
Carbon dioxide reduction by lanthanide(III) complexes supported by redox-active Schiff base ligands†
Nadir
Jori
 ,
Davide
Toniolo
,
Bang C.
Huynh
,
Rosario
Scopelliti
and
Marinella
Mazzanti
*
,
Davide
Toniolo
,
Bang C.
Huynh
,
Rosario
Scopelliti
and
Marinella
Mazzanti
*
Institut des Sciences et Ingénierie Chimiques Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
First published on 19th August 2020
Abstract
Here we have explored the ability of Schiff bases to act as electron reservoirs and to enable the multi-electron reduction of small molecules by lanthanide complexes. We report the reductive chemistry of the Ln(III) complexes of the tripodal heptadentate Schiff base H3trensal (2,2′,2′′-tris(salicylideneimino)triethylamine), [LnIII(trensal)], 1-Ln (Ln = Sm, Nd, Eu). We show that the reduction of the [EuIII(trensal)] complex leads to the first example of a Eu(II) Schiff base complex [{K(μ-THF)(THF)2}2{EuII(trensal)}2], 3-Eu. In contrast the one- and two-electron reduction of the [NdIII(trensal)] and [SmIII(trensal)] leads to the intermolecular reductive coupling of the imino groups of the trensal ligand and to the formation of one and two C–C bonds leaving the metal center in the +3 oxidation state. The resulting one- and two electron reduced complexes [{K(THF)3}2Ln2(bis-trensal)], 2-Ln, and [{K(THF)3}2{K(THF)}2Ln2(cyclo-trensal)], 4-Ln (Ln = Sm, Nd) are able to effect the reductive disproportionation of carbon dioxide by transferring the electrons stored in the C–C bonds to CO2 to selectively afford carbonate and CO. The selectivity of the reaction contrasts with the formation of multiple CO2 reduction products previously reported for a U(IV)-trensal system.
Introduction
The redox-chemistry of lanthanide complexes has attracted increasing attention in recent years because of the unique reactivity of Ln(II) ions in small molecule activation and in particular their ability to reduce N2, CO2 or CO.1 However, lanthanides can only undergo one-electron transfer processes and therefore reduction of CO2 or other small molecules (N2, CO) can only occur by simultaneous electron transfer by several mononuclear complexes2 or by suitably designed polynuclear compounds.3A novel alternative approach for implementing multielectron transfer in lanthanide complexes takes advantage of redox-active ligands for the storage and release of electrons. Redox-active ligands are increasingly used across the periodic table to facilitate metal-based multielectron transfer reactivity and catalytic activity.4 Redox-active ligands can either directly store electrons during reactivity by forming stable radicals or through the formation of new reversible C–C bonds in the ligand backbone.5
Recently it has been shown that multiple redox states can become accessible to lanthanides when associated to redox active ligands.4c,d,5a,c,6 However, only two systems were reported that show that the electrons stored in lanthanide complexes supported by redox active ligands can be used for the reductive transformation of substrates such as S8, Se or 9,10-phenanthrenequinone.7
Redox active ligands were also found to enable redox-switchable catalysis in rare-earth promoted polymerization chemistry.8 However, the use of redox-active ligands to implement multi-electron transfer in f elements chemistry remains significantly rarer compared to the d block. Moreover, there are so far no examples of the ability of such systems to reduce less reactive molecules such as carbon dioxide.
The ability of Schiff bases to act as electron reservoirs and to enable the multi-electron reduction of small molecules such as carbon dioxide has been identified in complexes of d block metals9 and more recently in complexes of uranium.5d,10
In contrast, the use of Schiff base ligands in lanthanide chemistry has been essentially limited to Ce(IV)11 and Ln(III) complexes that have found application as efficient Near-IR and visible luminescence emitters12,13 and have shown attractive physical properties.14
The reductive chemistry of lanthanide Schiff base complexes remains so far largely unexplored.6f,7b,15
Our group demonstrated that the tetradentate salophen Schiff base salophen (N,N′-disalicylidene-o-phenylenediaminate) acts as redox active ligand also when combined to lanthanide ions.7b Notably we showed that the reduction of monoligand and bis-ligand Ln(III) complexes of salophen results in the reduction of the imino groups of the ligand followed by formation of intermolecular or intramolecular C–C bonds(Scheme 1). The electrons stored in the C–C bonds could be used in the reduction of I2 or 9,10-phenanthrenequinone but could not effect the reduction of less reactive substrates such as carbon dioxide.
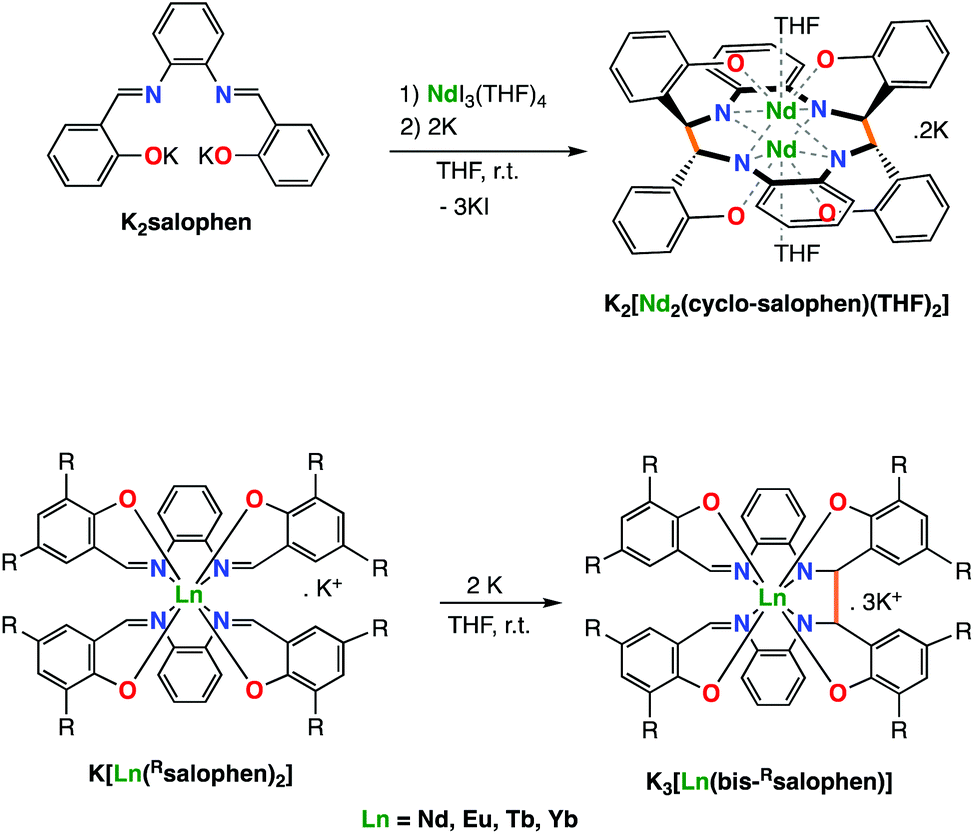 | ||
| Scheme 1 The two-electron reduction of mono- (top) and bis-(bottom) Schiff base complexes of Nd(III) with alkali metals leads to intermolecular (top) or intramolecular C–C bond formation (bottom). | ||
Here we report the reductive chemistry of the Ln(III) complexes of the tripodal heptadentate Schiff base H3trensal (2,2′,2′′-tris(salicylideneimino)triethylamine), [LnIII(trensal)], 1-Ln (Ln = Sm, Nd, Eu). [LnIII(trensal)] complexes have been previously synthesised and crystallographically characterized for a wide range of ions.16 These complexes have attracted significant interest for their magnetic properties14b,c,17 but their reductive chemistry was never explored. Here we show that the reduction of the [EuIII(trensal)] leads to the Eu(II) analogue. In contrast the one- and two-electron reduction of the [NdIII(trensal)] and [SmIII(trensal)] leads to the intermolecular reductive coupling of the imino groups of the trensal ligand and to the formation of one and two C–C bonds leaving the metal center in the +3 oxidation state. The resulting reduced complexes are able to effect the reductive disproportionation of carbon dioxide by transferring the electrons stored in the C–C bonds to CO2 to selectively afford carbonate and CO. The selectivity of the reaction contrasts with the formation of multiple CO2 reduction products previously reported for a U(IV)-trensal system.10c
Experimental section
Materials and methods
Synthetic procedures
1-Ln were prepared by reacting 1 equiv. of K3trensal salt with the anhydrous [LnX3] (Ln = Sm, Eu, Nd; X = OTf, I) precursors in THF. Because of the low solubility of the 1-Ln complexes in THF we did not attempt the separation of the potassium salts and the resulting crude mixture was used as such for further reduction. The 1-Ln complexes were characterized by 1H NMR spectroscopy.[Nd(trensal)], 1-Nd. 1H NMR (pyr-d5, 400 MHz, 298 K) δ = 27.20 (s, 3H), 11.28 (s, 3 H), 9.29 (s, 3H), 8.14 (s, 3H), 7.28 (s, 3H), 4.15 (s, 6H) and −5.28 (s, 6H).
