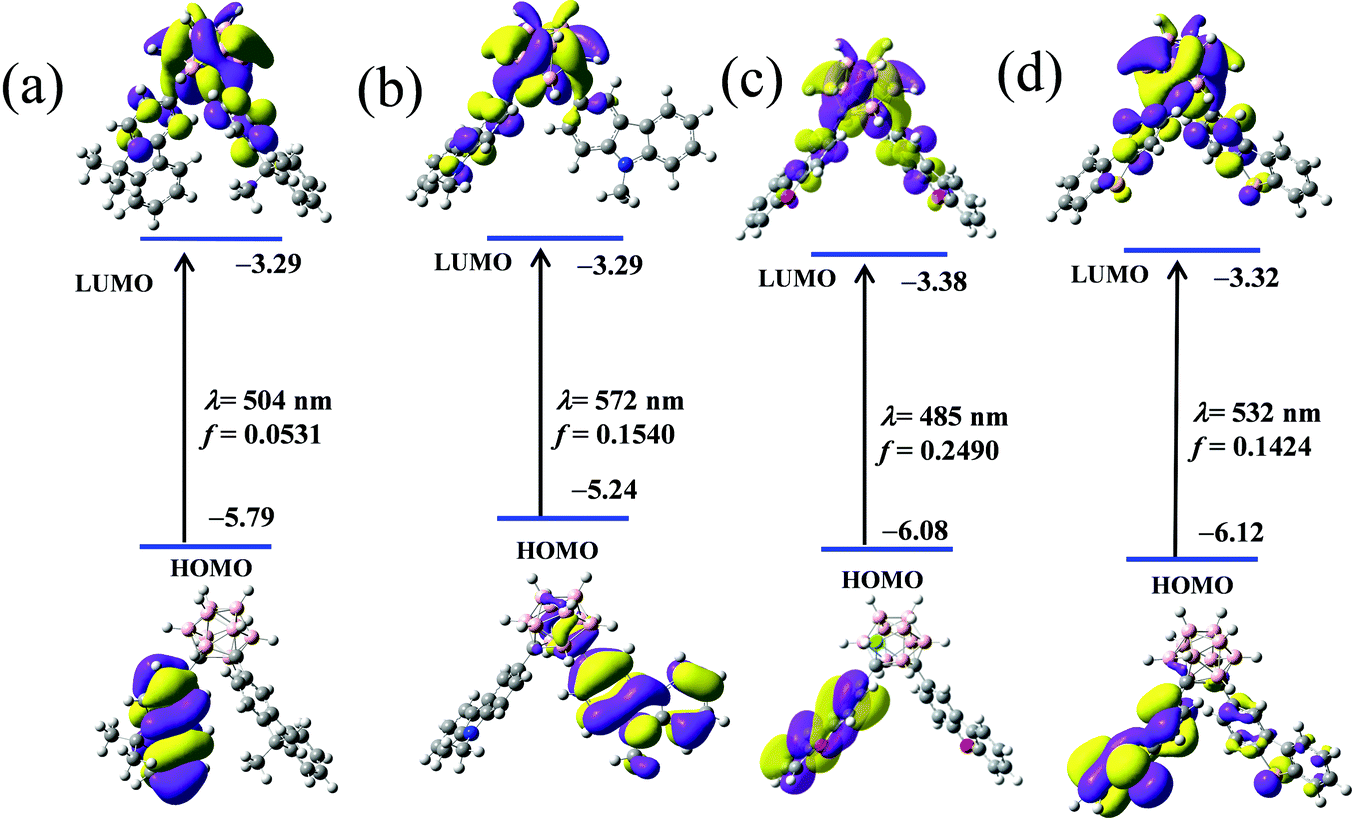 | ||
| Fig. 9 Frontier molecular orbitals in the first singlet excited state (S1) optimized geometries of (a) CDC, (b) CDN, (c) CDO and (d) CDS in the gas state and the lowest-energy electronic transitions from the TD-DFT calculation at the B3LYP/6-31G(d,p) level by Gaussian 09. | ||
In the ground state, the ESP maps reveal that the electron-deficient regions are mainly located on the o-carborane groups (yellow color area) in the o-carborane-containing heterocyclic compounds due to the strong electron-withdrawing properties. The strongest positive electron areas were around the heterocyclic compounds (blue color area) (Fig. 10). Besides, the DFT-calculated dipole moments were 8.05, 10.16, 6.56 and 6.06 D for the S0 states of CDC, CDN, CDO and CDS, respectively. In the excited state, the ESP maps show that the electron-deficient regions are mainly located on the o-carborane groups (yellow color area) as well as in the o-carborane-containing heterocyclic compounds. However, the changes in the electron density cannot be ignored in the excited state. The electron density of the π-conjugated organic aryl moiety in the compounds changed from electron-rich in the ground state to electron-deficient in the excited state, which strongly confirmed the charge transfer in the formed compounds (Fig. 10). Besides, the degree of change in the electron density is related to the different electron-rich or deficient attributes of the heteroatoms. We observed a large excited-state dipole moment as compared with the ground-state one, which was also supported by the PL spectra. The positive bathochromic shift in the PL spectra, as opposed to negligible solvatochromism in the absorption spectra, is indicative of efficient ICT from the center of the heterocyclic compound to the o-carborane moiety through the bond. The DFT calculated Δμ (Δμ = μE − μG) of 15.81 Debye for CDO is larger than 11.77 Debye for CDN (Table S4, ESI†); the result can be well correlated with the ICT emission bands in CDN and CDO, showing the fluorescence color from blue to yellow emissions (λem = 465–570 nm).
 | ||
| Fig. 10 Electrostatic potential of CDC, CDN, CDO and CDS at 0.001 a.u. isosurface of electron density by (TD-) DFT computed at the B3LYP/6-31G(d,p) level in the ground (μG) and excited (μE at the Franck–Condon geometry) dipole moments (blue color indicates electron-rich, red color indicates electron-deficient). | ||
