
Anion replacement in silver chlorobromide nanocubes: two distinct hollowing mechanisms†
Sasitha C.
Abeyweera
 ,
Shea
Stewart
and
Yugang
Sun
*
,
Shea
Stewart
and
Yugang
Sun
*
Department of Chemistry, Temple University, 1901 North 13th Street, Philadelphia, Pennsylvania 19122, USA. E-mail: ygsun@temple.edu
First published on 9th October 2019
Abstract
Silver chlorobromide nanocubes have shown promise as a class of sacrificial template for the synthesis of silver-based hollow nanostructures through chemical transformation reactions. Strong chemical bonding between silver and sulfur enables the transformation of AgCl0.5Br0.5 nanocubes into hollow nanoshells of silver–sulfur compounds. During these transformations, factors such as the crystalline structure and nature of the shell material influence the reaction kinetics, and thus the structural and morphological evolution of the hollow structures. Herein, we systematically investigate the hollowing mechanism and associated reaction kinetics for the controlled conversion of AgCl0.5Br0.5 nanocubes into hollow nanostructures. The use of sulfide anions (S2−) and benzenethiolate anions (BT−) for the anion exchange reaction with halide ions has been compared to illustrate the hollowing mechanisms. The S2− ions are comparable in size to the halide ions, resulting in a nano-scale Kirkendall effect that is mainly responsible for the formation of hollow Ag2S nanoshells. In contrast, the Kirkendall effect is absent from forming hollow nanoshells of silver benzenethiolate (AgBT) because the much larger size of BT− ions relative to halide ions hinders their diffusion in the solid AgBT lattice. The molecular-type layered structure of AgBT also disfavors both the inward and outward diffusion of ions through the AgBT shells, thus leading to an overall reduction in the kinetics of the transformation reaction. The transformation reaction is driven only by the dissolution of AgCl0.5Br0.5 and reprecipitation of Ag+ and BT− ions. These results allow for understanding of the underlying factors in hollowing mechanisms to enable the controlled synthesis of well-defined hollow nanostructures with desired properties for a broad range of potential applications.
Introduction
Hollow nanostructures are of great interest because their unique geometrical configurations lead to properties different from their solid counterparts.1,2 For example, a hollow nanoshell exposes both the inner surface and outer surface to offer a surface area much larger than that of a solid nanoparticle formed from the same amount of material. The enlarged surface area of hollow nanoshells is beneficial for applications requiring exposed surfaces such as catalysis3–5 and surface plasmon resonance.6,7 In addition to the exposed surfaces, the hollow interior spaces confined in nanoshells are useful for the development of controlled drug delivery,8–10 sensing devices,11,12 nanoscale reactors,13,14 and electrode materials for electrochemical energy storage.15–18A common strategy when synthesizing hollow nanostructures relies on the use of sacrificial template nanostructures, which can either transform into the materials of the resulting hollow nanostructures through appropriate chemical reactions or serve as a physical support to receive materials of the resulting hollow nanostructures.19–22 The former templated chemical transformation represents the most promising approach, which has been widely adopted in the past decades. Typical mechanisms include nanoscale galvanic replacement reactions23 and the Kirkendall process.24 Galvanic replacement reactions involve redox reactions of the sacrificial template nanostructures, which are usually composed of metals25 or metal oxides with varying oxidation states.26,27 The nanoscale Kirkendall process is responsible for the formation of hollow nanoshells when the outward diffusion of the template nanostructure species is larger than the inward diffusion of the reactant species across the newly developed interfacial boundaries. During this process, the unbalanced diffusion of materials results in vacancies in the nanoparticles, which can further grow and coalescence to form hollow interiors in nanoparticles. The Kirkendall hollowing process can be triggered by a broad range of reactions including redox reactions28 and ion exchange reactions.29 In a typical Kirkendall hollowing process, the initial reaction deposits a thin layer of the desired shell material on the surfaces of the solid sacrificial template nanoparticles, creating interfacial boundaries to facilitate the following Kirkendall hollowing process.
Herein, uniform ionic nanocrystals of silver chlorobromide (AgCl0.5Br0.5) with well-defined cubic morphology are studied as a sacrificial template to react with sulfur-containing reagents including sodium sulfide and benzenethiol. Due to the strong binding interaction between silver and sulfur, both sulfide and benzenethiol replace the halide ions, transforming the silver chlorobromide nanocubes to hollow nanoshells made of silver sulfide (Ag2S) and silver benzenethiolate (AgBT), respectively. The hollowing mechanism and the dimensionality of the hollow nanoshells are different when different sulfur-containing reagents are used. The difference of the hollow nanoshells originates from the size of the S-containing species and the crystallinity of the nanoshells. The similarity of the ionic crystalline structure of AgCl0.5Br0.5 and Ag2S and the size of the anions favors the hollowing process to follow the Kirkendall process, in which the outward diffusion of halide ions is faster than the inward diffusion of S2− ions. In contrast, the molecular-type feature and layered crystalline structure of AgBT result in a hollowing process that is different from the Kirkendall process and exhibits slower reaction kinetics. Comparative studies highlight the importance of the reactant species and the crystalline structure of the nanoshell materials in determining the hollowing process and properties of the resulting nanoshells.
Experimental
Synthesis of AgCl0.5Br0.5 nanocubes
The synthesis of AgCl0.5Br0.5 nanocubes was performed through a controlled precipitation reaction between Ag+ ions and mixed halide ions with [Cl−]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Br−] = 1:1.30,31 In a typical synthesis, 2.5 g of poly(vinylpyrrolidone) (PVP, Mw ≈ 55000, Sigma Aldrich) powder was added to a 50 mL three-necked flask containing 12 mL of ethylene glycol (EG, Fisher Chemical). The mixture was stirred vigorously for 20 minutes under an N2 atmosphere until the PVP powder was completely dissolved. To this solution was added 10.2 mg (17.5 mmol) of NaCl (Fisher Chemical) and 18.2 mg (17.7 mmol) of NaBr (Acros Organics). The flask was then sealed by capping the three necks with rubber septa, through which syringe needles were used to flow N2 gas and purge the mixture. The N2 purging continued for 20 minutes at 60 °C to dissolve the salt crystals completely while the stirring was maintained. The flask was wrapped with aluminum foil to block light. 1 mL of EG solution of 0.34 mol L−1 AgNO3 was then added to the EG solution containing PVP and halide ions at a rate of 1 mL min−1 using a syringe pump, triggering a precipitation reaction between Ag+ cations and halide anions. The stirring rate was maintained at 340 rpm (revolutions per minute), and the temperature was maintained at 60 °C throughout the reaction of 2 hours.30,32 The product solution was then centrifuged, and the product nanocubes were washed with ethanol. For example, 7 mL of the product solution was mixed with 14 mL of ethanol (Fisher) in a 50 mL centrifuge tube, followed by centrifugation at 6000 rpm for 20 minutes. The settled nanocubes were washed by repeated dispersion/centrifuging cycles three times, each of which involved adding 20 mL of ethanol to the nanocubes and centrifuging the dispersion at 6000 rpm for 20 minutes. The washed AgCl0.5Br0.5 nanocubes were then dispersed in 20 mL ethanol, forming the stock solution for further use.