[Sm(trensal)], 1-Sm. 1H NMR (pyr-d5, 400 MHz, 298 K) δ = 8.11 (d, 3H), 7.68 (s, 3H), 7.13 (t, 3H), 6.83 (t, 3H), 6.79 (d, 3H), 3.02 (s, 6H) and −0.42 (s, 6H).
[Eu(trensal)], 1-Eu. 1H NMR (pyr-d5, 400 MHz, 298 K) δ = 9.12 (s, 6H), 7.18 (s, 3 H), 5.68 (s, 3H), 3.84 (s, 3H), 3.09 (s, 3H), 1.36 (s, 6H) and −23.78 (s, 3H).
Elem. anal. calc. (%) for [K4Nd2(cyclo-trensal)(THF)1.5]: C, 49.22%; H, 4.54%; N, 7.65%. Found: C, 48.82%; H, 5.04%; N, 7.65%. Crystals suitable for X-ray diffraction were obtained by slow diffusion of hexane into a THF solution of 4-Nd.
Reactivity with CO2
Upon removal of the solvent in vacuo and after dissolution in basic D2O (pD = 13.4), quantitative 13C{1H} NMR experiments were performed (13C-labelled sodium acetate as reference).
The yields in carbonate correspond to 96% and 98% for Nd and Sm, respectively.
The characterization of the product was prevented by its low solubility in organic solvents. The 1H NMR spectrum of the reaction mixture taken immediately after addition of 4 equiv. of 13CO2 showed only signals assigned to complex 1. The 13C{1H} NMR spectrum of the reaction mixtures (THF-d8, 100 MHz, 298 K) showed a signal at δ = 181.30 ppm of 13CO.
Upon removal of the solvent in vacuo and after dissolution in basic D2O (pD = 13.4), quantitative 13C{1H} NMR experiments were performed (13C-labelled sodium acetate as reference).
The yields in carbonate correspond to 100% and 97% for Nd and Sm, respectively.
X-ray crystallography
The diffraction data for the analysed crystal structures were collected at low temperature using Cu (2-Sm, 3-Eu) or Mo (2-Nd, 4-Sm, 4-Nd) Kα radiation on a Rigaku SuperNova dual system in combination with Atlas type CCD detector. The data reduction and correction were carried out by CrysAlisPro.21The solutions and refinements were performed by SHELXT22 and SHELXL,23 respectively. The crystal structures were refined using full-matrix least-squares based on F2 with all non-H atoms defined in anisotropic manner. Hydrogen atoms were placed in calculated positions by means of the “riding” model.
Compound 2-Nd displayed 2 disordered THF molecules. Their treatment was carried out by the split method; some similarity restraints (SADI and SIMU cards) were applied to bond distances and to the ADPs. Compound 2-Sm showed a quite similar disorder but the entire K(THF)3+ moiety was involved; it was rather easy to identify two different positions of K but the splitting of the THFs did not work and this explains why their occupancies is less than 1 and why some restraints were used (ISOR, DFIX and RIGU cards). The crystal structure of 3-Eu showed the disorder of one THF. DFIX and SIMU restraints were used to get reasonable parameters. The crystal structures of 4-Sm and 4-Nd were also affected by the same kind of disorder, e.g. 2 and 3 disordered THF molecules; some similarity restraints were imposed on the bond distances (SADI and DFIX commands), some were used for the ADPs (SIMU and RIGU commands).
Owing to the high degree of displayed disorder, the free solvent molecules (about 1 THF) were removed by applying the SQUEEZE algorithm of PLATON.24
Electrochemical methods
Cyclic voltammetry data were carried out at room temperature in an argon-filled glovebox described above. Data were collected using a Biologic SP-300 potentiostat connected to a personal computer. All samples were saturated in complex with 0.1 M [Bu4N][PF6] supporting electrolyte in THF solution. The experiments were carried out with a platinum disk (d = 5 mm) working electrode, a platinum wire counter electrode, and an Ag/AgCl reference electrode. The experiments were repeated on independently synthesized samples to assess the reproducibility of the measurement. Potential calibration was performed at the end of each data collection cycle using the ferrocene/ferrocenium [(C5H5)2Fe]+/0 couple as an internal standard.Results and discussion
Reduction of [Ln(trensal)] complexes
The previously reported 1-Ln complexes were prepared “in situ” using a salt metathesis procedure previously used in the synthesis of the [UIV(trensal)I] complex.19This procedure avoids the presence of protic solvent or residual water in the final product that would be difficult to remove. The addition of one equivalent of KC8 to the 1-Ln complexes in THF afforded the complexes [{K(THF)3}2Ln2(bis-trensal)], 2-Ln in 70% (Ln = Sm) and 53% (Ln = Nd) yield (Scheme 2). Alternatively, 2-Sm can also be obtained from the reaction of the divalent [SmI2] with one equiv. of the trensalK3 ligand salt (Fig. S3†). The 1H NMR spectra of 2-Nd and 2-Sm (Fig. S1 and S2† respectively) in THF-d8 show the presence of 27 overlapping signals in the paramagnetic range, consistent with the presence of fully asymmetric solution species and in agreement with the solid state structure.
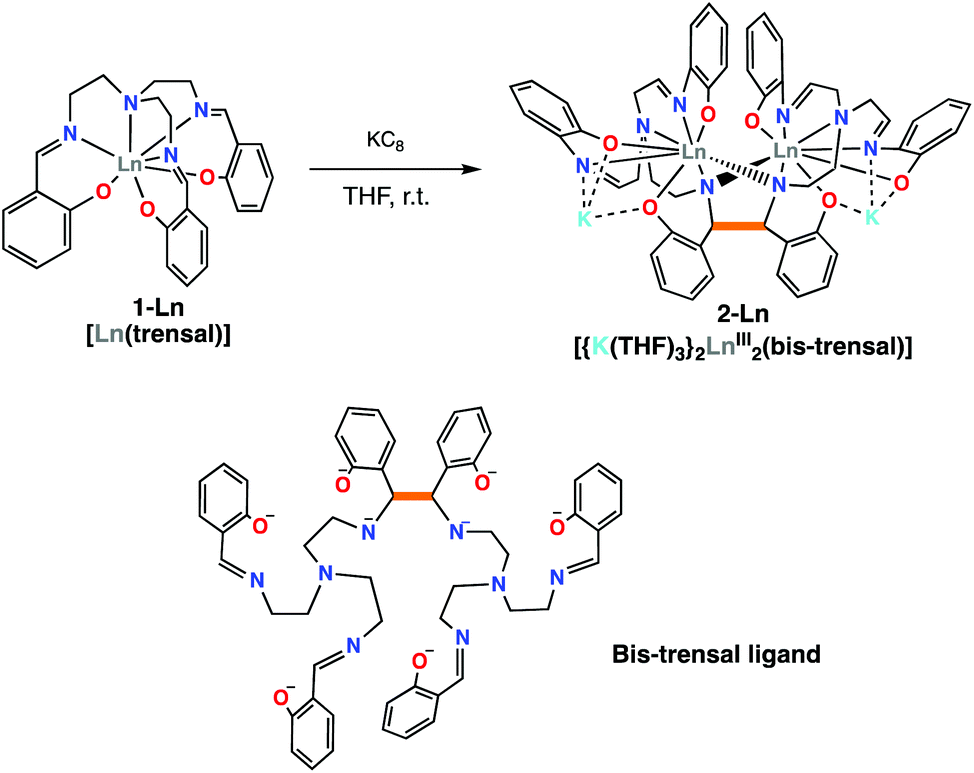 | ||
| Scheme 2 Synthesis of [{K(THF)3}2Ln2(bis-trensal)], 2-Ln, Ln = Nd, Sm. | ||
The two dinuclear 2-Nd and 2-Sm complexes crystallize in the C2/c group and are isostructural. Their solid-state molecular structure shows respectively the presence of two neodymium(III) (Fig. 1) and samarium(III) (Fig. S12†) cations coordinated by the octa-anionic bis-trensal ligand, formed by C–C coupling of two imino groups of the original trensal ligands. The formation of the bis-trensal ligand was previously observed by our group during the reduction of the trensal ligand by UI3 yielding the [UIV2(bis-trensal)] complex.10c The resulting dinucleating ligand in 2-Ln holds the two metal centres in close proximity (Nd–Nd distance = 3.8922(3) Å and Sm–Sm distance = 3.8389(4) Å) (Table 1).