Conclusions
In summary, four AIE luminogens with tunable fluorescence colors were obtained when different electron-rich or electron-deficient heteroatoms were introduced into the carbon atoms in o-carborane by a modified nickel-catalyzed cross-coupling reaction. The results showed that the extent of the electron-rich or electron-deficient heteroatoms and the distance effect as well as the intermolecular interactions were responsible for the resultant excited states with varied emission colors (λem = 465–570 nm) and excellent emission quantum efficiencies in the solid state (ΦF > 70%). The ground and excited states calculated by DFT and TD-DFT correlated with the corresponding absorption spectra to illustrate the electronic perturbation in a systematic fashion, suggesting that the heteroatoms lowered the electronic perturbations and disturbed the excited states. Our findings not only demonstrate an efficient strategy for constructing o-carborane-functionalized AIE luminogens containing heterocyclic moieties, but also indicate that they are promising as advanced optoelectronic materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21571152 and U1703251), Program for Tianshan Innovative Research Team of Xinjiang Uygur Autonomous Region (2018D14002), the Open Fund of the Key Laboratory of Xinjiang Uygur Autonomous Region (2017D04014), Scientific Research Program of the Higher Education Institution of Xinjiang (XJEDU2017A001).Notes and references
- O. Ostroverkhova, Chem. Rev., 2016, 116, 13279 CrossRef CAS.
- L. X. Pan, Y. J. Cai, H. Z. Wu, F. Zhou, A. J. Qin, Z. M. Wang and B. Z. Tang, Mater. Chem. Front., 2018, 2, 1310 RSC.
- H. Sasabe and J. Kido, Chem. Mater., 2011, 23, 621 CrossRef CAS.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC.
- J. Mei, N. Leung, R. Kwok, J. W. Lam and B. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- M. Yamaguchi, S. Ito, A. Hirose, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2017, 1, 1573 RSC.
- G. Feng, R. T. K. Kwok, B. Z. Tang and B. Liu, Appl. Phys. Rev., 2017, 4, 021307 Search PubMed.
- H. Wang, E. Zhao, J. W. Y. Lam and B. Z. Tang, Mater. Today, 2015, 18, 365 CrossRef CAS.
