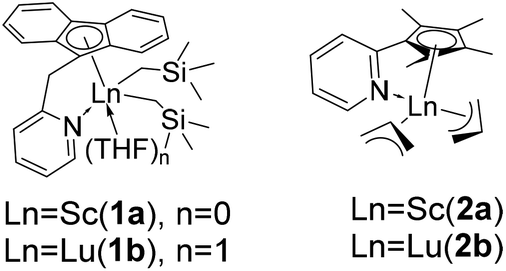
DOI:
10.1039/D0QI00719F
(Research Article)
Inorg. Chem. Front., 2020,
7, 3961-3968
Syndio-and cis-1,4 dually selective copolymerization of polar fluorostyrene and butadiene using rare-earth metal catalysts†
Received
18th June 2020
, Accepted 21st August 2020
First published on 25th August 2020
Abstract
Synthesizing functional butadiene–styrene rubber through coordination polymerization is a theoretical challenge for polymer science, since functional monomers usually deactivate to the applied catalyst. Herein, we report the coordination copolymerization of polar para-fluorostyrene (pFS) and butadiene (BD) using pyridyl–methylene–fluorenyl supported complexes [(Py-CH2-Flu)Ln(CH2SiMe3)2(THF)n (Ln = Sc (1a), n = 0; Ln = Lu (1b), n = 1)] and pyridyl–cyclopentadienyl supported complexes [(Py-Cp)Ln(η3-C3H5)2 (Ln = Sc (2a), Lu (2b))]. Strikingly, complexes 2a and 2b exhibited dually >99% syndio- and >95% cis-1,4 regio- selectivities and showed obvious characteristics of living polymerization. The insertion of pFS can be facilely tuned in the full range of 0–100% by changing the pFS-to-BD feed ratio. Diblock P(pFS-BD) copolymers were isolated by concurrent addition of monomers and the kinetics study of the copolymerization reaction revealed that BD had the privilege to coordinate to the active metal center. Interestingly, the polymerizations of BD and pFS via pulse loading of BD afforded multi-block copolymers of a novel type of fluoro styrene–butadiene rubber with high thermal stability (Td = 368 °C). The microstructures of resultant copolymers were confirmed by 1H and 13C NMR measurements and different phase morphologies of the di- and multi-block polymers were displayed through atomic force microscopy (AFM).
Introduction
Synthetic rubbers have played an irreplaceable role in our modern society since we are living in a world “on wheels”.1 On the other hand these materials are difficult to reuse and recycle and it is nearly impossible for them to be bio-degraded, and so they are the main source of “black pollution”.2,3 For the sustainable development of tyre and rubber industries, fabricating “green tyres” has been the challenging project in the past three decades.2 One of the strategies is to synthesize functional rubbers to improve their compatibility with polar fillers, anticipated to endow tyres with excellent wear resistance (prolonged use time) and low rolling resistance without compromising the wet-skid resistance.4,5 Styrene–butadiene rubber (SBR) is the most widely used synthetic rubber in passenger cars, to fabricate green tyres, by mixing SBR with functionalized fillers.6–9 End-functionalizing SBR prepared by anionic solution polymerization with a halogen group is a convenient method to achieve this target; however, obviously, the number of the incorporated functional groups is rather limited.10,11 Copolymerization with polar group modified monomers is an alternative, anticipated to control the amount and distribution of polar groups in the main chain.12–16 Radical copolymerization of styrene with nitrile-functionalized butadiene,17 2,3-bis(4-ethoxy-4-oxobutyl)-1,3-butadiene18 or N,N-diethyl-2-methylene-3-butenamide (DEA),19 respectively, provides functionalized poly(styrene–butadiene) rubbers, where the cis-1,4 selectivity of the functional butadiene is rather low and its incorporation also needs to improve. Coordination copolymerization is the most powerful method to adjust the regio- and stereo-regularity, when the active species are prone to be deactivated by functional groups.20–22 We reported for the first time the coordination homo- and copolymerizations of polar styrene monomers including methoxystyrene (MOSt) and halogen substituted styrenes with the use of side-armed half-sandwich rare-earth metal complexes, with styrene exhibiting high activity and stereoselectivity.23–25 Thereafter copolymers composed of polar styrenes/ethylene and anisylpropylenes/ethylene were obtained.26–28 With respect to polar dienes, copolymerization of 2-(2-methylidenebut-3-enyl)furan (MBEF) with isoprene without a masking reagent was achieved by using the bis(phosphino)carbazolideyttrium precursor.29 The amino-functionalized styrene and butadiene were mediated to copolymerize; however, a solvent fractionation experiment was needed to obtain pure copolymers.30 Guo et al. recently realized the copolymerization of 4-(N,N-diphenylamino)styrene and isoprene to produce functionalized poly(isoprene-styrene).31
Despite remarkable characteristics such as low surface energy and superior chemical resistance of fluoropolymers32 as well as the excellent properties of thermoplastic syndiotactic poly(p-fluorostyrene) and elastic cis-1,4-PBD,33 incorporating all these factors into one macromolecular chain, such as fluorinated butadiene–styrene copolymers, with highly regulated microstructures has remained a promising but challenging subject for academic and industrial fields.
Herein, we report the coordination copolymerization of p-fluorostyrene (pFS) and butadiene (BD) by using rare-earth metal precursors bearing various sterics and electronics (Chart 1). When the sterically less hindered and Lewis acidic rare-earth metal precursors are used, copolymerization of pFS and BD occurs rather successfully, in particular in the living mode. The incorporation of polar pFS can be adjusted facilely by adjusting the feed-ratio, and its distribution can be di-block and multi-block according to the loading mode. The resultant P(pFS-BD) copolymers have >99% syndioregularity for P(pFS) sequences and >95% cis-1,4-tacticity for PBD sequences, which show a much higher thermal stability (Td = 368 °C) than that of commercial SBR.