:[Br−] = 1:1.30,31 In a typical synthesis, 2.5 g of poly(vinylpyrrolidone) (PVP, Mw ≈ 55000, Sigma Aldrich) powder was added to a 50 mL three-necked flask containing 12 mL of ethylene glycol (EG, Fisher Chemical). The mixture was stirred vigorously for 20 minutes under an N2 atmosphere until the PVP powder was completely dissolved. To this solution was added 10.2 mg (17.5 mmol) of NaCl (Fisher Chemical) and 18.2 mg (17.7 mmol) of NaBr (Acros Organics). The flask was then sealed by capping the three necks with rubber septa, through which syringe needles were used to flow N2 gas and purge the mixture. The N2 purging continued for 20 minutes at 60 °C to dissolve the salt crystals completely while the stirring was maintained. The flask was wrapped with aluminum foil to block light. 1 mL of EG solution of 0.34 mol L−1 AgNO3 was then added to the EG solution containing PVP and halide ions at a rate of 1 mL min−1 using a syringe pump, triggering a precipitation reaction between Ag+ cations and halide anions. The stirring rate was maintained at 340 rpm (revolutions per minute), and the temperature was maintained at 60 °C throughout the reaction of 2 hours.30,32 The product solution was then centrifuged, and the product nanocubes were washed with ethanol. For example, 7 mL of the product solution was mixed with 14 mL of ethanol (Fisher) in a 50 mL centrifuge tube, followed by centrifugation at 6000 rpm for 20 minutes. The settled nanocubes were washed by repeated dispersion/centrifuging cycles three times, each of which involved adding 20 mL of ethanol to the nanocubes and centrifuging the dispersion at 6000 rpm for 20 minutes. The washed AgCl0.5Br0.5 nanocubes were then dispersed in 20 mL ethanol, forming the stock solution for further use.
Synthesis of hollow Ag2S nanoshells
A stock solution of 12 mM Na2S was prepared by dissolving 14.6 mg of Na2S·9H2O (Sigma Aldrich) in 5 mL ethanol. A 100 µL aliquot of the Na2S stock solution was added to 1 mL of the AgCl0.5Br0.5 nanocube stock solution in a 2 mL Eppendorf centrifuge tube followed by prompt vortexing. The reaction lasted 10 minutes in the dark to ensure the complete transformation of the AgCl0.5Br0.5 nanocubes. The product nanoparticles were collected through centrifuging and washing cycles similar to those applied to the AgCl0.5Br0.5 nanocubes.Synthesis of hollow AgBT nanoshells
In the synthesis of hollow silver benzenethiolate (AgBT) nanoshells, 1 mL of the AgCl0.5Br0.5 nanocube stock solution was mixed with 0.5 mL ethanolic solution of 0.49 M benzenethiol (HBT, 99%, Acros Organics) in a 2 mL Eppendorf centrifuge tube followed by prompt vortexing. The reaction continued for 12 hours in the dark under ambient conditions. The product nanoparticles were collected through centrifuging and washing cycles similar to those applied to the AgCl0.5Br0.5 nanocubes.Characterization of nanoparticles
Scanning electron microscopy (SEM) images were obtained with an FEI Quanta 450 scanning electron microscope operating in high vacuum mode. The SEM samples were prepared by drop-casting 5 µL aliquots of nanoparticle dispersions on clean silicon wafers with a size of 5 mm × 5 mm, followed by drying in a vacuum desiccator in the dark. Backscattered electron images were recorded using a concentric backscattering detector as part of the same SEM. An X-MaxN 50 spectrometer as part of the SEM was used to perform energy-dispersive X-ray spectroscopy (EDS). Transmission electron microscopy (TEM) images were obtained using a JEOL JEM-1400 electron microscope. X-ray diffraction (XRD) patterns were obtained with a Bruker D8 X-ray diffractometer consisting of a Cu Kα (λ = 0.154 nm) source. The XRD samples were prepared in a similar way to preparing the SEM samples except that more nanoparticles were deposited on the silicon wafers. The optical UV-visible absorption spectra of the nanoparticle dispersions were recorded using an Evolution 220 photospectrometer (Thermo Scientific).Kinetics studies of transforming AgCl0.5Br0.5 nanocubes to hollow nanoshells
The transformation kinetics was probed using time-dependent UV-visible absorption spectroscopy. In a typical study, 40 µL of the AgCl0.5Br0.5 nanocube stock solution was added to a quartz cuvette containing 3 mL of ethanol. The solution was mixed thoroughly via air agitation using a glass pipet. With ethanol as the background reference, the absorbance at a single wavelength that corresponds to the characteristic feature of the resulting hollow nanoshells made of Ag2S (490 nm) and AgBT (360 nm) was continuously recorded at appropriate time intervals depending on the reaction kinetics. The hollowing reactions were performed using either 40 µL ethanolic solution of 12 mM Na2S or 0.49 M HBT. The absorption spectra in the range of 250–600 nm were also obtained using the same procedure to track any noticeable changes.Evaluation of the photocatalytic activity