 | ||
| Fig. 1 Molecular structure of complex 2-Nd (C–C bond between imine highlighted in yellow, 50% probability ellipsoids). Hydrogen atoms and THF molecules were omitted for clarity. | ||
| Complex | C–C | C–Nim | C–Nam | M–Nim | M–Nam | M–M |
|---|---|---|---|---|---|---|
| 1-Nd | — | 1.280(6) | — | 2.573(4) | — | — |
| 1-Sm | — | 1.274(8) | — | 2.531(4) | — | — |
| 1-Eu | — | 1.277(16) | — | 2.535(7) | — | — |
| 2-Nd | 1.573(6) | 1.287(2) | 1.487(4) | 2.67(4) | 2.48(7) | 3.8922(3) |
| [Nd(bis-salophen)]3− | 1.56(2) | 1.29(2) | 1.45(1) | 2.64(4) | 2.45(2) | — |
| 2-Sm | 1.586(7) | 1.288(6) | 1.485(5) | 2.63(5) | 2.50(7) | 3.8389(4) |
| 3-Eu | — | 1.272(6) | — | 2.63(5) | — | — |
| 4-Nd | 1.558(3) | 1.273(3) | 1.472(4) | 2.6895(16) | 2.6(1) | 3.42619(19) |
| [Nd2(cyclo-salophen)]2− | 1.622(3) | — | 1.459(3) | — | 2.416(9) | 3.54(1) |
| 4-Sm | 1.560(6) | 1.263(6) | 1.470(3) | 2.679(9) | 2.61(12) | 3.4077(4) |
The 2-Ln complexes present two equivalent Ln centers octacoordinated by two imino nitrogen (avg Ln–Nim = 2.67(4) Å for Nd and 2.63(5) Å for Sm), one amino nitrogen (Ln–Nam = 2.782(2) Å for Nd and 2.745(4) Å for Sm), two bridging amido nitrogen atoms (Ln–Namido = 2.478(2) Å for Nd and 2.50(7) Å for Sm) and three phenoxide oxygen atoms (avg Ln–O = 2.38(4) Å for Nd and 2.36(4) Å) of the bis-trensal ligand. Two of the phenoxide oxygen atoms (O1 and O3) coordinate a potassium cation, which is also bound to three molecules of THF.
The structural parameters of 2-Ln, as summarized in Table 1, clearly show that the reduction has occurred on the imino groups of the ligands rather than on the metal ions. The values of the C–C bond distance (1.573(6) Å for 2-Nd and 1.586(7) for 2-Sm) compare well with the intermolecular C–C bonds found in the previously reported [Na2U(bis-salophen)] (1.559(7) Å),5d [Nd(bis-salophen)]3− (1.56(2) Å)7b and [U2(bis-tBuTrensal)] (1.59(2) Å)10c complexes. The C–Namido bond distances in 2-Nd (1.487(4) Å) and in 2-Sm (1.485(5) Å) are longer than the C–Nimino bond distances (1.287(2) Å in 2-Nd and 1.288(6) Å in 2-Sm) which remain similar to those reported16a,b for 1-Nd (1.280(6) Å) and 1-Sm (1.274(8) Å), respectively. The Ln–Namido bond distances for 2-Nd (2.48(7) Å) and 2-Sm (2.50(7) Å) are shorter than the Ln–Nimino distances (2.573 (4) Å for 1-Nd and 2.531(4) Å 1-Sm) found in the 1-Ln complexes. These values are in agreement with the presence of the bis-amido, tetra-imino, hexaphenolate bis-trensal ligand resulting from the reductive coupling of two imino moieties from two different trensal units. These distances also compare well with the values reported7b for the complex [Nd(bis-salophen)]3− (Nd–Nam (2.45(2) Å) and Nd–Nim (2.64(4) Å) which also presents a reduced Schiff base ligand.
In contrast with what observed with Nd and Sm the reduction of 1-Eu did not afford the Eu(III) bis-trensal complex, but led instead to the reduction of the metal center and formation of the Eu(II) complex of the trensal ligand, [{K(μ-THF)(THF)2}2{EuII(trensal)}2], 3-Eu (Scheme 3). No 1H NMR spectrum was observed as expected for the 4f7 configuration of Eu(II).
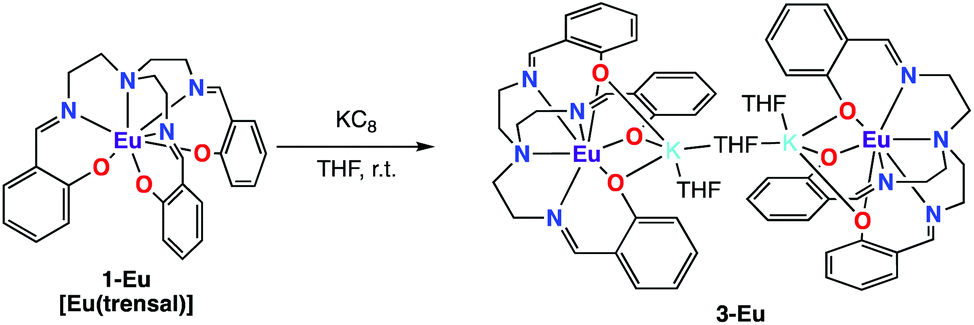 | ||
| Scheme 3 Reduction of 1-Eu. | ||
The molecular structure of 3-Eu (Fig. 2) shows two potassium-bound Eu(II)(trensal) complexes bridged by a THF molecule. Each Eu(II)(trensal) moiety binds a potassium counterion trough three phenoxide oxygen atoms, the latter being also coordinated by one terminal THF molecule and one bridging THF.
 | ||
| Fig. 2 Molecular structure of complex 3-Eu (50% probability ellipsoids). Hydrogen atoms were omitted for clarity. | ||
The europium centers are heptacoordinated by the amino nitrogen atoms (Eu–Namino = 2.826(4) Å), the three imino nitrogen atoms (avg Eu–Nimino = 2.66(4) Å) and the three phenoxide oxygen atoms (avg Eu–O = 2.45(1) Å) from the trensal ligand. The average Eu–N and Eu–O distances are consistent with the formation of a divalent europium complex,21 and the C–N distances (1.272(6) Å) remain unchanged compared to those found22 in the crystal structure of the H3trensal ligand (average 1.266(3) Å). The Eu–N distances in 3-Eu are longer compared to those reported16b for the [EuIII(trensal)] complex (Eu–Namino = 2.761(9) Å, Eu–Nimino = 2.535(7) Å, Eu–O = 2.233(6) Å), as expected due to the larger radius of the reduced metal center. The structural parameters show that the reduction occurred at the metal center rather than in the ligand like in 2-Nd and 2-Sm.
These results indicate that while for 1-Nd and 1-Sm the reduction of the ligand occurs before the reduction of the metal center, in the case of 1-Eu reduction occurs first on the metal center probably due to its lower redox potential (Eu3+/Eu2+ = −0.35 V, Sm3+/Sm2+ = −1.55 V, Nd3+/Nd2+ = −2.6 V, referenced vs. NHE).23
This result differs significantly from what was observed with the Eu(III) complex of the salophen Schiff base K[Eu(salophen)2], where the reduction following the addition of alkali metal occurs on the ligand scaffold.7b These results indicate that the trensal ligand is better suited than the salophen ligand to stabilize the divalent oxidation state in europium compounds.
Complex 3-Eu is the first example of a divalent europium complex supported by a Schiff base ligand. Only one complex of a divalent lanthanide (Sm(II)) supported by a Schiff base ligand had been reported so far.6f
Complexes 2-Nd and 2-Sm can be further reduced in a controlled fashion by the addition of excess KC8 (three equiv.) to a white suspension of [Ln(trensal)], 1-Ln in THF affording the complexes [{K(THF)3}Ln2(cyclo-trensal)], 4-Ln in 77% (Ln = Sm) and 78% (Ln = Nd) yield (Scheme 4). When the reduction was conducted with only 2 equiv. of KC8 it yielded a mixture of 2-Ln and 4-Ln. The 1H NMR spectrum of 4-Nd (Fig. S4†) shows rather broad signals suggesting the presence of fluxional solution species. In contrast the 1H NMR spectrum of 4-Sm (Fig. S5†) show only one set of 14 well resolved signals suggesting the presence of D2h symmetric solution species in agreement with the observed D2h pseudo-symmetry of the solid state structure.