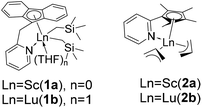 |
| | Chart 1 Structures of rare-earth metal catalysts 1 and 2. | |
Experimental section
General experimental procedures
All experiments were performed under a dry and oxygen-free nitrogen atmosphere using standard high vacuum Schlenk techniques or in a glove box. All solvents were purified via a solvent purification system. p-Fluorostyrene was purchased from Aldrich and dried by CaH2 under stirring for 48 hours and distilled before use. Catalysts 1 and 2 were synthesized according to the literature.34,351H, 13C NMR spectra were recorded on a Bruker AV400 (FT, 400 MHz for 1H; 100 MHz for 13C) or AV500 (FT, 500 MHz for 1H; 125 MHz for 13C) at 25 °C and CDCl3 was used as the solvent with CHCl3 as the internal standard (7.26 ppm in 1H NMR and 77.16 ± 0.06 ppm in 13C NMR). The molecular weights (Mn) and molecular weight distributions of the polymers (Mw/Mn) were measured by TOSOH HLC-8220 GPC at 40 °C using THF as the eluent (the flow rate was 0.35 mL min−1) against polystyrene standards. Differential scanning calorimetry analyses were carried out on a Q100 DSC from TA Instruments under a nitrogen atmosphere at heating and cooling rates of 10 °C min−1. Thermo-gravimetric analysis (TGA) was carried out on the TA Instruments SDT Q600 under a nitrogen atmosphere to characterize the thermal stability. AFM was used to study the surface topography of the spin-coated film. Images were obtained using a SPI3800N AFM (Seiko Instruments Inc., Japan). The cantilevers were operated slightly below their resonance frequency of around 20–150 kHz. Image acquisition was performed under ambient conditions. AFM was used in the tapping mode to reduce tip-induced surface degradation and sample damages. Imaging was conducted in the height and phase modes. The sample was prepared by dipping a silicon wafer into a tetrahydrofuran solution of a polymer sample and then placing it on a flat poly(tetrafluoroethylene) plate to allow the solvent to gradually evaporate. Next, the wafer was heated in a vacuum oven at 40 °C to eradicate the solvent before test.
Homopolymerization of pFS
A typical procedure for polymerizing pFS is as follows (Table 2, entry 1): under a nitrogen atmosphere and at 20 °C, a toluene (0.5 mL) solution of [Ph3C][B(C6F5)4] (4.8 mg, 5 μmol) and a toluene solution (0.5 mL) of complex 2a (1.9 mg, 5 μmol) were added to a 10 mL flask. Then purified pFS (0.122 g, 1.0 mmol) was added under stirring to initiate the polymerization. The reaction was terminated after 20 min by the addition of a small amount of acidic methanol and the mixture was poured into methanol (50 mL). The precipitated polymer was collected by filtration and dried under vacuum at 40 °C to a constant weight (0.122 g, >99%). All other polymerization data were obtained by following the same procedure but with different pFS or catalyst feed amounts.
Table 1 Copolymerization of para-fluorostyrene and butadienea
| Entry |
Cat |
pFS fed (mol %) |
Time/min |
Conv. (%) |
pFS b (mol%) |
rrrr
(%) |
Selectivity b (%) |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c × 10−4 c × 10−4 |
M
w/Mnc |
|
cis-1,4:trans-1,4:1,2 |
|
Conditions: [Sc] 5 μmol, [pFS] + [BD]/[Sc]/[Ph3C][B(C6F5)4] = 1000:1:1 (mol/mol/mol), toluene 2 mL, T = 20 °C.
Measured by 1H NMR and 13C NMR in CDCl3 at 25 °C.
Determined by GPC in THF at 40 °C against polystyrene standard.
[Sc]/[AliBu] = 1:10.
|
| 1d |
1a
|
50 |
10 |
60.4 |
n. d |
n. d |
n. d |
n. d |
n. d |
| 2d |
1b
|
50 |
60 |
32.8 |
34.2 |
n. d |
n. d |
16.7/1.5 |
1.1/1.8 |
| 3 |
2a
|
90 |
10 |
90.7 |
89.1 |
>99 |
95:3:2 |
25.7 |
1.3 |
| 4 |
2a
|
70 |
10 |
76.0 |
63.8 |
>99 |
95:3:2 |
21.7 |
1.2 |
| 5 |
2a
|
50 |
10 |
66.1 |
38.7 |
>99 |
96:3:1 |
15.8 |
1.1 |
| 6 |
2a
|
30 |
10 |
47.4 |
4.3 |
>99 |
97:2:1 |
15.5 |
1.1 |
| 7 |
2a
|
10 |
10 |
79.3 |
3.6 |
>99 |
97:2:1 |
13.8 |
1.1 |
| 8 |
2b
|
90 |
60 |
84.6 |
89.6 |
>99 |
96:2:2 |
24.1 |
1.3 |
| 9 |
2b
|
70 |
60 |
61.2 |
56.8 |
>99 |
96:2:2 |
13.4 |
1.5 |
| 10 |
2b
|
50 |
60 |
41.1 |
16.9 |
>99 |
96:2:2 |
12.4 |
1.2 |
| 11 |
2b
|
30 |
60 |
49.5 |
6.8 |
>99 |
96:2:2 |
15.2 |
1.2 |
| 12 |
2b
|
10 |
60 |
80.0 |
3.5 |
>99 |
96:2:2 |
17.0 |
1.2 |
Table 2 Polymerization of para-fluorostyrenea
| Entry |
Cat |
pFS/Cat |
Time (min) |
Conv. (%) |
rrrr
(%) |
M
nc × 10−4 |
M
w/Mnc |
T
md (°C) |
|
Conditions: [Sc] 5 μmol, [Sc]/[Ph3C][B(C6F5)4] = 1:1 (mol/mol), Toluene 1 mL, T = 20 °C.
Measured by 1H NMR and 13C NMR in CDCl3 at 25 °C.