In a typical experiment, 40 µL of benzyl alcohol (BAL, 99%, Acros Organics) and 100 µL of the Ag2S nanoshell stock solution were added to 3 mL of benzotrifluoride (BTF, 99%, Alfa Aesar) that was saturated with O2. The mixture was transferred to a 4 mL glass vial enclosed with a rubber septum. Light illumination was provided using a high-intensity LED illuminator (Fiber-lite MI LED B1, a wavelength of 350–750 nm) at room temperature. Aliquots of 50 µL were taken out from the reaction dispersion at different times and analyzed using an Agilent 7820A gas chromatograph (GC) equipped with a flame ionization detector (FID). Control experiments were done in the dark. The reference Ag2S solid nanoparticles were synthesized using the following procedure. 0.0773 g of AgNO3 was mixed with 0.1044 g of thiourea (Sigma-Aldrich) in 15 mL of water. After the powders were dissolved completely, to the solution was added a 15 mL aqueous solution of cetyltrimethylammonium bromide (CTAB, 0.0422 g), followed by heating up to and maintaining at 160 °C for 1 hour in a microwave reaction vessel using a microwave reactor (Anton Paar, Monowave 300).Results and discussion
The low solubility constants of silver sulfide (Ag2S with Ksp = 6.3 × 10−50 mol3 L−3 at 20 °C)33 and silver thiolates (AgBT with Ksp = 2.4 × 10−21 mol−2 L−2)34 usually drive fast precipitation reactions between Ag+ cations and the corresponding anions in solution, resulting in difficulty in synthesizing nanoparticles of Ag2S and AgBT in a controlled manner. Slowing the precipitation reaction kinetics requires the concentrations of precursor ions to be extremely low at the micromolar or even nanomolar levels. The low concentrations make it challenging to supply enough precursor ions to synthesis nanoparticles in the desired quantities. In this work, ternary silver halide nanocubes made of AgCl0.5Br0.5 are used as an alternative precursor that can provide and maintain freestanding Ag+ at a low concentration. The continuous release of Ag+ ions from the AgCl0.5Br0.5 nanocubes ensures the availability of precursor Ag+ ions to synthesize nanoparticles of Ag2S and AgBT in large quantities using Na2S and HBT as anion precursors, respectively. The reactions convert the precursor AgCl0.5Br0.5 nanocubes to hollow nanoshells of Ag2S and AgBT. Fig. 1 compares the key steps involved in the conversion from AgCl0.5Br0.5 nanocubes to Ag2S and AgBT nanoshells.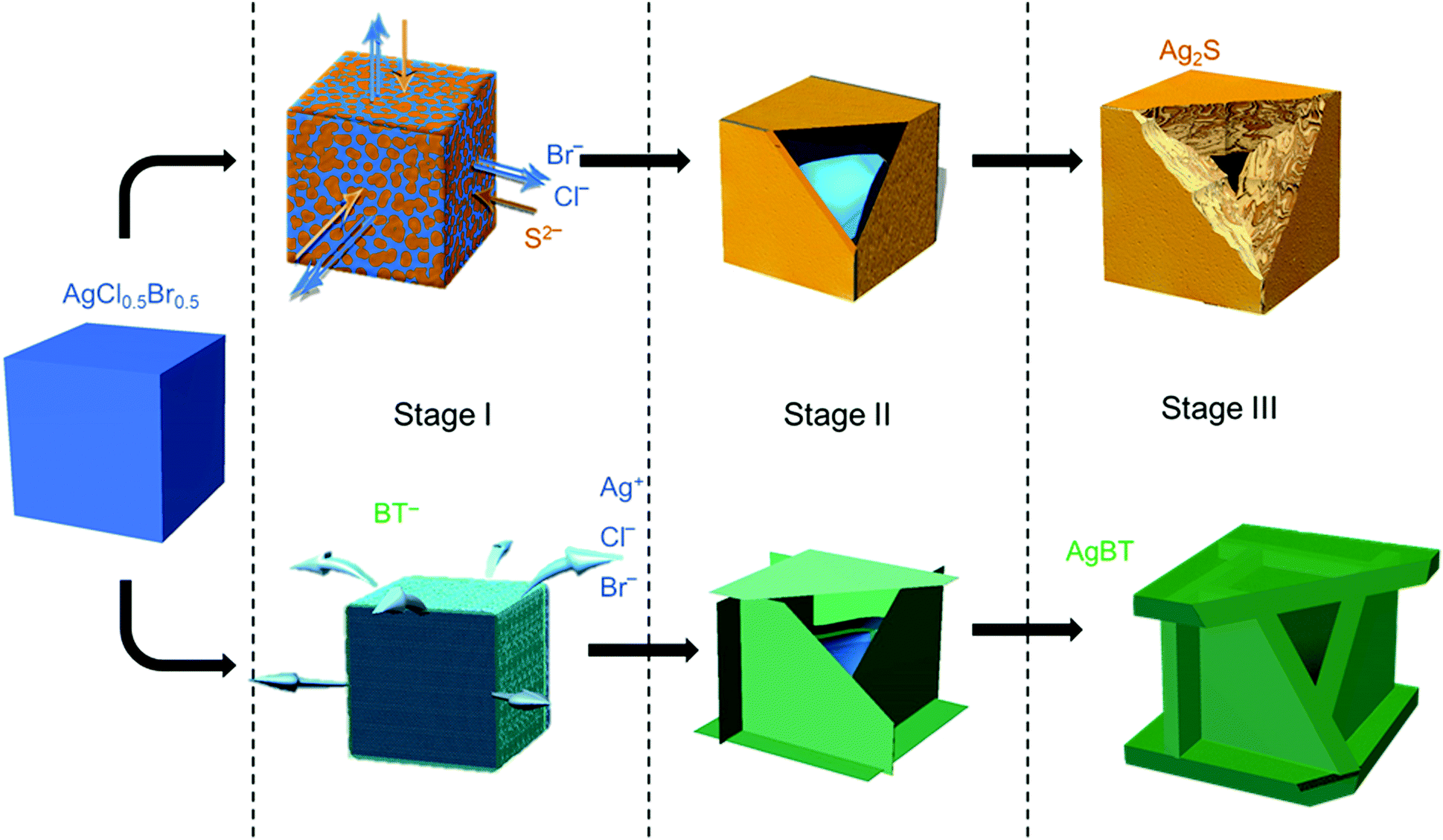 | ||
| Fig. 1 Schematic illustration highlighting the major steps involved in transforming AgCl0.5Br0.5 nanocubes to hollow nanoshells of Ag2S (top) and silver benzenethiolate (AgBT, bottom). For clarity, different materials are coded with different colors: blue-AgCl0.5Br0.5, green-AgBT, yellow-Ag2S. The arrows highlight the diffusion directions of various ions involved in the anion exchange reactions between the template AgCl0.5Br0.5 nanocubes and the reaction solutions. The edges of the nanostructures of stage II and III are partially opened to portray the hollow geometry and wall thickness of the nanoshells. | ||
The AgX (X denotes the halide ions) crystals exhibit low values of Ksp (e.g., 1.8 × 10−10 and 5.0 × 10−13 mol2 L−2 for AgCl and AgBr at 25 °C in aqueous solution, respectively),33 resulting in a low and constant concentration of Ag+ in liquid solution at room temperature. When an ethanolic dispersion of the AgCl0.5Br0.5 nanocubes is mixed with a solution of either Na2S or HBT, a precipitation reaction is initiated on the surfaces of the AgCl0.5Br0.5 nanocubes (Stage I, Fig. 1). Since Na2S can directly dissociate into freestanding S2− anions and Na+ cations in an ethanolic solution, the concentration of S2− in a Na2S solution is high to react with the available freestanding Ag+ at a large rate. The high-rate precipitation of Ag2S favors heterogeneous nucleation on the surfaces of the AgCl0.5Br0.5 nanocubes with the formation of high-density nuclei. The crystalline orientations of individual Ag2S nuclei are random because of the different crystalline structures of Ag2S and AgCl0.5Br0.5 (hexagonal lattice versus cubic lattice).31,35 Therefore, the growth of the nuclei in the following reaction results in the transformation of the AgCl0.5Br0.5 nanocubes into nanostructured Ag2S with polycrystallinity. In contrast, benzenethiol molecules only release freestanding benzenethiolate anions (BT−) with low concentration in solution because of the weak acidity of the HBT molecules. The reaction of the AgCl0.5Br0.5 nanocubes with benzenethiol slowly precipitates AgBT to nucleate on the surfaces of the AgCl0.5Br0.5 nanocubes, forming a low density of nuclei. Since AgBT crystals exhibit a layered structure consisting of parallel conjugated molecular layers,36 the AgBT nucleus on each surface of the nanocube prefers anisotropic in-plane growth to form two-dimensional (2D) AgBT nanoplates.