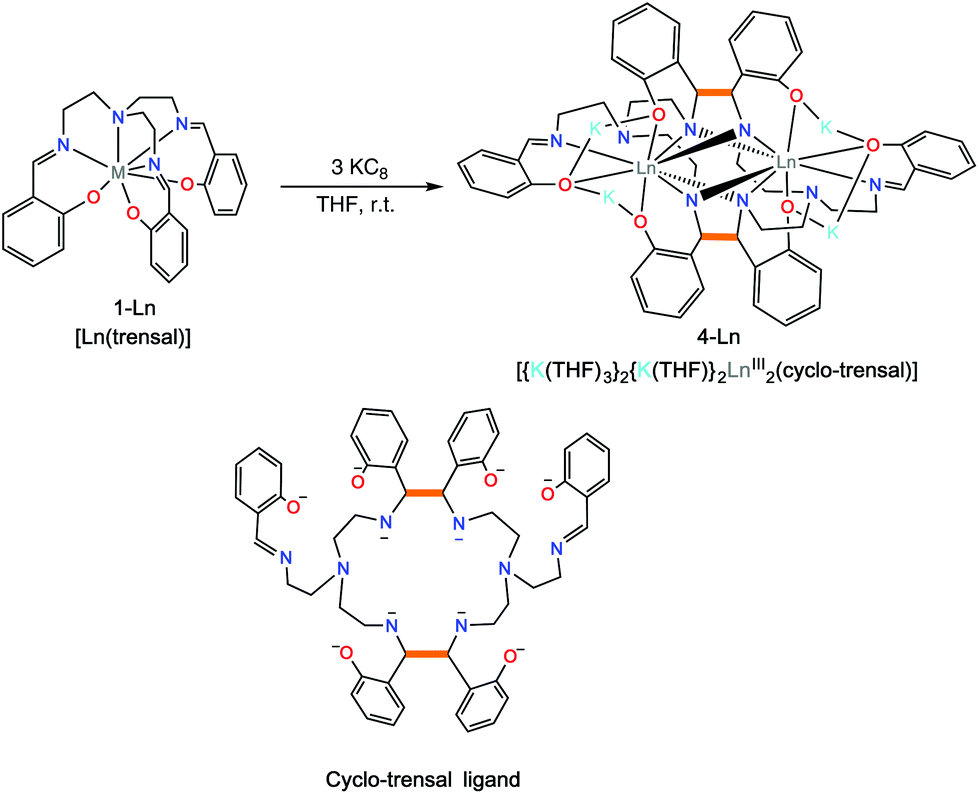 | ||
| Scheme 4 Synthesis of [{K(THF)3}2{K(THF)}2Ln2(cyclo-trensal)], 4-Ln, Ln = Nd, Sm. | ||
The two complexes 4-Nd and 4-Sm crystallize in the P21/n group and are isostructural. The molecular structure of the 4-Ln complexes shows the presence of two Nd(III) (Fig. 3) or Sm(III) cations (Fig. S13†) bound by the dodecadentate decaanionic amidophenolate macrocyclic ligand cyclo-trensal produced in the reductive coupling of two imino groups of the trensal ligand. The dinucleating cyclo-trensal ligand in 4-Ln holds the two Ln centers in close proximity, at Ln–Ln distances (3.42619(19) Å for Nd and 3.4077(4) Å for Sm) shorter than those found in 2-Ln (3.8922(3) Å for Nd and 3.8389(4) Å for Sm) or in the macrocyclic complex in [Nd2(cyclo-salophen)]4− (Nd–Nd = 3.54(1) Å)7b (Table 1).
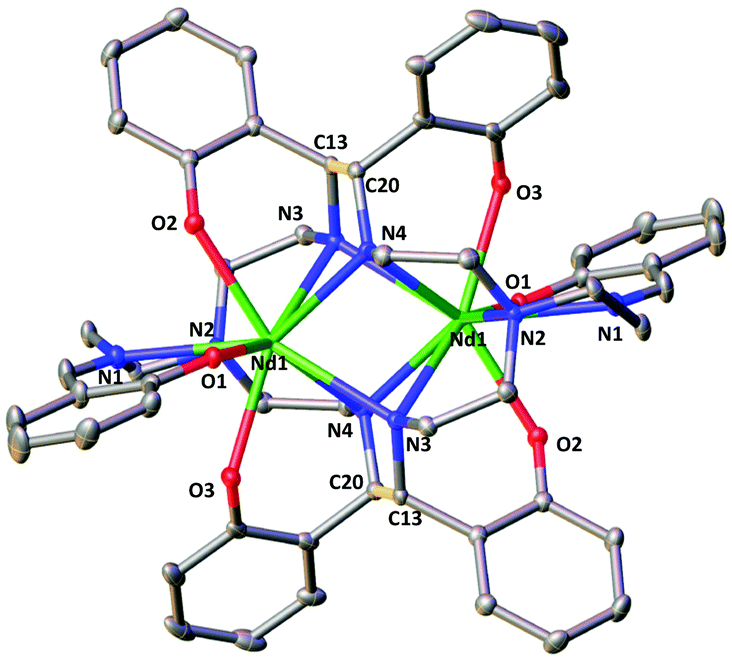 | ||
| Fig. 3 Molecular structure of complex 4-Nd (C–C bonds between imine highlighted in yellow, 50% probability ellipsoids Hydrogen atoms, potassium atoms and THF molecules were omitted for clarity. | ||
The 4-Ln complexes present two equivalent Ln(III) centers in a tricapped trigonal prismatic geometry nonacoordinated by four amido nitrogens (avg Ln–Namido = 2.6 (1) Å for Nd and 2.61(12) Å for Sm), one imino nitrogen atom (Ln–Nimino = 2.6895(16) Å for Nd and 2.679(9) Å for Sm), one amino nitrogen atoms (Ln–Namino = 2.7209(15) Å for Nd and 2.706(3) Å) and three phenoxide oxygen atoms (avg Ln–O = 2.47(1) Å for Nd and 2.45(1) Å for Sm). The six phenoxide oxygen atoms coordinate also four potassium counterions, whose coordination sphere is completed by THF molecules.
When comparing the selected bond distances shown in Table 1, the C–Namido bond distances (mean C–Nam = 1.472(4) Å for Nd and 1.470(3) Å for Sm) of the ligand backbone are much longer than those of the remaining imino group (C–Nimino = 1.273(3) Å for Nd and 1.263(6) Å for. Sm) or the values observed for the free ligand (avg. C–Nim = 1.266(3) Å),.22 These data are in agreement with the presence of four amido groups. The value of the C–C bond distances (1.558(3) Å for Nd and 1.560(3) Å for Sm) fall in the range of those reported for the intermolecular and intramolecular C–C bonds in [Na2U(bis-salophen)] (1.559(7) Å),5d [Nd2(cyclo-salophen)]4− (1.622(3) Å)7b and [Nd(bis-salophen)]3− (1.56(2) Å).7b
These studies show that the lanthanide complexes of the flexible trensal ligand can be reduced in a controlled manner by the addition of stoechiometric amounts of strong reducing agent (KC8) affording well defined reduced species in high yield. The one electron reduction leads to the reduction of the metal center only for the Eu cation. The one- and two-electron reduction of the Sm(III) and Nd(III) trensal complexes leads to the reductive coupling of the imino groups of the trensal ligands leaving the metal in the +3 oxidation state. The resulting Ln(III) dinuclear complexes contain two or four electrons stored in C–C bonds that could become available for substrate reduction.