Determined by GPC in THF at 40 °C against polystyrene standard.
Determined by DSC.
|
| 1 |
2a
|
200 |
20 |
>99 |
>99 |
5.2 |
1.7 |
323 |
| 2 |
2a
|
400 |
20 |
>99 |
>99 |
10.3 |
2.3 |
323 |
| 3 |
2b
|
200 |
60 |
51 |
>99 |
3.7 |
1.6 |
323 |
| 4 |
2b
|
400 |
60 |
50 |
>99 |
5.7 |
2.0 |
323 |
Copolymerization of pFS and BD
A typical procedure for butadiene and pFS copolymerization by the concurrent addition of both monomers is as follows (Table 1, entry 5): a toluene (1 mL) solution of [Ph3C][B(C6F5)4] (4.8 mg, 5 μmol) and a toluene solution (1 mL) of complex 2a (1.9 mg, 5 μmol) were added to a 10 mL flask under a nitrogen atmosphere and the mixture was kept under stirring for a few minutes. Then a mixture of pFS (0.30 g, 2.5 mmol) and butadiene (0.135 g, 2.5 mmol, 19%wt in toluene) was added. The reaction was terminated after 10 min by the addition of a small amount of acidic methanol and the mixture was poured into methanol (50 mL). The precipitated copolymer was collected by filtration and dried under vacuum at 40 °C to a constant weight (0.294 g, 66%). All other polymerization data were obtained following the same procedure but with different pFS-to-BD feed ratios or catalyst.
Synthesis of multi-block P(pFS-BD)
A typical procedure for synthesizing a multi-block P(pFS-BD) is as follows: a toluene (0.5 mL) solution of [Ph3C][B(C6F5)4] (4.8 mg, 5 μmol) and a toluene solution (0.5 mL) of complex 2a (1.9 mg, 5 μmol) were added to a 10 mL flask under a nitrogen atmosphere. The mixture was stirred at room temperature for a few minutes. As soon as purified pFS (0.3 g, 2.5 mmol) was added into the above system, butadiene (0.26 g, 4.8 mmol, 13%wt in toluene) was added dropwise over 10 min. Then the reaction was terminated by the addition of a small amount of acidic methanol followed by pouring the mixture into methanol (50 mL) to precipitate the copolymer product. The copolymer product was collected by filtration and dried under vacuum at 40 °C to a constant weight (0.47 g, 84%).
Kinetics studies for pFS and BD copolymerization
To a 50 mL flask were added a toluene solution (5 mL) containing [Ph3C][B(C6F5)4] (24 mg, 25 μmol) and complex 2a (9.5 mg, 25 μmol), under a nitrogen atmosphere at 20 °C. After vigorous stirring for a few minutes, a mixture of pFS (0.61 g, 5.0 mmol) and butadiene (0.27 g, 5.0 mmol, 17%wt in toluene) was added to the above system, which was evenly divided into five portions, and the reaction was terminated by adding acidic methanol at the set time (44s, 61s, 75s, 92s, 122s), respectively. Each copolymer was collected by filtration and was dried under vacuum at 40 °C and weighed for calculating the conversions of BD and pFS. The kinetics study for the copolymerization of pFS and BD by complex 2b was performed following the same procedure as described above.
Results and discussion
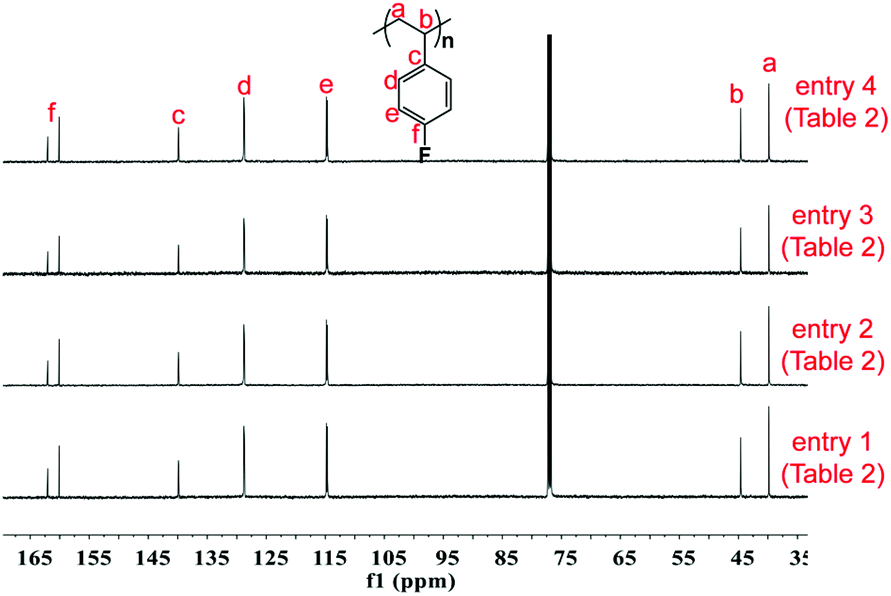
First, the copolymerization of p-fluorostyrene (pFS) and butadiene (BD) in toluene at room temperature was investigated by using the catalytic systems of 1/[Ph3C][B(C6F5)4]/[AliBu], which are highly active towards the homopolymerization of pFS and the copolymerization with styrene and ethylene, respectively.25,26 The polymerization catalyzed by 1a (Flucent–Sc–N (96.26°)) occurred fast but gave a copolymer insoluble in many solvents even under a high temperature of 160 °C. We proposed that cross-linking took place, since the scandium active species is Lewis acidic and can initiate polymerization of the dangling C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of the 1,2-regulated PBD segments. When switched to catalyst 1b (Flucent–Lu–N (92.16°)), the polymerization activity turned out to be very slow. The GPC curve of the isolated polymer shows multi-peaks, suggesting poor controllability of catalyst 1b during polymerization (Table 1, entries 1 and 2). Catalytic systems based on complexes 2a and 2b with smaller bite angles (Cpcent–Sc–N (86.6°) and Cpcent–Lu–N (84.1°)) possess dual cis-1,4 regio- and perfect stereo- selectivities for the polymerization of butadiene and styrene, respectively; however, whether they are tolerant to polar fluorine substituted monomers is still unknown.36 Thus the homopolymerization of pFS using complex 2a or 2b upon activation with [Ph3C][B(C6F5)4] was first attempted. Complex 2a showed a high activity to consume pFS completely in 20 min and provided perfect syndiotactic P(pFS) (rrrr > 99%, Fig. 1, entry 1). Changing the pFS-to-scandium molar ratio from 200:1 to 400:1, the molecular weight (Mn) of the resultant poly(pFS) increased from 5.2 × 104 to 10.3 × 104 while the molecular weight distribution remained constant and narrow (Table 2 entries 1 and 2). In addition, the Mn of P(pFS) increased linearly in proportion to the monomer conversion ([pFS]/[Complex 2a]/[[Ph3C][B(C6F5)4]] = 1000/1/1) as shown in Fig. 2, suggesting a living polymerization mode. On the other hand, complex 2b also showed a perfect syndioselectivity albeit in a low activity, probably owing to the less acidic lutetium metal center.