- Y. J. Cai, A. J. Qin and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 7375 RSC.
- X. Gu, R. T. Kwok, J. W. Y. Lam and B. Z. Tang, Biomaterials, 2017, 146, 115–135 CrossRef CAS PubMed.
- D. Li, J. Liu, R. T. K. Kwok, Z. Liang, B. Z. Tang and J. H. Yu, Chem. Commun., 2012, 48, 7167 RSC.
- Y. Liu, Y. Tang, N. N. Barashkov, I. S. Irgibaeva, J. W. Lam, R. Hu, D. Birimzhanova, Y. Yu and B. Z. Tang, J. Am. Chem. Soc., 2010, 132, 13951 CrossRef CAS PubMed.
- M. Gon, K. Tanaka and Y. Chujo, Polym. J., 2018, 50, 109 CrossRef CAS.
- D. S. Tu, S. Z. Cai, C. Fernandez, H. L. Ma, X. Wang, H. Wang, C. Q. Ma, H. Yan, C. S. Lu and Z. F. An, Angew. Chem., Int. Ed., 2019, 58, 9129 CrossRef CAS PubMed.
- K.-R. Wee, Y.-J. Cho, S. Jeong, S. Kwon, J.-D. Lee, I. H. Suh and S. O. Kang, J. Am. Chem. Soc., 2012, 134, 17982 CrossRef CAS.
- G. Schwartz, S. Reineke, T. Conrad Rosenow, K. Walzer and K. Leo, Adv. Funct. Mater., 2009, 19, 1319 CrossRef CAS.
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Chem. – Asian J., 2018, 12, 2134 CrossRef PubMed.
- Y. J. Xie, J. Tu, T. Q. Zhang, J. Q. Wang, Z. L. Xie, Z. G. Chi, Q. Peng and Z. Li, Chem. Commun., 2017, 53, 11330 RSC.
- Z. J. Wang, T. Y. Wang, C. Zhang and M. G. Humphrey, ChemPhotoChem, 2018, 2, 369 CrossRef CAS.
- W. J. Zhao, T. S. Cheung, N. Jiang, W. B. Huang, J. W. Y. Lam, X. P. Zhang, Z. K. He and B. Z. Tang, Nat. Commun., 2019, 10, 1595 CrossRef PubMed.
- W. Jiang, Y. Li and Z. H. Wang, Chem. Soc. Rev., 2013, 42, 6113 RSC.
- Y. M. Yang, Q. Zhao, W. Feng and F. Y. Li, Chem. Rev., 2013, 113, 192 CrossRef CAS PubMed.
- G. R. Hutchison, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 16866 CrossRef CAS PubMed.
- Z. Zhao, H. Ni, C. W. Ge, Y. J. Cai, Y. Xiong, J. Qi, W. T. Wu, R. T. K. Kwok, X. K. Gao, A. J. Qin, J. W. Y. Lam and B. Z. Tang, Adv. Sci., 2017, 4, 1700005 CrossRef PubMed.
- M. Yamaguchi, K. Tanaka and Y. Chujo, Chem. – Asian J., 2018, 13, 1342 CrossRef CAS PubMed.
- X.-H. Jin, D. Sheberla, L. J. W. Shimon and M. Bendikov, J. Am. Chem. Soc., 2014, 136, 2592 CrossRef CAS PubMed.
- C. Tang and Z. Xie, Angew. Chem., Int. Ed., 2015, 54, 7662 CrossRef CAS PubMed.
- Y. Chen, J. X. Guo, X. Y. Wu, D. Z. Jia and F. L. Tong, Dyes Pigm., 2018, 148, 180 CrossRef CAS.
- X. Y. Wu, J. X. Guo, J. Z. Zhao, Y. Y. Che, D. Z. Jia and Y. Chen, Dyes Pigm., 2018, 154, 44 CrossRef CAS.
- X. Y. Wu, J. X. Guo, Y. L. Cao, J. Z. Zhao, W. Jia, Y. Chen and D. Z. Jia, Chem. Sci., 2018, 9, 5270 RSC.
