
Anion replacement in silver chlorobromide nanocubes: two distinct hollowing mechanisms†

Abstract
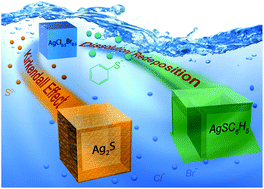
Silver chlorobromide nanocubes have shown promise as a class of sacrificial template for the synthesis of silver-based hollow nanostructures through chemical transformation reactions. Strong chemical bonding between silver and sulfur enables the transformation of AgCl0.5Br0.5 nanocubes into hollow nanoshells of silver–sulfur compounds. During these transformations, factors such as the crystalline structure and nature of the shell material influence the reaction kinetics, and thus the structural and morphological evolution of the hollow structures. Herein, we systematically investigate the hollowing mechanism and associated reaction kinetics for the controlled conversion of AgCl0.5Br0.5 nanocubes into hollow nanostructures. The use of sulfide anions (S2−) and benzenethiolate anions (BT−) for the anion exchange reaction with halide ions has been compared to illustrate the hollowing mechanisms. The S2− ions are comparable in size to the halide ions, resulting in a nano-scale Kirkendall effect that is mainly responsible for the formation of hollow Ag2S nanoshells. In contrast, the Kirkendall effect is absent from forming hollow nanoshells of silver benzenethiolate (AgBT) because the much larger size of BT− ions relative to halide ions hinders their diffusion in the solid AgBT lattice. The molecular-type layered structure of AgBT also disfavors both the inward and outward diffusion of ions through the AgBT shells, thus leading to an overall reduction in the kinetics of the transformation reaction. The transformation reaction is driven only by the dissolution of AgCl0.5Br0.5 and reprecipitation of Ag+ and BT− ions. These results allow for understanding of the underlying factors in hollowing mechanisms to enable the controlled synthesis of well-defined hollow nanostructures with desired properties for a broad range of potential applications.
- This article is part of the themed collection: Hollow Structures for Energy Applications
 Please wait while we load your content...
Please wait while we load your content...