CO2 reduction studies
The 2-Ln complexes present two electrons stored in the C–C bond of the bis-trensal ligand as found for the previously reported K3[Ln(bis-Rsalophen)] complexes.7b It was shown that the electrons in the K3[Ln(bis-Rsalophen)] are available for the reduction of strongly oxidizing substrates such as Ag+, molecular iodine or 9,10-phenanthrenequinone. However, the K3[Ln(bis-Rsalophen)] complexes could not effect the reduction of carbon dioxide. The lack of reactivity was in part attributed to the lack of accessible coordination sites at the sterically hindered 8-coordinated metal center.With the 2-Ln complexes in hand, we investigated if the higher flexibility of the reduced trensal ligand would lead to an increased reactivity. It should be noted that the complex 3-Eu did not show any reactivity with CO2. We found that 2-Ln react rapidly and irreversibly with carbon dioxide. The addition of 2 equiv. of CO2 to a THF solution 2-Ln resulted in a color change of the solution from yellow/orange to pale yellow, with the concomitant formation of an insoluble product. The 1H NMR spectra recorded after the formation of the solid products showed the disappearance of the signals assigned to 2-Ln. The low solubility of the reduction product prevented its structural characterization.
However, the removal of the solvent under vacuum and the dissolution of the residue in basic D2O (pD = 13.4) allowed the identification of the water soluble products. Quantitative 13C{1H} NMR spectra recorded in presence of 13AcO− as internal standard showed the formation of 13CO32− in 96(2)% yield (with respect to the value expected from the conversion of 2 CO2 molecules into 13CO32− and CO) for both 2-Ln complexes (Fig. S8 and S10†) (Scheme 5 top).
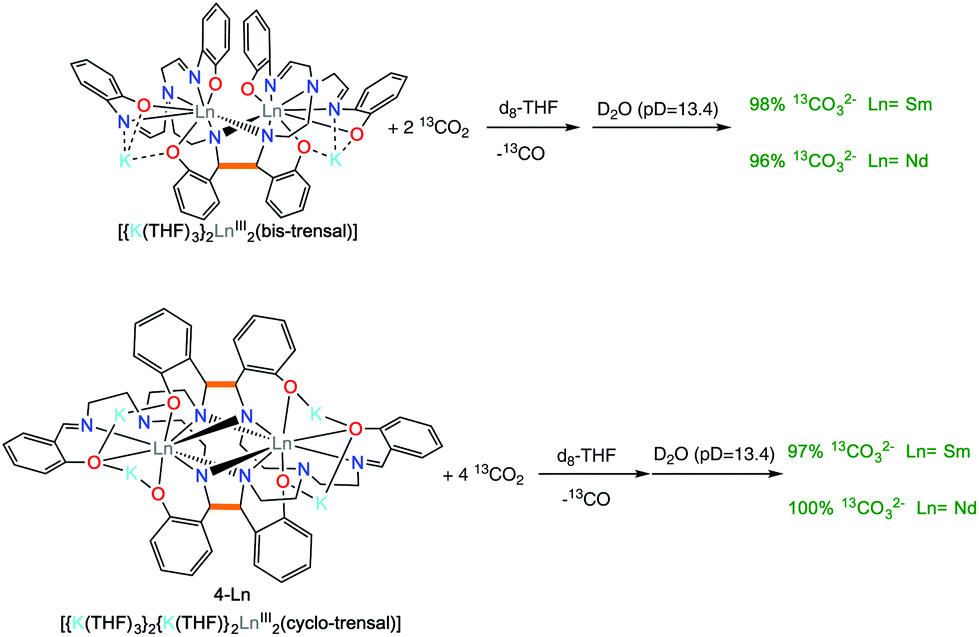 | ||
| Scheme 5 Reactivity of 2-Ln (top) and 4-Ln (bottom) with 2 and 4 eq. 13CO2, respectively. | ||
The presence of carbonate indicated that the electrons stored in the C–C bonds in complexes 2-Ln can effect the reductive disproportionation of CO2 to afford carbonate and CO. Thus the bis-trensal complexes show a dramatic increase in reactivity towards CO2 compared to the previously reported K3[Ln(bis-Rsalophen)] complexes.7b The reactivity of the 2-Ln complexes also differs from that of the recently reported U(IV) analogue, [U2(bis-trensal)],10c that only undergoes insertion of the CO2 into the U–Namide bond. In spite of the presence of the same binucleating bis-trensal ligand the molecular structure of the 2-Ln complexes differs significantly from that of the U(IV) complex [U2(bis-trensal)]. Notably in the structure of [U2(bis-trensal)] the amido nitrogen atoms do not bridge the two metal centers as found in the 2-Ln complexes resulting in a large U–U separation at 7.311(2) Å. Such difference is probably originating from the smaller size of the U(IV) cation which prevents amido bridging to occur.
The difference in reactivity between the 2-Ln complexes and the U(IV) complex [U2(bis-trensal)] is probably due to both structural and electronic differences. The bridging mode adopted by the amido nitrogen in 2-Ln probably results in a less basic character compared to the non-bridging amido nitrogen in [U2(bis-trensal)] preventing the insertion of CO2 into the Ln–Namido bond. Moreover, the close proximity of the Ln centers in the 2-Ln should promote CO2 activation via cooperative binding to the two metal centers.
The 4-Ln complexes contain four electrons stored in the two C–C bonds of the cyclo-trensal ligand similarly to the previously reported cyclo-salophen complexes K2[Nd2(cyclo-salophen)] and [U2(cyclo-salophen)] isolated from the reduction of salophen complexes. The cyclo-salophen complexes were found able to transfer the four electrons stored in the two C–C bonds to strongly oxidizing substrates but were not able to reduce CO2 or CS2. In contrast we found that the cyclo-trensal complexes 4-Ln display a dramatically different reactivity towards CO2. Notably, the addition of 4 equiv. of 13CO2 to a solution of 4-Ln in THF leads to an immediate color change of the solution from red/orange to pale yellow with concomitant formation of a precipitate. The 1H NMR spectra recorded after formation of the solid products showed the disappearance of the signals assigned to 4-Ln. The 13C{1H} NMR spectra recorded after one day showed the formation of 13CO (181 ppm) as only visible product in THF solution (Fig. S6†). The low solubility of the reduction product prevented its structural characterization.
However, the removal of the solvent under vacuum and the dissolution of the residue in basic D2O (pD = 13.4) allowed the identification of the water soluble products. The quantitative 13C{1H} NMR spectra recorded in presence of 13AcO− as internal standard showed the formation of 13CO32− in 97% (Ln = Sm) and 100% yield (Ln = Nd)((yields given with respect to the value expected from the conversion of 4 13CO2 molecules into 2 13CO32− and 2 13CO) (Scheme 5 bottom).
These results show that the four electrons stored in the two C–C bonds of the cyclo-trensal ligands are quantitatively used for the reductive disproportionation of CO2. The clean quantitative reductive disproportionation effected by the 4-Ln complexes contrasts remarkably with the multiple reaction pathways previously reported for the reaction of the analogue U(IV) complex [{K(THF)3}2U2(cyclo-trensal)] with CO2. Such pathways included reductive disproportionation of CO2, insertion of CO2 in the U–Namido bonds, CO2 cleavage and further addition of the formed CO to the U–Namido bonds.