The layers of reaction products, i.e., Ag2S and AgBT, initially formed on the surfaces of the template AgCl0.5Br0.5 nanocubes create boundaries between the sacrificial AgCl0.5Br0.5 solid phase and the liquid solution phase containing precursor anions. As the reaction proceeds, the unreacted AgCl0.5Br0.5 underneath the product layers constantly releases freestanding Ag+ to precipitate with the precursor anions, accumulating more product material on the outer surfaces of the nanoparticles to thicken the product layers and enlarge the lateral dimensions of the cubic nanoparticles (Stage II, Fig. 1). The crystalline nature of the product layers and the size of the precursor anions determine whether solid-state anion exchange can occur across the product layers. The Ag2S product layers are ionic crystals, in which both halide anions (Cl− and Br−) and S2− anions can diffuse. However, the outward diffusion of halide ions is faster than the inward diffusion of S2− ions. This difference of ionic diffusion in the Ag2S layers is ascribed to the lower charge of halide ions (1 negative charge per halide ion versus 2 negative charges per sulfide ion) since the size of the S2− anion (184 pm) is comparable to those of the Cl− (181 pm) and Br− (185 pm) anions.37 Both the unbalanced ionic diffusion and the continuous supply of Ag+ from the AgCl0.5Br0.5 nanocubes result in the formation of hollow shells of Ag2S. The inward diffusion of S2− cations across the Ag2S shells also thickens the Ag2S shells by forming more Ag2S on the inner surfaces of the shells. The observed morphological transformation is consistent with the classic nanoscale Kirkendall hollowing process.19 Hollow Ag2S nanoshells are formed when the Ag+ cations in the template AgCl0.5Br0.5 nanocubes are completely converted to Ag2S (Stage III, Fig. 1). The involvement of the Kirkendall process results in hollow interiors with sizes smaller than the size of the AgCl0.5Br0.5 nanocubes. In contrast, the AgBT product layers are molecular-type 2D sheets, which prevent the benzenethiolate anions with large size from penetrating the AgBT layers. This property eliminates the occurrence of the Kirkendall process in the reaction of the AgCl0.5Br0.5 nanocubes and HBT. Therefore, the transformation of the AgCl0.5Br0.5 nanocubes to hollow AgBT nanoshells is dominated only by the precipitation reaction between benzenethiolate anions and Ag+ cations released from the AgCl0.5Br0.5 nanocubes, which produces AgBT to deposit on the outer surfaces of the AgBT shells. The slow release of Ag+ from the AgCl0.5Br0.5 nanocrystals supports the continuous growth of the AgBT films, which extends the AgBT layers even beyond the surfaces of the AgCl0.5Br0.5 nanocubes due to the 2D layered crystalline structure of AgBT. The 2D layered structure prefers the growth along the side edges of the AgBT layers rather than their basal surfaces.32,36 When the size of individual layers is too large, the low concentration of available Ag+ cations cannot support their diffusion to the edges of the AgBT layers. Instead, the precipitation reaction of the available Ag+ cations and BT− anions then favors deposition of AgBT on the basal surfaces of the AgBT layers, thickening the AgBT layers. Continuous reaction eventually transforms the AgCl0.5Br0.5 nanocubes to hollow boxes, each of which is composed of six AgBT nanoplates (Stage III, Fig. 1). Due to the absence of the Kirkendall process, the AgBT nanoshells exhibit hollow interiors with dimensions similar to those of the template AgCl0.5Br0.5 nanocubes.
The morphological and dimensional differences of the hollow nanoshells of Ag2S and AgBT were characterized with SEM imaging. Fig. 2 presents the SEM images of the template AgCl0.5Br0.5 nanocubes, which have an average edge length of 157.6 nm (Fig. 2a and b), and the corresponding hollow nanoshells of Ag2S (Fig. 2d and e), and AgBT (Fig. 2g and h). The absence of XRD peaks of AgCl0.5Br0.5 (Fig. 2c) in the XRD patterns of the product nanoshells (Fig. 2f and i) confirms that the anion replacement reactions consume the template AgCl0.5Br0.5 nanocubes completely (the SEM images taken during the conversion of nanocubes to nanoshells are shown in Fig. S2, ESI†). The AgCl0.5Br0.5 nanocubes exhibit smooth surfaces. Although the cubic morphology remains, the polycrystalline Ag2S nanoshells exhibit rough and uneven surfaces, which is ascribed to the fast precipitation reaction kinetics and the formation of high-density nuclei at the early heterogeneous nucleation stage. The EDS mapping images (Fig. S3, ESI†) show the uniform distributions of both Ag and S elements in the nanoshells. Each wall consists of many Ag2S nanocrystals with an