C bonds of the 1,2-regulated PBD segments. When switched to catalyst 1b (Flucent–Lu–N (92.16°)), the polymerization activity turned out to be very slow. The GPC curve of the isolated polymer shows multi-peaks, suggesting poor controllability of catalyst 1b during polymerization (Table 1, entries 1 and 2). Catalytic systems based on complexes 2a and 2b with smaller bite angles (Cpcent–Sc–N (86.6°) and Cpcent–Lu–N (84.1°)) possess dual cis-1,4 regio- and perfect stereo- selectivities for the polymerization of butadiene and styrene, respectively; however, whether they are tolerant to polar fluorine substituted monomers is still unknown.36 Thus the homopolymerization of pFS using complex 2a or 2b upon activation with [Ph3C][B(C6F5)4] was first attempted. Complex 2a showed a high activity to consume pFS completely in 20 min and provided perfect syndiotactic P(pFS) (rrrr > 99%, Fig. 1, entry 1). Changing the pFS-to-scandium molar ratio from 200:1 to 400:1, the molecular weight (Mn) of the resultant poly(pFS) increased from 5.2 × 104 to 10.3 × 104 while the molecular weight distribution remained constant and narrow (Table 2 entries 1 and 2). In addition, the Mn of P(pFS) increased linearly in proportion to the monomer conversion ([pFS]/[Complex 2a]/[[Ph3C][B(C6F5)4]] = 1000/1/1) as shown in Fig. 2, suggesting a living polymerization mode. On the other hand, complex 2b also showed a perfect syndioselectivity albeit in a low activity, probably owing to the less acidic lutetium metal center.
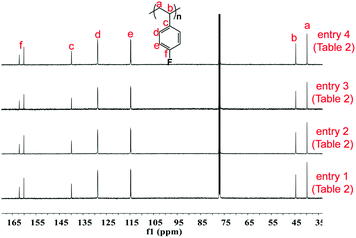 |
| | Fig. 1
13C NMR spectra of poly(pFS) (125 MHz, CDCl3, 25 °C). | |
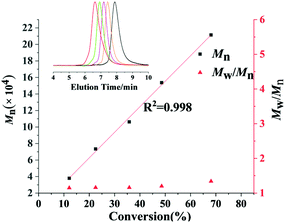 |
| | Fig. 2 Linear growth of molecular weight (Mn) as a function of pFS conversion. Inset: GPC traces as detected from the retention time. | |
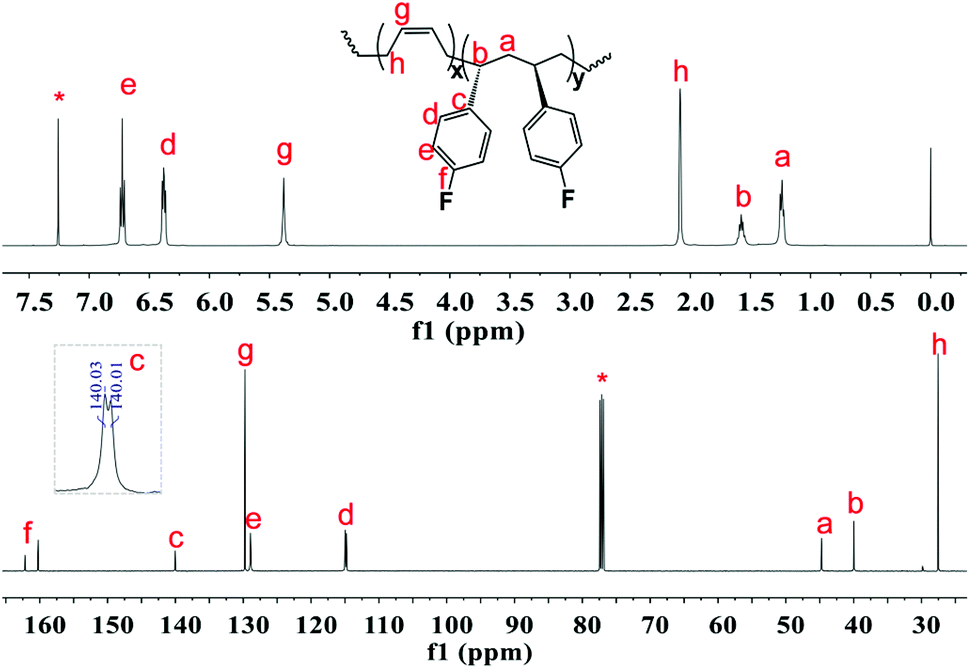
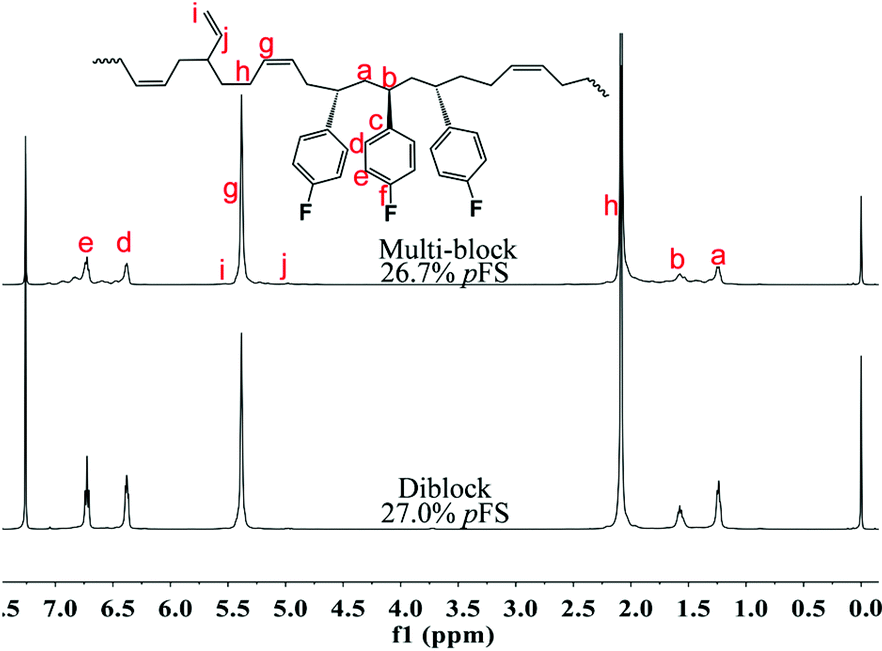
The excellent catalysis of 2/[Ph3C][B(C6F5)4] for highly selective polymerization of pFS prompted us to explore its copolymerization behavior with BD. The copolymerization with 2a was performed in toluene at 20 °C for 10 min by the concurrent addition of the two monomers under the pFS molar fraction varying from 10 to 90 mol%. As shown in Table 1, high conversions were obtained in the full range of monomer feed ratios, indicating distinguished high activities. The 1H NMR spectrum of the resultant polymer (Fig. 3 (top) (Table 2, entry 4)) shows that the peaks assigned to phenyl protons in P(pFS) units appear at δ = 6.29–6.89 ppm, whilst the peaks arising from olefinic protons in the 1,4-tactic PBD segments appear at δ = 5.30–5.46 ppm. According to the integration intensity ratio of the phenyl and olefinic protons, it was found that the pFS molar fraction in the copolymers varies corresponding to the loaded one, reaching up to 89.1 mol% when the pFS feed ratio was 90 mol%. Catalyst 2b showed a parallel catalytic behavior to 2a albeit with lower activity, which is much better than its performance for the homopolymerization of pFS.