Electrochemistry
Cyclic voltammetry data were measured for complexes 1-Ln in ∼0.1 M THF solution of [Bu4N][PF6] and are presented in Fig. 4. All redox potentials are referenced against the [(C5H5)2Fe]+/0 redox couple. Complexes 1-Nd and 1-Sm showed similar behavior, but the voltammogram of 1-Eu did not show any clear reduction event at any scan rates (see Fig. S22†).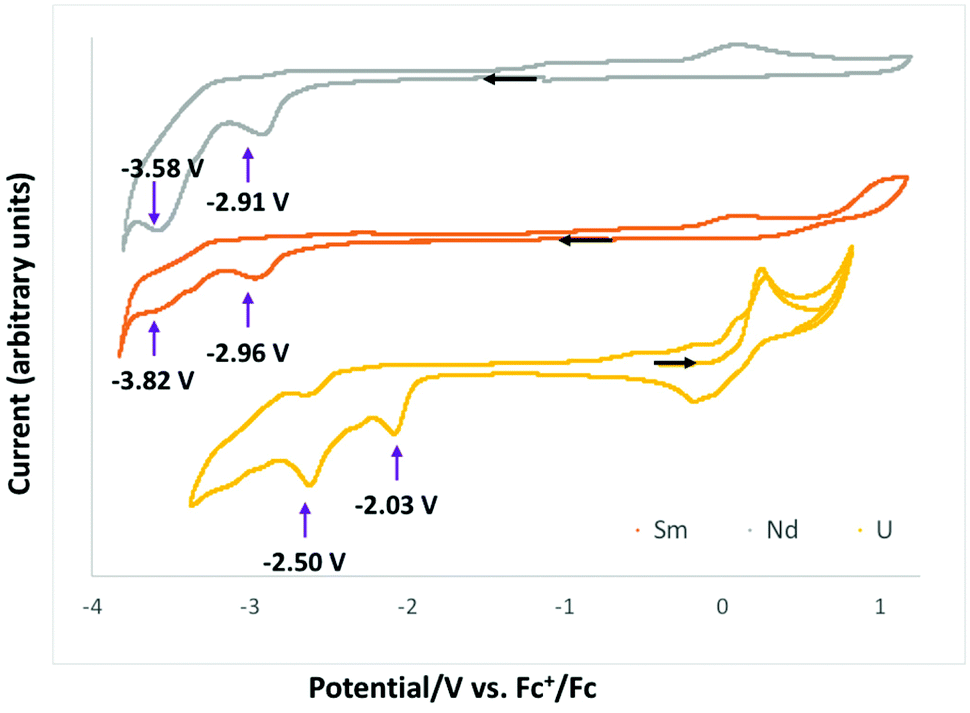 | ||
| Fig. 4 Cyclic voltammogram of complexes 1-Nd (grey), 1-Sm (orange) and 1-U (yellow) from ref. 10c in ∼0.1 M [Bu4N][PF6] THF solution at a 100 mV s−1 scan rate. | ||
The voltammograms of complexes 1-Nd and 1-Sm show two distinct irreversible reduction events at Epc = −2.91 V and −3.58 V (Fig. S16–S18†) and Epc = −2.96 V and −3.82 V (Fig. S19–S21†), respectively associated with a series of irreversible redox processes in the range −0.55 to 0.3 V. The irreversible reduction events observed for 1-Sm and 1-Nd can be assigned to ligand based redox processes resulting in formation of the first C–C bond in 2-Ln and to the formation of a second C–C bond in 4-Ln. However, the voltammograms of 2-Ln and 4-Ln complexes measured in the same conditions (in the presence or absence of cryptand) did not show clear redox events probably due to the strong interaction with potassium cations.
Previously reported electrochemistry studies10c of the analogous U(IV) complex [U(Trensal)][OTf], showed two similar irreversible ligand-based reduction events occurring at higher potentials (Epc = −2.03 V and −2.50 V for the formation of the first and the second C–C bond, respectively).
The ability of complex 2-Ln to reduce CO2 compared to the lack of redox reactivity towards CO2 reduction reported for the analogous mono-reduced U(IV) complex is probably due to its lower redox potential associated to a higher accessibility to the electrons stored in the ligand framework.
Conclusions
In summary we have reported the reductive chemistry of Ln(III) complexes of the tripodal heptadentate trensal3− ligand. We showed that, depending on the metal, the reduction can occur at the metal center affording a stable Eu(II)-trensal complex or can lead to the reductive coupling of two imino groups of two trensal ligands and to the formation of an intermolecular C–C bond where two electrons are stored. In the case of the Nd and Sm trensal complexes the one-electron reduction leads to the isolation of dinuclear Ln(III) complexes of the bis-trensal ligand. Further reduction results in the formation of a second C–C bond from the intramolecular reductive coupling of two imino groups of the bis-trensal ligand to afford a dinuclear macrocyclic complex of the cyclo-trensal ligand where two Ln(III) ions are held together in close proximity. These results show that the reductive chemistry of Ln(III) Schiff bases provide an attractive tool for the synthesis of polynuclear Ln(III) complexes. Remarkably the electrons stored in the C–C bonds of the bis-trensal and cyclo-trensal complexes become available for the reduction of carbon dioxide to selective afford CO and carbonate.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Swiss National Science Foundation grant number 178793 and the Ecole Polytechnique Fédérale de Lausanne (EPFL). We thank Dr Euro Solari for carrying out the elemental analyses, Farzaneh Fadaei-Tirani for important contributions to the X-ray single crystal structure analyses.Notes and references
- (a) K. A. Grice, Carbon dioxide reduction with homogenous early transition metal complexes: Opportunities and challenges for developing CO2 catalysis, Coord. Chem. Rev., 2017, 336, 78–95 CrossRef CAS; (b) M. D. Walter, Recent Advances in Transition Metal-Catalyzed Dinitrogen Activation, Adv. Organomet. Chem., 2016, 65, 261–377 CrossRef CAS.
- (a) M. Xemard, V. Goudy, A. Braun, M. Tricoire, M. Cordier, L. Ricard, L. Castro, E. Louyriac, C. E. Kefalidis, C. Clavaguera, L. Maron and G. Nocton, Reductive Disproportionation of CO2 with Bulky Divalent Samarium Complexes, Organometallics, 2017, 36, 4660–4668 CrossRef CAS; (b) N. W. Davies, A. S. P. Frey, M. G. Gardiner and J. Wang, Reductive disproportionation of carbon dioxide by a Sm(II) complex: Unprecedented f-block element reactivity giving a carbonate complex, Chem. Commun., 2006, 4853–4855 RSC; (c) G. B. Deacon, P. C. Junk, J. Wang and D. Werner, Reactivity of Bulky Formamidinatosamarium(II or III) Complexes with C=O and C=S Bonds, Inorg. Chem., 2014, 53, 12553–12563 CrossRef CAS PubMed; (d) W. J. Evans, C. A. Seibel and J. W. Ziller, Organosamarium-mediated transformations of CO2 and COS: Monoinsertion and disproportionation reactions and the reductive coupling of CO2 to [O2CCO2](2-), Inorg. Chem., 1998, 37, 770–776 CrossRef CAS; (e) W. J. Evans, G. Zucchi and J. W. Ziller, Dinitrogen reduction by Tm(II), Dy(II), and Nd(II) with simple amide and aryloxide ligands, J. Am. Chem. Soc., 2003, 125, 10–11 CrossRef CAS PubMed; (f) J. Andrez, J. Pecaut, P.-A. Bayle and M. Mazzanti, Tuning Lanthanide Reactivity Towards Small Molecules with Electron-Rich Siloxide Ligands, Angew. Chem., Int. Ed., 2014, 53, 10448–10452 CrossRef CAS PubMed; (g) D. Heitmann, C. Jones, D. P. Mills and A. Stasch, Low coordinate lanthanide(II) complexes supported by bulky guanidinato and amidinato ligands, J. Chem. Soc., Dalton Trans., 2010, 39, 1877–1882 RSC.
- (a) A. Willauer, D. Toniolo, F. Fadaei-Tirani, Y. Yang, S. Laurent and M. Mazzanti, Carbon dioxide reduction by dinuclear Yb(ii) and Sm(ii) complexes supported by siloxide ligands, J. Chem. Soc., Dalton Trans., 2019, 48, 6100–6110 RSC; (b) D. Toniolo, A. Willauer, R. Scopelliti, J. Andrez, Y. Yang and L. Maron, CS2 Reductive Coupling to Acetylenedithiolate by a Dinuclear Ytterbium(II) Complex, Chem. – Eur. J., 2019, 25, 7831–7834 CrossRef CAS PubMed; (c) N. F. M. Mukthar, N. D. Schley and G. Ung, Alkali-metal- and halide-free dinuclear mixed- valent samarium and europium complexes, J. Chem. Soc., Dalton Trans., 2020 10.1039/d0dt01095b; (d) J. I. Song and S. Gambarotta, The first dinuclear low-valent samarium complex with a short Sm-Sm contact, Angew. Chem., Int. Ed. Engl., 1995, 34, 2141–2143 CrossRef CAS.