average grain size of 23 nm. The backscattered electron images highlight the wall thickness of the nanoshells of 27.9 nm (Fig. S1c, ESI†). On the other hand, the anisotropic in-plane growth of individual AgBT plates enlarges them beyond the edges of the template AgCl0.5Br0.5 nanocubes, consistent with the observations of the SEM images (Fig. 2g and h). Each AgBT nanoplate has a uniform thickness throughout the entire plate and exhibits smooth surfaces. The formation of only one AgBT nanoplate on each surface of the AgCl0.5Br0.5 nanocubes indicates that the initial slow nucleation forms only one nucleus on each surface. Otherwise, forming multiple nuclei on each surface could grow multiple nanoplates to exhibit a flower-like morphology.32 The XRD pattern of the AgBT nanoshells exhibits a quintuplet of peaks that are different orders of reflection of the same lattice, confirming the layered crystalline structure of the AgBT nanoplates with an interlayer distance of 14.1 Å (Fig. 2i). Furthermore, the EDX mapping (Fig. S4, ESI†) shows that Ag, S, and C in the thiol molecules are distributed uniformly in the nanoshell.
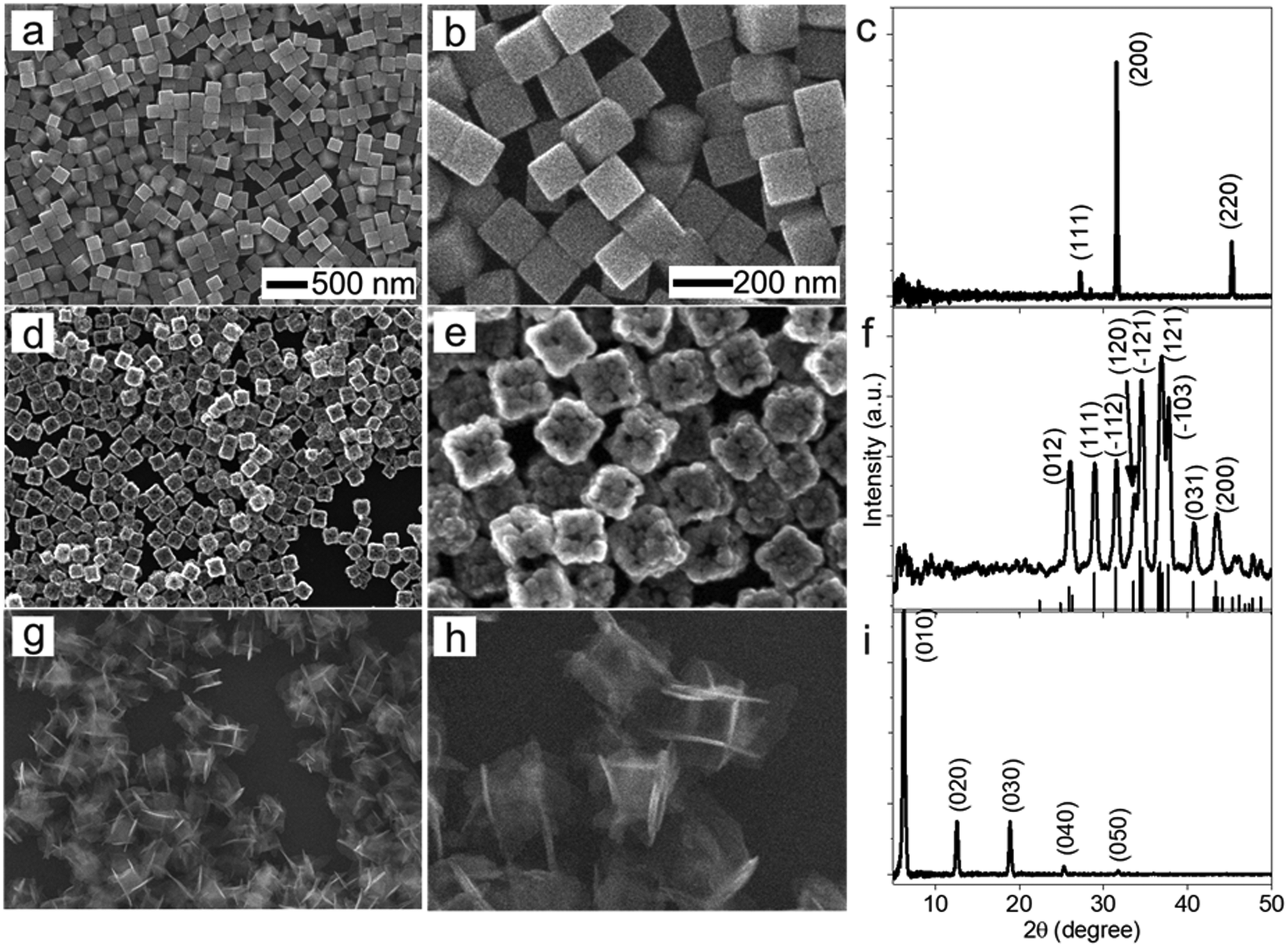 | ||
| Fig. 2 SEM images of (a and b) AgCl0.5Br0.5 nanocubes and the corresponding hollow nanoshells made of (d and e) Ag2S and (g and h) AgBT formed from the complete anion exchange reactions with Na2S and benzenethiol (HBT) solutions, respectively. The scale bar in (a) also applies to (d) and (g). The scale bar in (b) also applies to (e) and (h). The right column presents the XRD patterns of (c) the template AgCl0.5Br0.5 nanocubes, (f) the hollow Ag2S nanoshells, and (i) the hollow AgBT nanoshells. The standard XRD pattern of monoclinic acanthite Ag2S (PDF# 14-0072) is presented at the bottom of (f) as a reference. | ||
The hollowing mechanisms illustrated in Fig. 1 indicate that the involvement of the Kirkendall process produces hollow nanoshells with hollow interiors different from those formed in the absence of the Kirkendall process. Fig. 3 compares the average size and size distribution of the nanostructures shown in Fig. 2 in terms of their outer edge lengths and inner void edge lengths. Converting the AgCl0.5Br0.5 nanocubes to the hollow Ag2S nanoshells increases the outer edge length by 17.4 nm, i.e., from 157.6 nm to 175.0 nm. The average edge length of the interior voids of the Ag2S nanoshells is 119.2 nm, which is 38.4 nm smaller than the size of the template AgCl0.5Br0.5 nanocubes. Converting the AgCl0.5Br0.5 nanocubes to the AgBT hollow nanoshells increases the average lateral dimension from 157.6 nm to 189.2 nm, corresponding to a mean thickness of 15.8 nm for individual AgBT nanoplates. In contrast, the average edge length of the inner void is 154.7 nm, which is very close to the edge length of the template AgCl0.5Br0.5 nanocubes, indicating that the inward diffusion of anions barely occurs. The significant dimensional difference between the Ag2S nanoshells and the AgBT nanoshells confirms that the hollowing mechanisms involved in the formation of these two types of nanoshells are different, i.e., the nanoscale Kirkendall process makes a contribution to the formation of hollow Ag2S nanoshells with thick walls but not to the formation of AgBT nanoshells.
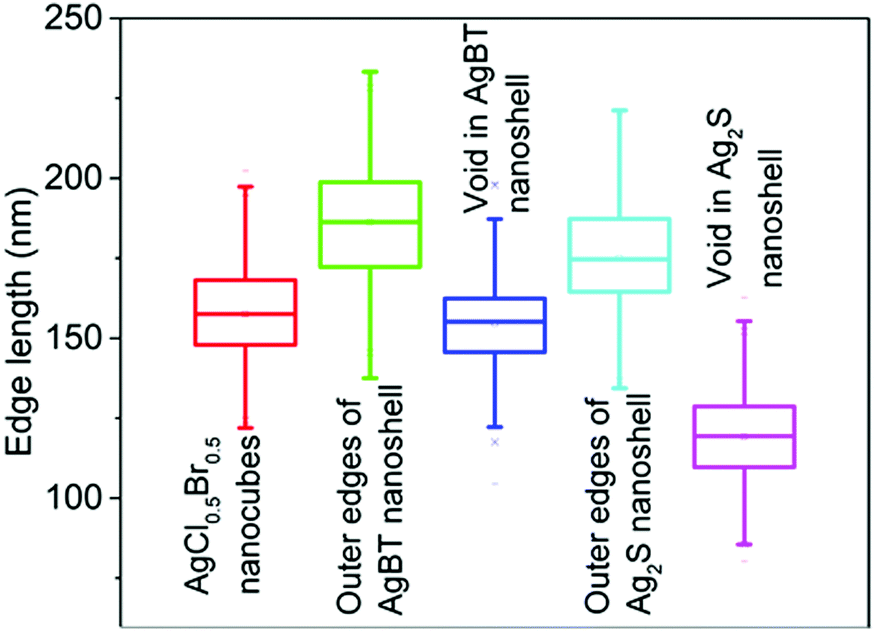 | ||
| Fig. 3 Average edge lengths of the template AgCl0.5Br0.5 nanocubes, and the resulting nanoshells of Ag2S and AgBT. The edge lengths of the inner voids of the hollow nanoshells are also presented. The statistical analysis was performed using the corresponding backscattered electron SEM images as shown in Fig. S1 (ESI†). | ||
The chemical reaction kinetics of transforming the AgCl0.5Br0.5 nanocubes to hollow nanoshells were analyzed by time-dependent UV-visible absorption spectroscopy because the product materials of the nanoshells exhibit different absorption spectra from those of the template AgCl0.5Br0.5 nanocubes. For example, Ag2S shows an absorption peak centered at 490 nm corresponding to its direct bandgap,38,39 while the absorption peak of AgCl0.5Br0.5 is located at 312 nm (Fig. 4a and Fig. S5, ESI†). In the course of transforming the AgCl0.5Br0.5 nanocubes to the Ag2S nanoshells, the absorbance at 490 nm quickly increases to a plateau, indicating that a fast reaction consumes the template AgCl0.5Br0.5 nanocubes quickly. The fast reaction kinetics are ascribed to the synergy between the Kirkendall process and the precipitation reaction of freestanding Ag+ cations with high-concentration freestanding S2− anions. The reaction completes the transformation within ∼5 seconds at an initial reaction rate of 0.066 s−1 under the experimental conditions (Fig. 4b).