- L. J. Deng, J. Y. Li, G.-X. Wang and L.-Z. Wu, J. Mater. Chem. C, 2013, 1, 8140 RSC.
- J. Y. Li, T. Zhang, Y. J. Liang and R. X. Yang, Adv. Funct. Mater., 2013, 23, 619 CrossRef CAS.
- S.-Y. Kim, Y.-J. Cho, H.-J. Son, C. H. Kim and S. O. Kang, J. Organomet. Chem., 2018, 865, 152 CrossRef CAS.
- S.-Y. Kim, Y.-J. Cho, G. Jin, W.-S. Han, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2015, 17, 15679 RSC.
- G. F. Jin, Y.-J. Cho, K.-R. Wee, S. A. Hong, I.-H. Suh, H.-J. Son, J.-D. Lee, W.-S. Han, D. W. Cho and S. O. Kang, Dalton Trans., 2015, 44, 2780 RSC.
- J. Ochi, K. Tanaka and Y. Chujo, Eur. J. Org. Chem., 2019, 2984 CrossRef CAS.
- R. Núńez, M. Tarrés, A. Ferrer-Ugalde, F. F. de Biani and F. Teixidor, Chem. Rev., 2016, 116, 14307 CrossRef.
- K. Nishino, H. Yamamoto, K. Tanaka and Y. Chujo, Org. Lett., 2016, 18, 4064 CrossRef CAS PubMed.
- K. Nishino, K. Hashimoto, K. Tanaka, Y. Morisaki and Y. Chujo, Tetrahedron Lett., 2016, 57, 2025 CrossRef CAS.
- J. Llop, C. Viñas, J. M. Oliva, F. Teixidor, M. A. Flores, R. Kivekäs and R. Sillanpää, J. Organomet. Chem., 2002, 657, 232–238 CrossRef CAS.
- M. Tominaga, H. Naito, Y. Morisaki and Y. Chujo, New J. Chem., 2014, 38, 5686 RSC.
- K. Kokado and Y. Chujo, Macromolecules, 2009, 42, 1418 CrossRef CAS.
- J. Mei, Y. Hong, J. W. Y. Lam, A. J. Qin, Y. H. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS.
- Y.-N. Jing, S.-S. Li, M. Q. Su, H. L. Bao and W.-M. Wan, J. Am. Chem. Soc., 2019, 141, 16839–16848 CrossRef CAS.
- H. Naito, K. Nishino, Y. Morisaki, K. Tanaka and Y. Chujo, Angew. Chem., Int. Ed., 2017, 56, 254 CrossRef CAS PubMed.
- K. Nishino, K. Tanaka and Y. Chujo, Asian J. Org. Chem., 2019 DOI:10.1002/ajoc.201900537.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef.
- K.-R. Wee, W.-S. Han, D. W. Cho, S. Kwon, C. Pac and S. O. Kang, Angew. Chem., Int. Ed., 2012, 51, 2677 CrossRef CAS.
- B. W. Hutton, F. MacIntosh, D. Ellis, F. Herisse, S. A. Macgregor, D. McKay, V. Petrie-Armstrong, G. M. Rosair, D. S. Perekalin, H. Tricas and A. J. Welch, Chem. Commun., 2008, 5345 RSC.
- K.-R. Wee, Y.-J. Cho, J. K. Song and S. O. Kang, Angew. Chem., Int. Ed., 2013, 52, 9682 CrossRef CAS PubMed.
- I. García-Benito, I. Zimmermann, J. Urieta-Mora, J. Aragó, J. Calbo, J. Perles, A. Serrano, A. Molina-Ontoria, E. Ortí, N. Martín and M. K. Nazeeruddin, Adv. Funct. Mater., 2018, 28, 1801734 CrossRef.
- H. L. Jia, X. H. Ju, M. D. Zhang, Z. M. Ju and H. G. Zheng, Phys. Chem. Chem. Phys., 2015, 17, 16334 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng,J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09W (Revision A.01), Gaussian Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Molecular structure characterization data, absorption and fluorescence emission data and crystallographic data. CCDC 1838965 (CDO) and 1843555 (CDS). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qm00621d |
| This journal is © the Partner Organisations 2020 |