- (a) C. Camp and J. Arnold, On the non-innocence of “Nacnacs”: ligand-based reactivity in beta-diketiminate supported coordination compounds, J. Chem. Soc., Dalton Trans., 2016, 45, 14462–14498 RSC; (b) P. J. Chirik and K. Wieghardt, Radical Ligands Confer Nobility on Base-Metal Catalysts, Science, 2010, 327, 794–795 CrossRef CAS PubMed; (c) S. S. Galley, S. A. Pattenaude, R. F. Higgins, C. J. Tatebe, D. A. Stanley, P. E. Fanwick, M. Zeller, E. J. Schelter and S. C. Bart, A reduction series of neodymium supported by pyridine(diimine) ligands Dedicated to Professor Geoff Cloke on the occasion of his 65(th) birthday, J. Chem. Soc., Dalton Trans., 2019, 48, 8021–8025 RSC; (d) I. L. Fedushkin, A. N. Lukoyanov and E. V. Baranov, Lanthanum Complexes with a Diimine Ligand in Three Different Redox States, Inorg. Chem., 2018, 57, 4301–4309 CrossRef CAS PubMed; (e) E. J. Coughlin, Y. S. Qiao, E. Lapsheva, M. Zeller, E. J. Schelter and S. C. Bart, Uranyl Functionalization Mediated by Redox-Active Ligands: Generation of O-C Bonds via Acylation, J. Am. Chem. Soc., 2019, 141, 1016–1026 CrossRef CAS PubMed; (f) N. H. Anderson, S. O. Odoh, Y. Y. Yao, U. J. Williams, B. A. Schaefer, J. J. Kiernicki, A. J. Lewis, M. D. Goshert, P. E. Fanwick, E. J. Schelter, J. R. Walensky, L. Gagliardi and S. C. Bart, Harnessing redox activity for the formation of uranium tris(imido) compounds, Nat. Chem., 2014, 6, 919–926 CrossRef CAS PubMed; (g) D. P. Cladis, J. J. Kiernicki, P. E. Fanwick and S. C. Bart, Multi-electron reduction facilitated by a trianionic pyridine(diimine) ligand, Chem. Commun., 2013, 49, 4169–4171 RSC.
- (a) G. Nocton, W. W. Lukens, C. H. Booth, S. S. Rozenel, S. A. Medling, L. Maron and R. A. Andersen, Reversible Sigma C-C Bond Formation Between Phenanthroline Ligands Activated by (C5Me5)(2)Yb, J. Am. Chem. Soc., 2014, 136, 8626–8641 CrossRef CAS PubMed; (b) M. J. Monreal and P. L. Diaconescu, Reversible C-C Coupling in a Uranium Biheterocyclic Complex, J. Am. Chem. Soc., 2010, 132, 7676–7683 CrossRef CAS PubMed; (c) G. Nocton and L. Ricard, Reversible C-C coupling in phenanthroline complexes of divalent samarium and thulium, Chem. Commun., 2015, 51, 3578–3581 RSC; (d) C. Camp, V. Mougel, P. Horeglad, J. Pecaut and M. Mazzanti, Multielectron Redox Reactions Involving C-C Coupling and Cleavage in Uranium Schiff Base Complexes, J. Am. Chem. Soc., 2010, 132, 17374–17377 CrossRef CAS PubMed; (e) A. A. Trifonov, E. A. Fedorova, G. K. Fukin, E. V. Baranov, N. O. Druzhkov and M. N. Bochkarev, Bridging mu-eta(5):eta(4)-coordination of an indenyl ligand and reductive coupling of diazabutadienes in the assembly of di- and tetranuclear mixed-valent ytterbium indenyldiazabutadiene complexes, Chem. – Eur. J., 2006, 12, 2752–2757 CrossRef CAS PubMed.
- (a) K. Vasudevan and A. H. Cowley, Synthesis and structures of 1,2-bis(imino)acenaphthene (BIAN) lanthanide complexes that involve the transfer of zero, one, or two electrons, Chem. Commun., 2007, 3464–3466 RSC; (b) I. L. Fedushkin, O. V. Maslova, A. G. Morozov, S. Dechert, S. Demeshko and F. Meyer, Genuine Redox Isomerism in a Rare-Earth-Metal Complex, Angew. Chem., Int. Ed., 2012, 51, 10584–10587 CrossRef CAS PubMed; (c) I. L. Fedushkin, D. S. Yambulatov, A. A. Skatova, E. V. Baranov, S. Demeshko, A. S. Bogomyakov, V. I. Ovcharenko and E. M. Zueva, Ytterbium and Europium Complexes of Redox-Active Ligands: Searching for Redox Isomerism, Inorg. Chem., 2017, 56, 9825–9833 CrossRef CAS PubMed; (d) J. E. Kim, P. J. Carroll and E. J. Schelter, Bidentate nitroxide ligands stable toward oxidative redox cycling and their complexes with cerium and lanthanum, Chem. Commun., 2015, 51, 15047–15050 RSC; (e) H. Sugiyama, I. Korobkov, S. Gambarotta, A. Moller and P. H. M. Budzelaar, Preparation, characterization, and magnetic behavior of the in derivatives (Ln = Nd, La) of a 2,6-diiminepyridine ligand and corresponding dianion, Inorg. Chem., 2004, 43, 5771–5779 CrossRef CAS PubMed; (f) C. D. Berube, S. Gambarotta, G. P. A. Yap and P. G. Cozzi, Di- and trivalent dinuclear samarium complexes supported by pyrrole-based tetradentate Schiff bases, Organometallics, 2003, 22, 434–439 CrossRef CAS; (g) I. L. Fedushkin, O. V. Maslova, M. Hummert and H. Schumann, One- and Two-Electron-Transfer Reactions of (dpp-Bian)Sm(dme)(3), Inorg. Chem., 2010, 49, 2901–2910 CrossRef CAS PubMed.
- (a) E. J. Coughlin, M. Zeller and S. C. Bart, Neodymium(III) Complexes Capable of Multi-Electron Redox Chemistry, Angew. Chem., Int. Ed., 2017, 56, 12142–12145 CrossRef CAS PubMed; (b) C. Camp, V. Guidal, B. Biswas, J. Pecaut, L. Dubois and M. Mazzanti, Multielectron redox chemistry of lanthanide Schiff-base complexes, Chem. Sci., 2012, 3, 2433–2448 RSC.
- (a) W. L. Huang and P. L. Diaconescu, Reactivity and Properties of Metal Complexes Enabled by Flexible and Redox-Active Ligands with a Ferrocene Backbone, Inorg. Chem., 2016, 55, 10013–10023 CrossRef CAS PubMed; (b) J. N. Wei and P. L. Diaconescu, Redox-Switchable Ring-Opening Polymerization with Ferrocene Derivatives, Acc. Chem. Res., 2019, 52, 415–424 CrossRef CAS PubMed.
- (a) S. DeAngelis, E. Solari, E. Gallo, C. Floriani, A. ChiesiVilla and C. Rizzoli, Formation of carbon-carbon-bonded dimers in the reduction of [Co(II)salophen] [salophen equals N,N’-o-phenylenebis(salicylideneaminato)]: Their reactivity with electrophiles to form Co-C bonds, Inorg. Chem., 1996, 35, 5995–6003 CrossRef CAS; (b) F. Franceschi, E. Solari, R. Scopelliti and C. Floriani, Metal-mediated transfer of electrons between two different C-C single bonds that function as electron-donor and electron-acceptor units, Angew. Chem., Int. Ed., 2000, 39, 1685–1686 CrossRef CAS; (c) C. Floriani, E. Solari, F. Franceschi, R. Scopelliti, P. Belanzoni and M. Rosi, Metal-metal and carbon-carbon bonds as potential components of molecular batteries, Chem. – Eur. J., 2001, 7, 3052–3061 CrossRef CAS PubMed; (d) E. Solari, C. Maltese, F. Franceschi, C. Floriani, A. ChiesiVilla and C. Rizzoli, Geometrical isomerism and redox behaviour in zirconium-Schiff base complexes: the formation of C-C bonds functioning as two-electron reservoirs, J. Chem. Soc., Dalton Trans., 1997, 2903–2910 RSC; (e) S. Gambarotta, M. Mazzanti, C. Floriani and M. Zehnder, A Tetranuclear Polyfunctional Sodium-Vanadium(III) Complex Containing a Vanadium(III)-Vanadium(III) Double-Bond, J. Chem. Soc., Chem. Commun., 1984, 1116–1118 RSC; (f) J. Andrez, V. Guidal, R. Scopelliti, J. Pecaut, S. Gambarelli and M. Mazzanti, Ligand and Metal Based Multielectron Redox Chemistry of Cobalt Supported by Tetradentate Schiff Bases, J. Am. Chem. Soc., 2017, 139, 8628–8638 CrossRef CAS PubMed; (g) M. B. Solomon, B. Chan, C. P. Kubiak, K. A. Jolliffe and D. M. D'Alessandro, The spectroelectrochemical behaviour of redox-active manganese salen complexes, J. Chem. Soc., Dalton Trans., 2019, 48, 3704–3713 RSC; (h) A. W. Nichols, S. Chatterjee, M. Sabat and C. W. Machan, Electrocatalytic Reduction of CO2 to Formate by an Iron Schiff Base Complex, Inorg. Chem., 2018, 57, 2111–2121 CrossRef CAS PubMed; (i) D. Toniolo, R. Scopelliti, I. Zivkovic and M. Mazzanti, Assembly of High-Spin [Fe3] Clusters by Ligand-Based Multielectron Reduction, J. Am. Chem. Soc., 2020, 142, 7301–7305 CrossRef CAS PubMed.