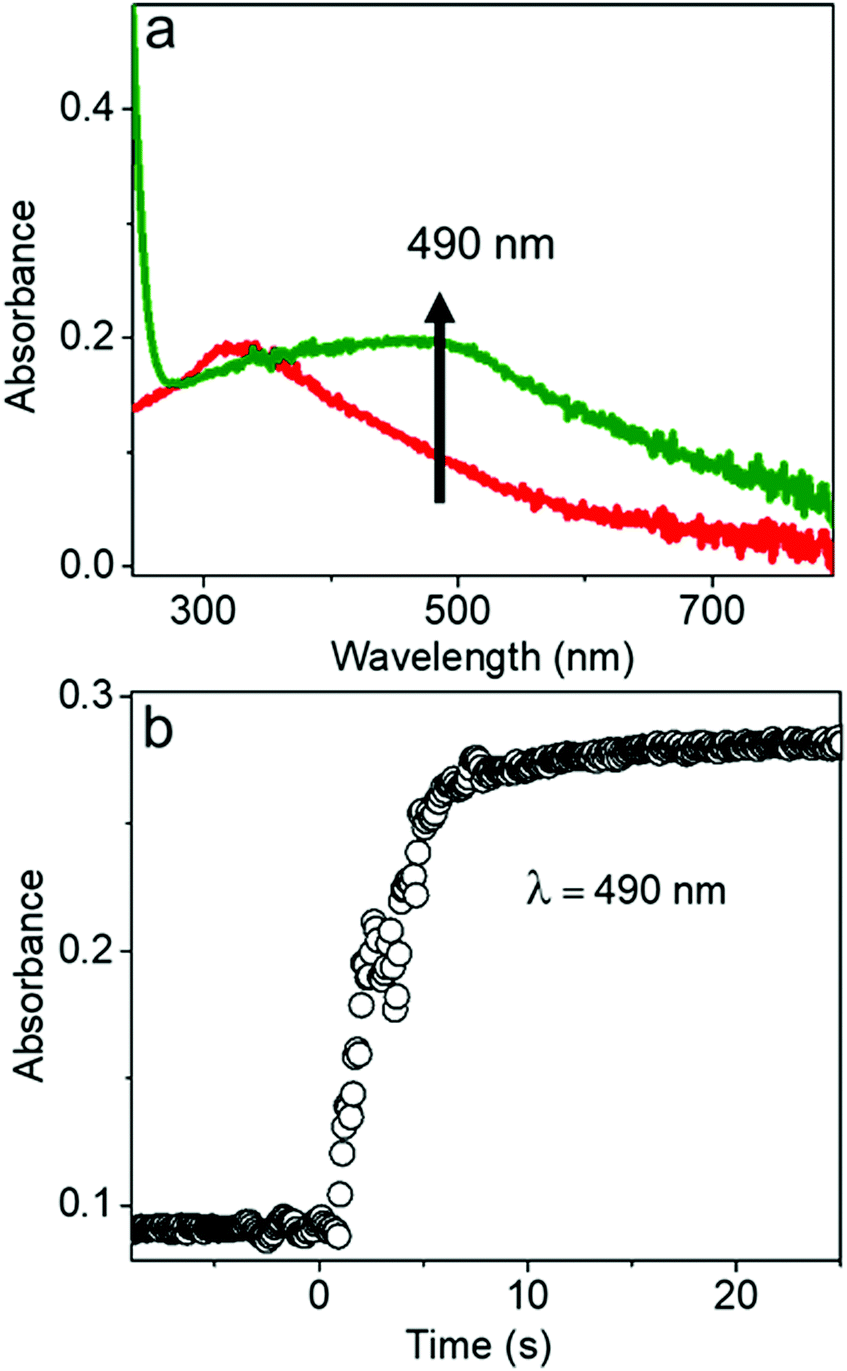 | ||
| Fig. 4 (a) UV-visible absorption spectra of dispersions of the AgCl0.5Br0.5 nanocubes (red) and the corresponding Ag2S hollow nanoshells (green). (b) Time-dependent variation of optical absorbance at a wavelength of 490 nm, which corresponds to the characteristic absorption peak of Ag2S, in the course of the anion exchange reaction between the AgCl0.5Br0.5 nanocubes and the Na2S solution. | ||
The characteristic absorption peak of AgBT, which increases as the reaction of the AgCl0.5Br0.5 nanocubes and HBT proceeds, appears at 360 nm (Fig. 5a). The sharp absorption peak originates from ligand-to-metal charge transfer. The time-dependent increase of the absorbance at 360 nm determines that the initial reaction rate for forming AgBT nanoshells is only 0.000051 s−1 (Fig. 5b), which is approximately 1300 times lower than that of forming Ag2S nanoshells. The slow reaction kinetics is consistent with the absence of the Kirkendall hollowing process in transforming the AgCl0.5Br0.5 nanocubes to the hollow AgBT nanoshells. The lower concentration of available freestanding BT− anions is also responsible for the slow reaction kinetics. The comparison of reaction kinetics highlights the importance of precursor anions in determining the morphology and dimensions of the product hollow nanoshells that are synthesized through anion replacement reactions using the template AgCl0.5Br0.5 nanocubes.
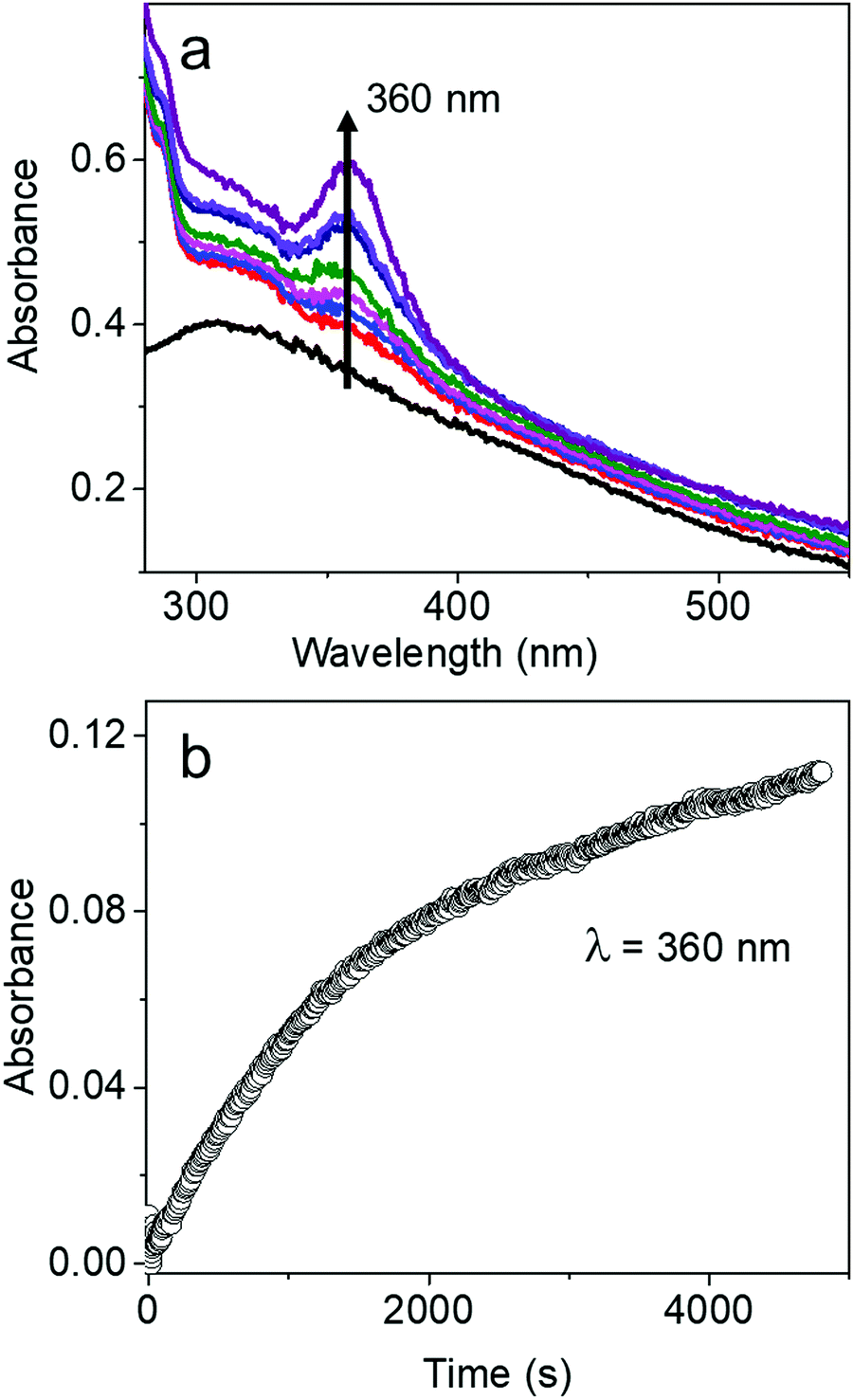 | ||
| Fig. 5 (a) A series of UV-visible absorption spectra of the AgCl0.5Br0.5 nanocubes before (black curve) and after reaction with HBT for varying times. (b) Time-dependent variation of optical absorbance at 360 nm, the absorption peak position of AgBT, during the anion exchange reaction. | ||
Hollow nanoshells of assembled small-size nanograins usually exhibit much higher colloidal stability than the freestanding nanocrystals with a size similar to the nanograins. The geometry of the hollow nanoshells exposes both the outer and inner surfaces, promoting applications requiring large surface areas such as photocatalysis. A model reaction, i.e., photocatalytic oxidation of BAL to BAD (benzaldehyde), is used to evaluate the colloidal stability of the hollow Ag2S nanoshells (Fig. 6a) and freestanding Ag2S nanocrystals (Fig. 6b and Fig. S6, ESI†) under operation conditions.40–42 The results show that the hollow Ag2S nanoshells maintain the photocatalytic activity over 48 hours despite a slight drop at long reaction time (Fig. 6c). The rate decrease of the photocatalytic reaction could be attributed to catalyst deactivation after prolonged irradiation with light. In contrast, the photocatalytic activity of the freestanding Ag2S nanocrystals quickly drops to nearly zero (corresponding to the plateau of the blue curve in Fig. 6d) after the reaction lasts 6 hours. The complete deactivation of the freestanding Ag2S nanocrystals in a short time is ascribed to the quick aggregation and fusion of the small Ag2S nanocrystals into large particles (Fig. S7, ESI†), highlighting the low colloidal stability of the Ag2S nanocrystals with small sizes. The enhanced colloidal stability of small nanocrystal domains with exposure of large surface areas represents one of the unique properties of hollow nanoshells. The results shown in Fig. 6 justify the importance of synthesizing hollow nanoshells with controlled parameters for exploring unique properties and improving the performance of current applications.