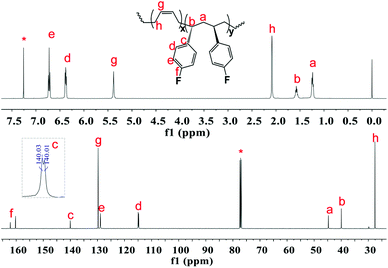 |
| | Fig. 3
1H and 13C NMR spectra of poly(pFS-BD) (Table 1, entry 4) (500 MHz for 1H NMR and 125 MHz for 13C NMR, CDCl3, 25 °C). | |
Gel permeation chromatography (GPC) analyses of these crude P(pFS-BD) copolymers without extraction show that the molecular weights are high (Mn = 12.4–25.7 × 104) and the molecular weight distributions are unimodal and narrow (PDI = 1.1–1.5), suggesting that copolymers were obtained successfully instead of a mixture of homopolymers, which is consistent with the single-sited catalytic behavior and obvious characteristics of the living copolymerization fashion.
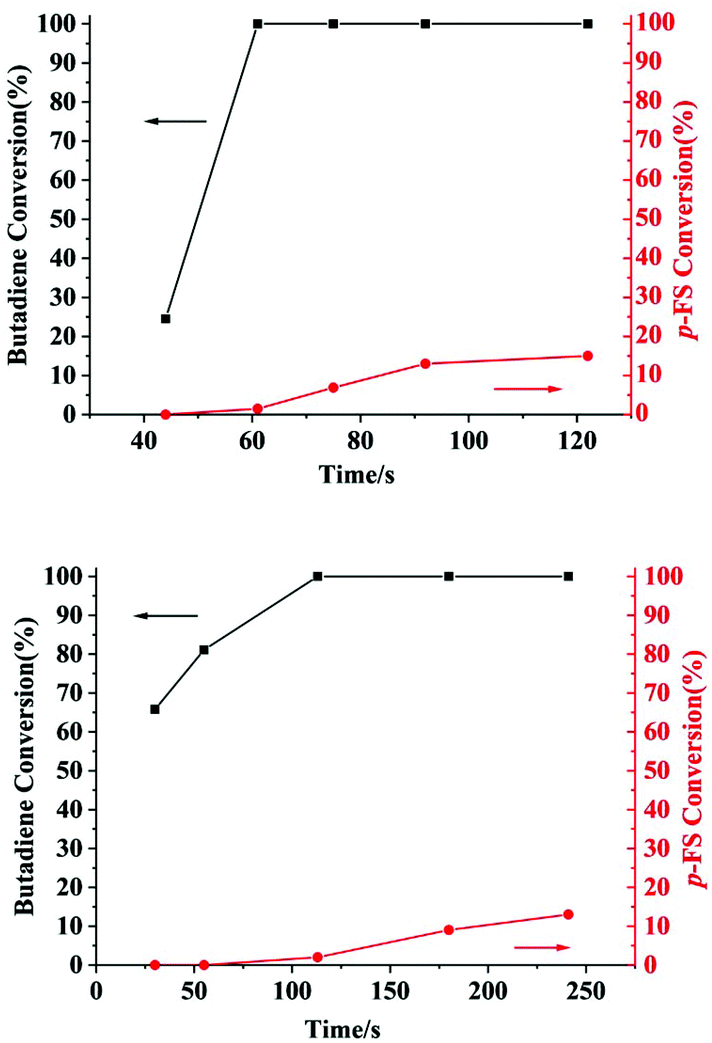
The microstructures of these copolymers were defined by 13C NMR spectroscopy analyses. The representative spectrum of a poly(pFS-BD) product with a moderate pFS content of 63.8 mol% is shown in Fig. 3 (bottom) (Table 2, entry 4). The ipso-carbon Cc giving a doublet at δ = 140.02 ppm with a coupling constant 4JC–F = 2.5 Hz indicates the syndiotactic (rrrr > 99%) P(pFS) block. The strong singlet at δ = 27.42 ppm indicates a high degree of cis-1,4-PBD unit (>95%). However, the resonances arising from the carbon–carbon linkages of pFS-BD joints are not observed. The 13C NMR spectra of the copolymers with different pFS contents show similar topologies. Meanwhile the cis-1,4 tacticity of the PBD segments remains unchanged (95–97%) in all copolymers, which is not affected by the pFS content. All these characterizations suggest that the copolymers obtained by using 2a/[Ph3C][B(C6F5)4] might have a diblock sequence. To confirm this deduction, a kinetics study of the copolymerization reaction in toluene at 20 °C by the concurrent addition of both monomers was carried out. As shown in Fig. 4 (top), when the polymerization catalyzed by 2a/[Ph3C][B(C6F5)4] was carried out for 1 min, the isolated product was almost pure PBD corresponding to the almost complete conversion of BD (>99%), whereas no pFS incorporation was found. With the polymerization going on and consumption of BD monomer, the pFS fraction in the copolymer increased dramatically. A similar phenomenon was observed using the 2b/[Ph3C][B(C6F5)4] system (Fig. 4 (bottom)). These results indicate that in the presence of pFS, BD has the priority when coordinating to the active metal center and propagating into polymer. Until BD is almost fully consumed, pFS starts to insert into Sc-PBD active species to generate the diblock structure without homopolymer contaminants.