- (a) C. Camp, J. Andrez, J. Pecaut and M. Mazzanti, Synthesis of Electron-Rich Uranium(IV) Complexes Supported by Tridentate Schiff Base Ligands and Their Multi-Electron Redox Chemistry, Inorg. Chem., 2013, 52, 7078–7086 CrossRef CAS PubMed; (b) C. Camp, L. Chatelain, V. Mougel, J. Pecaut and M. Mazzanti, Ferrocene-Based Tetradentate Schiff Bases as Supporting Ligands in Uranium Chemistry, Inorg. Chem., 2015, 54, 5774–5783 CrossRef CAS PubMed; (c) N. Jori, M. Falcone, R. Scopelliti and M. Mazzanti, Carbon Dioxide Reduction by Multimetallic Uranium(IV) Complexes Supported by Redox-Active Schiff Base Ligands, Organometallics, 2020, 39, 1590–1601 CrossRef CAS.
- (a) B. E. Klamm, C. J. Windorff, M. L. Marsh, D. S. Meeker and T. E. Albrecht-Schmitt, Schiff-base coordination complexes with plutonium(IV) and cerium(IV), Chem. Commun., 2018, 54, 8634–8636 RSC; (b) B. E. Klamm, C. J. Windorff, C. Ceis-Barros, M. L. Marsh, D. S. Meeker and T. E. Albrecht-Schmitt, Experimental and Theoretical Comparison of Transition-Metal and Actinide Tetravalent Schiff Base Coordination Complexes, Inorg. Chem., 2018, 57, 15389–15398 CrossRef CAS PubMed; (c) M. Paul, S. Shirase, Y. Morimoto, L. Mathey, B. Murugesapandian, S. Tanaka, S. Itoh, H. Tsurugi and K. Mashima, Cerium-Complex-Catalyzed Oxidation of Arylmethanols under Atmospheric Pressure of Dioxygen and Its Mechanism through a Side-On -Peroxo Dicerium(IV) Complex, Chem. – Eur. J., 2016, 22, 4008–4014 CrossRef CAS PubMed.
- H. Uh, P. D. Badger, S. J. Geib and S. Petoud, Synthesis and Solid-State, Solution, and Luminescence Properties of Near-Infrared-Emitting Neodymium(3+) Complexes Formed with Ligands Derived from Salophen, Helv. Chim. Acta, 2009, 92, 2313–2329 CrossRef CAS.
- (a) R. D. Archer, H. Y. Chen and L. C. Thompson, Synthesis, characterization, and luminescence of europium(III) Schiff base complexes, Inorg. Chem., 1998, 37, 2089–2095 CrossRef CAS; (b) Y. H. Yao, H. Y. Yin, Y. Y. Ning, J. Wang, Y. S. Meng, X. Y. Huang, W. K. Zhang, L. Kang and J. L. Zhang, Strong Fluorescent Lanthanide Salen Complexes: Photophysical Properties, Excited-State Dynamics, and Bioimaging, Inorg. Chem., 2019, 58, 1806–1814 CrossRef CAS PubMed; (c) X. P. Yang, R. A. Jones and S. M. Huang, Luminescent 4f and d-4f polynuclear complexes and coordination polymers with flexible salen-type ligands, Coord. Chem. Rev., 2014, 273, 63–75 CrossRef.
- (a) R. Hussain, G. Allodi, A. Chiesa, E. Garlatti, D. Mitcov, A. Konstantatos, K. S. Pedersen, R. De Renzi, S. Piligkos and S. Carretta, Coherent Manipulation of a Molecular Ln-Based Nuclear Qudit Coupled to an Electron Qubit, J. Am. Chem. Soc., 2018, 140, 9814–9818 CrossRef CAS PubMed; (b) M. Perfetti, E. Lucaccini, L. Sorace, J. P. Costes and R. Sessoli, Determination of Magnetic Anisotropy in the LnTRENSAL Complexes (Ln = Tb, Dy, Er) by Torque Magnetometry, Inorg. Chem., 2015, 54, 3090–3092 CrossRef CAS PubMed; (c) E. Lucaccini, L. Sorace, M. Perfetti, J. P. Costes and R. Sessoli, Beyond the anisotropy barrier: slow relaxation of the magnetization in both easy-axis and easy-plane Ln(trensal) complexes, Chem. Commun., 2014, 50, 1648–1651 RSC.
- T. Dube, S. Gambarotta and G. Yap, Preparation and reactivity of a compartmental Schiff-base samarium dinuclear complex, Organometallics, 1998, 17, 3967–3973 CrossRef CAS.
- (a) P. V. Bernhardt, B. M. Flanagan and M. J. Riley, Completion of the isomorphous Ln(trensal) series, Aust. J. Chem., 2001, 54, 229–232 CrossRef CAS; (b) P. V. Bernhardt, B. M. Flanagan and M. J. Riley, Isomorphous lanthanide complexes of a tripodal N4O3 ligand, Aust. J. Chem., 2000, 53, 229–231 CrossRef CAS; (c) M. Kanesato, T. Yokoyama, O. Itabashi, T. M. Suzuki and M. Shiro, Synthesis and structural characterization of praseodymium(III) and neodymium(III) complexes of tripodal tris 2-(salicylideneamino)ethyl amine, Bull. Chem. Soc. Jpn., 1996, 69, 1297–1302 CrossRef CAS; (d) M. Kanesato, T. Yokoyama and T. M. Suzuki, Reaction of tris(2-aminoethyl)amine coordinated to lanthanum(III) and gadolinium(III) with salicylaldehyde, Chem. Lett., 1997, 93–94 CrossRef CAS.
- (a) K. S. Pedersen, A. M. Ariciu, S. McAdams, H. Weihe, J. Bendix, F. Tuna and S. Piligkos, Toward Molecular 4f Single-Ion Magnet Qubits, J. Am. Chem. Soc., 2016, 138, 5801–5804 CrossRef CAS PubMed; (b) K. S. Pedersen, L. Ungur, M. Sigrist, A. Sundt, M. Schau-Magnussen, V. Vieru, H. Mutka, S. Rols, H. Weihe, O. Waldmann, L. F. Chibotaru, J. Bendix and J. Dreiser, Modifying the properties of 4f single-ion magnets by peripheral ligand functionalisation, Chem. Sci., 2014, 5, 1650–1660 RSC.
- S. Mandal, D. K. Seth and P. Gupta, Encapsulating ruthenium and osmium with tris(2-aminoethyl)amine based tripodal ligands, Polyhedron, 2012, 31, 167–175 CrossRef CAS.
- R. Faizova, S. White, R. Scopelliti and M. Mazzanti, The effect of iron binding on uranyl(v) stability, Chem. Sci., 2018, 9, 7520–7527 RSC.
- W. J. Evans, J. W. Grate, H. W. Choi, I. Bloom, W. E. Hunter and J. L. Atwood, Solution synthesis and crystallographic characterization of the divalent organosamarium complexes (C5Me5)2Sm(THF)2 a nd [(C5Me5)2Sm (mu-I)(THF)2]2, J. Am. Chem. Soc., 1985, 107, 941–946 CrossRef CAS.
- J. Andrez, G. Bozoklu, G. Nocton, J. Pecaut, R. Scopelliti, L. Dubois and M. Mazzanti, Lanthanide(II) Complexes Supported by N,O-Donor Tripodal Ligands: Synthesis, Structure, and Ligand-Dependent Redox Behavior, Chem. – Eur. J., 2015, 21, 15188–15200 CrossRef CAS PubMed.
- N. Gunduz, T. Gunduz, M. B. Hursthouse, H. G. Parkes, L. S. Shaw, R. A. Shaw and M. Tuzun, X-ray crystallographic, h-1, and c-13 nuclear magnetic-resonance investigation of the potentially heptadentate ligand trensal, 2,2′,2′′-tris(salicylideneimino)triethylamine, J. Chem. Soc., Perkin Trans. 2, 1985, 899–902 RSC.
- L. R. Morss, Thermochemical Properties of Yttrium, Lanthanum, and Lanthanide Elements and Ions, Chem. Rev., 1976, 76, 827–841 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra. CCDC 2013179 (2-Nd), 2013180 (3-Eu), 2013181 (4-Sm), 2013182 (2-Sm), –2013183 (4-Nd). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qi00801j |
| This journal is © the Partner Organisations 2020 |