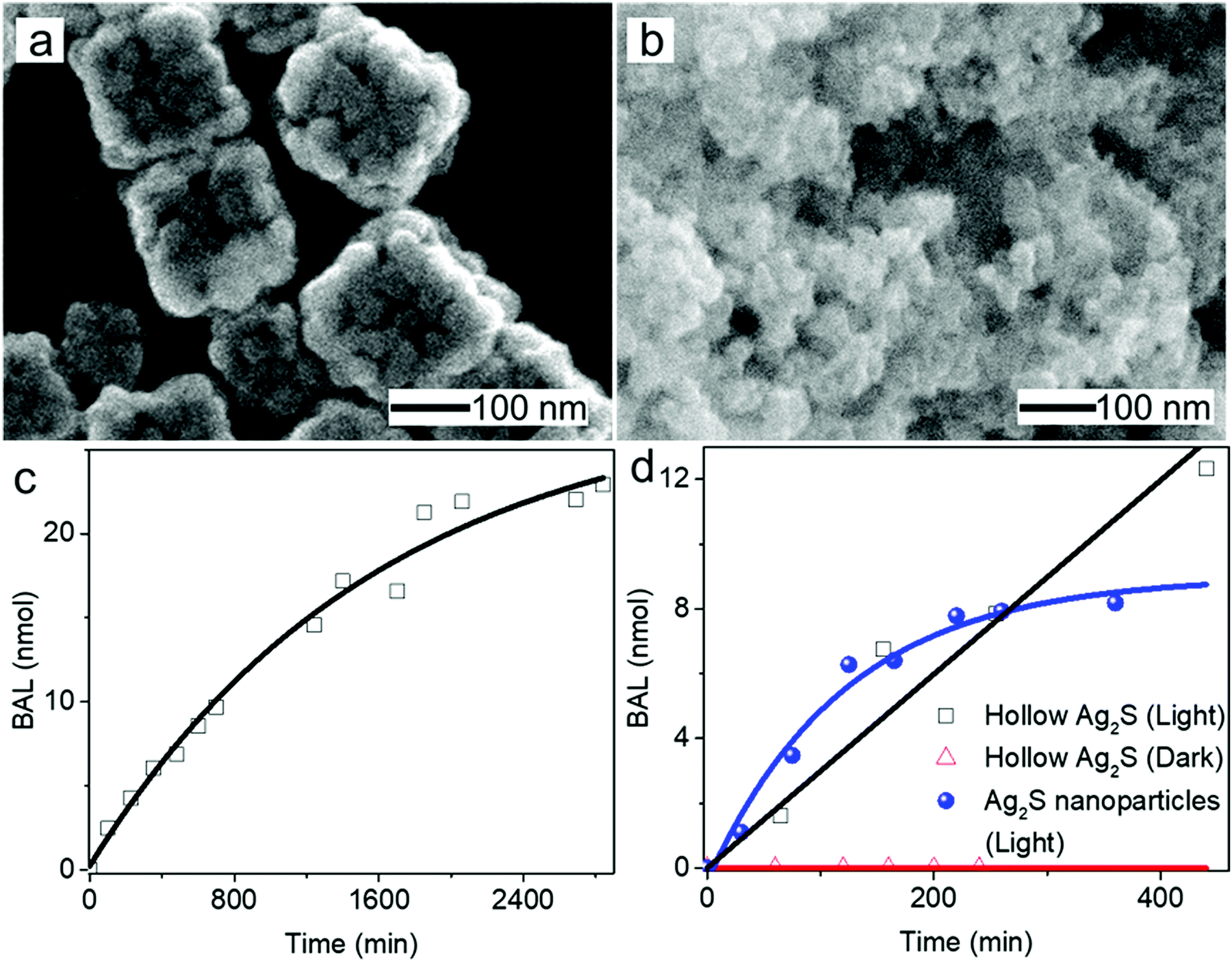 | ||
Fig. 6 High-magnification SEM images of (a) Ag2S nanoshells and (b) freestanding Ag2S nanoparticles (serving as a control sample). (c) Time-dependent yield of BAD produced from the photocatalytic oxidation of BAL using the hollow Ag2S nanoshells as a photocatalyst. (d) Comparison of the photocatalytic production of BAD using the hollow Ag2S nanoshells (□) and the freestanding Ag2S nanoparticles ( ) as photocatalysts for a short reaction time. The reaction in the presence of the hollow Ag2S nanoshells in the dark was also studied for comparison ( ) as photocatalysts for a short reaction time. The reaction in the presence of the hollow Ag2S nanoshells in the dark was also studied for comparison ( ). ). | ||
Conclusions
In summary, we have systematically demonstrated the structural, morphological, and compositional evolution of hollow nanostructures using well-defined AgCl0.5Br0.5 nanocubes as a sacrificial template, which react with sulfur-containing precursors including sodium sulfide and benzenethiol. The lower solubility of sulfur-containing silver salts (e.g., AgBT and Ag2S) compared to the silver halides governs the transformation of the AgCl0.5Br0.5 nanocubes to hollow nanoshells of Ag2S and AgBT in the presence of Na2S and HBT, respectively. The size of the sulfur-containing anions and the crystalline structure of the nanoshell materials represent two crucial factors in determining the hollowing transformation mechanism, which influences the physical dimensions of the nanoshells and the kinetics of the anion replacement of the hollowing process. For instance, due to the similarities in crystalline nature between AgCl0.5Br0.5 and Ag2S with the comparable sizes of S2− and halide ions, the transformation follows the nanoscale Kirkendall effect where the outward diffusion of halide ions and the inward diffusion of S2− occur across an initial Ag2S shell layer. The differences in charge between halide ions and S2− ions (i.e., −1 versus −2) leads to the faster outward diffusion of halide ions compared to the inward diffusion of S2− ions, corresponding to a net outward diffusion of mass and the formation of hollow Ag2S nanoshells. In contrast, the initial molecular-type AgBT shell layer does not allow inward diffusion of the relatively large BT− anions to exchange the core halide anions, preventing the Kirkendall process. The absence of the Kirkendall process in the reaction with HBT gives much slower reaction kinetics than the transformation of Ag2S. Overall, the size of the exchanging ionic species and the crystal structure of the shell material determine the hollowing mechanism and the morphological evolution. Thorough understanding of various hollowing mechanisms and associated kinetics benefits design and control of hollowing processes. Therefore, template-based synthesis of hollow nanostructures offers a simple and scalable approach towards well-defined hollow nanostructures with desirable properties and functions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We sincerely acknowledge the startup fund from Temple University and the Temple Materials Institute (TMI) for providing instrumentation for material characterization.Notes and references
- Y. Sun, B. Mayers and Y. Xia, Adv. Mater., 2003, 15, 641–646 CrossRef CAS.