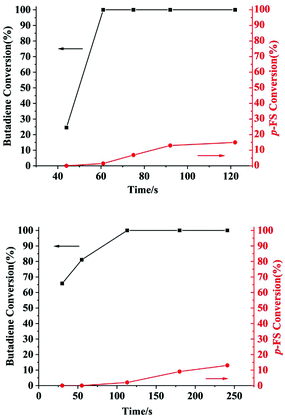 |
| | Fig. 4 Monomer conversions versus time with catalytic systems 2a (top) and 2b (bottom). | |
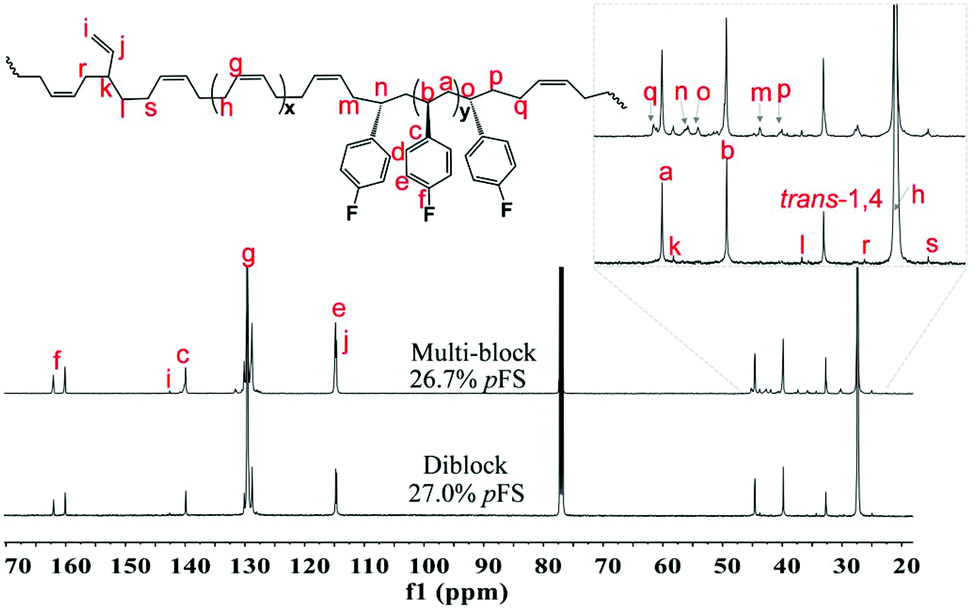
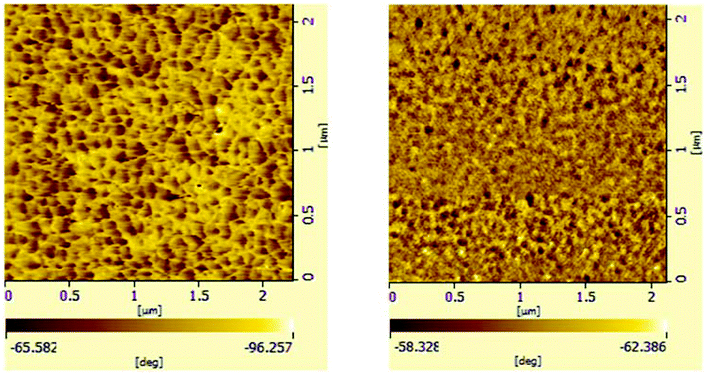
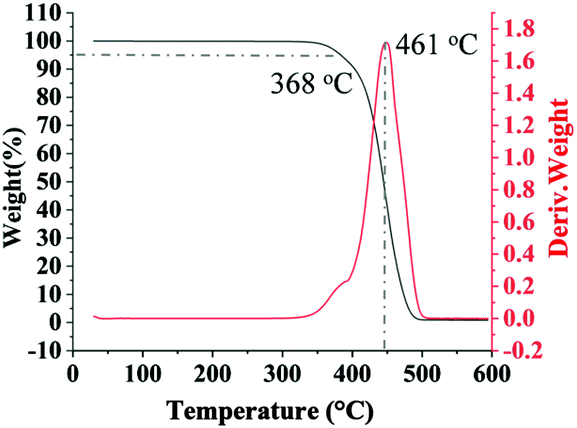
Multi-block copolymers as a kind of significant macromolecular materials exhibit outstanding properties. To reuse and strengthen the interfaces of abandoned PE and iPP, Coates et al. produced PE/iPP multi-block polymers by pyridylamidohafnium catalyst activated with B(C6F5)3.37 Miller and Ellison synthesized multi-block poly(ethylene terephthalate)-polyethylene, which enhances the ability to recycle PET/PE mixed waste streams.38 In other aspects, multi-block sulfonated poly(arylene ether sulfone)s are used in polymer electrolyte membrane fuel cells.39 According to the living polymerization mode of BD and pFS in the catalyst 2a system, a multiple BD feeding method was adopted to exclude the formation of long BD and pFS sequences for preparing multi-block P(pFS-BD) copolymers. A typical procedure was carried out in which pFS (0.3 g) was added in one portion into the polymerization system catalyzed by 2a/[Ph3C][B(C6F5)4] (5 μmol), and then a BD solution (2.0 g, 13%wt) was added over 10 min. As soon as the addition of the BD solution was completed, the copolymerization was terminated and a copolymer with 26.7 mol% pFS content was isolated (0.47 g, Mn = 13.9 × 104, PDI = 1.9). Its molecular weight distribution gives a unimodal peak (Fig. S32†), suggesting successful copolymerization. All the resonances in the 1H NMR spectrum of the multi-block copolymer are broadened as compared to the corresponding resonances in the diblock copolymer, especially at the aromatic region (Fig. 5). In the 13C spectrum, the resonances of carbons from the joints of the multi-block copolymer are observed (m, n, o, p and q points in Fig. 6). The averaged chain lengths of PBD and P(pFS) in the multi-block copolymer obtained by calculating the ratios of the integration intensities of the corresponding repeat units and the joints in the quantitative 13C NMR spectrum are 161 and 27, respectively (Fig. S21†). Since the highly syndiotactic P(pFS) is crystalline,35 the diblock copolymer with a 27.0 mol% P(pFS) content (one long sequence) has a microphase separation rather different from the multi-block copolymer with a similar P(pFS) content (26.7 mol%, many short sequences). As shown in Fig. 7, the AFM micrograph of the diblock copolymer (left) reveals a larger degree of phase separation than the multi-block one (right). In addition, there is no obvious melting point in the multi-block P(pFS-BD) as compared with the di-block one (Tm = 316 °C) in the curves of the differential scanning calorimetric analyses (Fig. S34†). The 5% mass loss temperature of the multi-block copolymer is at 368 °C, which is much higher than that of the commercial SBR (Fig. 8),40 indicating that it is a quite different and more thermally stable material.
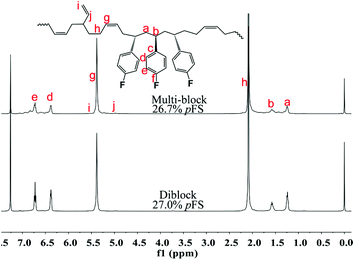 |
| | Fig. 5
1H NMR spectra of multi-block and diblock poly(pFS-BD) (500 MHz, CDCl3, 25 °C). | |
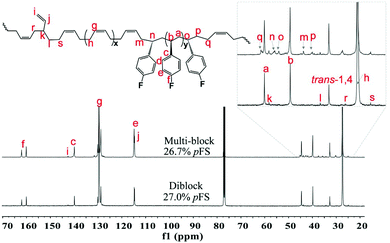 |
| | Fig. 6
13C NMR spectra of multi-block and diblock poly(pFS-BD) (125 MHz, CDCl3, 25 °C). | |
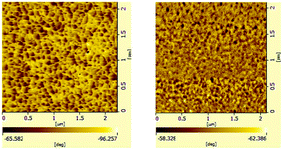 |
| | Fig. 7 AFM micrographs of a spin-coated thin film of diblock poly(pFS-BD)(left) (Table 2, entry 10, x(pFS) = 26.7 mol%) and multi-block poly(pFS-BD)(right)(x(pFS) = 27.0 mol%) (phase images, scan size: 2.0 μm). | |
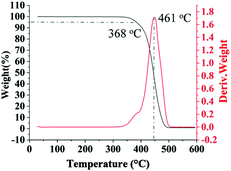 |
| | Fig. 8 TG and DTG curves of multi-block poly(pFS-BD). | |
Conclusions
In summary, we have demonstrated the unprecedented highly cis-1,4 selective (>95%) and perfectly syndiospecific (>99%) copolymerization of BD and pFS using pyridyl functionalized Cp ligated CGC rare-earth metal bis(allyl) complexes, which provide an active rare-earth metal center with an open coordination sphere to avoid low cis-1,4 selectivity and cross-linking process. In addition, the excellent tolerance for polar groups of these catalysts permits the insertion of pFS facilely tuned in the full range of 0–100% by changing the pFS-to-BD ratio. The diblock sequence copolymer is obtained by the concurrent addition of pFS and BD, with BD as the privilege, which is consistent with the kinetics study. By the merit of the living copolymerization characteristics, remarkably, a multi-block copolymer is isolated, for the first time, via pulse loading of butadiene. An unexpectedly high thermal stability was found for the multi-block copolymer during TGA testing. This work paves a new way to access functional styrene–butadiene copolymers with controllable regio- and stereo-regularities as well as sequence distribution. The mechanical properties of the di- and multi-block copolymers and the comparison of these copolymers with the SBR prepared by the traditional anionic mechanism are under investigation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the financial support from NSFC (projects No.21634007, 21774118, 21674108 and U1862108), and Department of Science and Technology of Jilin Province project No. 20190201067JC.