- D. Chen and J. Ye, Adv. Funct. Mater., 2008, 18, 1922–1928 CrossRef CAS.
- G. Prieto, H. Tüysüz, N. Duyckaerts, J. Knossalla, G.-H. Wang and F. Schüth, Chem. Rev., 2016, 116, 14056–14119 CrossRef CAS.
- J. Lan, K. Wang, Q. Yuan and X. Wang, Mater. Chem. Front., 2017, 1, 1217–1222 RSC.
- G. Wu, L. Zhao, H. Liang, Y. Yan and H. Tan, Mater. Chem. Front., 2019, 3, 922–930 RSC.
- M. A. Mahmoud and M. A. El-Sayed, Nano Lett., 2011, 11, 946–953 CrossRef CAS.
- H.-P. Liang, L.-J. Wan, C.-L. Bai and L. Jiang, J. Phys. Chem. B, 2005, 109, 7795–7800 CrossRef CAS.
- Y. Chen, H. Chen, D. Zeng, Y. Tian, F. Chen, J. Feng and J. Shi, ACS Nano, 2010, 4, 6001–6013 CrossRef CAS.
- Y. Li, N. Li, W. Pan, Z. Yu, L. Yang and B. Tang, ACS Appl. Mater. Interfaces, 2017, 9, 2123–2129 CrossRef CAS.
- D. H. M. Dam, L. Zhao, S. A. Jelsma, Y. Zhao and A. S. Paller, Mater. Chem. Front., 2019, 3, 1113–1122 RSC.
- M. A. Mahmoud, D. O’Neil and M. A. El-Sayed, Chem. Mater., 2014, 26, 44–58 CrossRef CAS.
- 12 X. Lai, J. Li, B. A. Korgel, Z. Dong, Z. Li, F. Su, J. Du and D. Wang, Angew. Chem., Int. Ed., 2011, 50, 2738–2741 CrossRef CAS.
- M. A. Mahmoud, F. Saira and M. A. El-Sayed, Nano Lett., 2010, 10, 3764–3769 CrossRef CAS.
- F. Zheng, M. He, Y. Yang and Q. Chen, Nanoscale, 2015, 7, 3410–3417 RSC.
- X. Lai, J. E. Halpert and D. Wang, Energy Environ. Sci., 2012, 5, 5604–5618 RSC.
- J. Wang, N. Yang, H. Tang, Z. Dong, Q. Jin, M. Yang, D. Kisailus, H. Zhao, Z. Tang and D. Wang, Angew. Chem., Int. Ed., 2013, 52, 6417–6420 CrossRef CAS.
- S. Xu, C. M. Hessel, H. Ren, R. Yu, Q. Jin, M. Yang, H. Zhao and D. Wang, Energy Environ. Sci., 2014, 7, 632–637 RSC.
- J. Wang, H. Tang, H. Wang, R. Yu and D. Wang, Mater. Chem. Front., 2017, 1, 414–430 RSC.
- W. Wang, M. Dahl and Y. Yin, Chem. Mater., 2013, 25, 1179–1189 CrossRef CAS.
- Y. Sun, B. T. Mayers and Y. Xia, Nano Lett., 2002, 2, 481–485 CrossRef CAS.
- L. Yang, B. Zhang, W. Ma, Y. Du, X. Han and P. Xu, Mater. Chem. Front., 2018, 2, 1523–1528 RSC.
- J. Zhao, X. Cheng, G. Zhao, Y. Lu, Y. Wang, H. Wang and H. Chen, Chem. Commun., 2019, 55, 5571–5574 RSC.
- Y. Sun and Y. Xia, Nano Lett., 2003, 3, 1569–1572 CrossRef CAS.
- S. Wu and Y. Sun, Small Methods, 2018, 2, 1800165 CrossRef.
- Y. Sun and Y. Xia, J. Am. Chem. Soc., 2004, 126, 3892–3901 CrossRef CAS.
- K. Cao, T. Jin, L. Yang and L. Jiao, Mater. Chem. Front., 2017, 1, 2213–2242 RSC.
- M. H. Oh, T. Yu, S.-H. Yu, B. Lim, K.-T. Ko, M.-G. Willinger, D.-H. Seo, B. H. Kim, M. G. Cho, J.-H. Park, K. Kang, Y.-E. Sung, N. Pinna and T. Hyeon, Science, 2013, 340, 964–968 CrossRef CAS.
- Y. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711–714 CrossRef CAS.
- C. An, J. Wang, J. Liu, S. Wang and Y. Sun, ChemSusChem, 2013, 6, 1931–1937 CrossRef CAS.
- Z. Li and Y. Sun, J. Mater. Chem. A, 2013, 1, 6786–6793 RSC.
- S. C. Abeyweera, K. D. Rasamani and Y. Sun, Acc. Chem. Res., 2017, 50, 1754–1761 CrossRef CAS.
- S. C. Abeyweera and Y. Sun, MRS Adv., 2019, 4, 2087–2094 CrossRef CAS.
- D. Ebbing and S. D. Gammon, General Chemistry, Cengage Learning, 2016, p. 586 Search PubMed.
- D. Wang, Flotation Reagents: Applied Surface Chemistry on Minerals Flotation and Energy Resources Beneficiation: Volume 1: Functional Principle, Springer, Singapore, 2016, p. 79 Search PubMed.
- H. Doh, S. Hwang and S. Kim, Chem. Mater., 2016, 28, 8123–8127 CrossRef CAS.
- I. G. Dance, K. J. Fisher, R. M. H. Banda and M. L. Scudder, Inorg. Chem., 1991, 30, 183–187 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- K. Akamatsu, S. Takei, M. Mizuhata, A. Kajinami, S. Deki, S. Takeoka, M. Fujii, S. Hayashi and K. Yamamoto, Thin Solid Films, 2000, 359, 55–60 CrossRef CAS.
- L. Dong, Y. Chu, Y. Liu and L. Li, J. Colloid Interface Sci., 2008, 317, 485–492 CrossRef CAS.
- L. Wang, ChemPhysChem, 2015, 16, 1542–1550 CrossRef CAS.
- N. Zhang, C. Han, Y.-J. Xu, J. J. Foley IV, D. Zhang, J. Codrington, S. K. Gray and Y. Sun, Nat. Photonics, 2016, 10, 473–482 CrossRef CAS.
- X. Dai, K. D. Rasamani, S. Wu and Y. Sun, Mater. Today Energy, 2018, 10, 15–22 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures including backscattered electron images, EDS mapping data and XRD spectra. See DOI: 10.1039/c9qm00544g |
| This journal is © the Partner Organisations 2020 |