Notes and references
- P. Calvert, The American Synthetic Rubber Research Program, Nature, 1990, 346, 328–328 Search PubMed.
- J. M. Horner, Environmental Health Implications of Heavy Metal Pollution from Car Tires, Rev. Environ. Health, 1996, 11, 175–178 CAS.
- A. A. Shah, F. Hasan, Z. Shah, N. Kanwal and S. Zeb, Biodegradation of Natural and Synthetic Rubbers: A Review, Int. Biodeterior. Biodegrad., 2013, 83, 145–147 CrossRef.
- F. Zhao, Q. W. Huang, H. N. Gao and S. G. Zhao, Development of Raw Materials for Green Tire, Chin. Sci. Bull., 2016, 61, 3348–3358 CrossRef.
- H. Zhu, S. Zhang and Y. X. Wu, Progress in the Synthesis of High-Performance Butadiene-Based Elastomer for Green Tires, Chin. Sci. Bull., 2016, 61, 3326–3337 CrossRef.
- L. M. Polgar, M. van Duin, A. A. Broekhuis and F. Picchioni, Use of Diels−Alder Chemistry for Thermoreversible Cross-Linking of Rubbers: The Next Step toward Recycling of Rubber Products?, Macromolecules, 2015, 48, 7096–7105 CrossRef CAS.
- Y. R. Chen, A. Yasin, Y. G. Zhang, X. J. Zan, Y. X. Liu and L. T. Zhang, Preparation and Modification of Biomass-Based Functional Rubbers for Removing Mercury(II) from Aqueous Solution, Materials, 2020, 13, 632–648 CrossRef CAS.
- R. Bonda, G. F. Morton and L. H. Krol, A Tailor-Made Polymer for Tyre Applications, Polymer, 1984, 25, 132–140 CrossRef.
- N. Naga and Y. Imanishi, Copolymerization of Styrene and Conjugated Dienes with Half-Sandwich Titanium(IV) Catalysts: The Effect of the Ligand Structure on the Monomer Reactivity, Monomer Sequence Distribution, and Insertion Mode of Dienes, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 939–946 CrossRef CAS.
- S. Zhang, S. H. Zhao, X. Y. Zhang, L. Q. Zhang and Y. P. Wu, Preparation, Structure, and Properties of End-Functionalized Miktoarms Star-Shaped Polybutadiene–Sn–Poly(styrene–butadiene) Rubber, J. Appl. Polym. Sci., 2014, 131 Search PubMed , 40002.
- M. Hassanabadi, M. Najafi, G. H. Motlagh and S. S. Garakani, Synthesis and Characterization of End-Functionalized Solution Polymerized Styrene-Butadiene Rubber and Study the Impact of Silica Dispersion Improvement on the Wear Behavior of the Composite, Polym. Test., 2020, 85, 106431–106443 CrossRef CAS.
- D. M. Cui, Studies on Homo- and Co-polymerizations of Polar and Non-polar Monomers Using Rare-earth Metal Catalysts, Acta Polym. Sin., 2020, 51, 12–29 Search PubMed.
- Y. N. Na and C. L. Chen, Catechol-Functionalized Polyolefins, Angew. Chem., Int. Ed., 2020, 59, 7953–7959 CrossRef CAS.
- Y. P. Zhang, F. Wang, L. Pan, B. Wang and Y. S. Li, Facile Synthesis of High-Molecular-Weight Vinyl Sulfone (Sulfoxide) Modified Polyethylenes via Coordination−Insertion Copolymerization, Macromolecules, 2020, 53(13), 5177–5187 CrossRef CAS.
- Y. X. Zhang, C. Q. Wang, S. Mecking and Z. B. Jian, Ultrahigh Branching of Main-Chain-Functionalized Polyethylenes by Inverted Insertion Selectivity, Angew. Chem., Int. Ed., 2020, 59, 14296–14302 CrossRef CAS.
- G. F. Liao, Z. F. Xiao, X. L. Chen, C. Du, L. Zhong, C. S. Cheung and H. Y. Gao, Fast and Regioselective Polymerization of para-Alkoxystyrene by Palladium Catalysts for Precision Production of High-Molecular Weight Polystyrene Derivatives, Macromolecules, 2020, 53, 256–266 CrossRef CAS.
- Y. Jing and V. V. Sheares, Polar, Free Radical Copolymerization Studies of 2-Cyanomethyl-1,3-butadiene with Styrene and Acrylonitrile, Macromolecules, 2000, 33, 6262–6268 CrossRef CAS.
- M. D. Beery, M. K. Rath and V. V. Sheares, Polymerization Studies of 2,3-Bis(4-ethoxy-4-oxobutyl)-1,3-butadiene and Copolymerization with Styrene, Macromolecules, 2001, 34, 2469–2475 CrossRef CAS.
- T. Yaegashi, S. Yodoya, M. Nakamura, H. Takeshita, K. Takenaka and T. Shiomi, Polymerization of 1,3-Dienes with Functional Groups. III. Free-Radical Polymerization of N, N-Diethyl-2-methylene-3-butenamide, J. Polym. Sci., Part
A: Polym. Chem., 2004, 42, 999–1007 CrossRef CAS.
- Z. C. Wang, D. T. Liu and D. M. Cui, Highly Selective Polymerization of 1,3-Conjugated Dienes by rare Earth Organometallic Complexes, Acta Polym. Sin., 2015, 9, 989–1009 Search PubMed.
- X. F. Li, J. Baldamus, M. Nishiura, O. Tardifand and Z. M. Hou, Cationic Rare-Earth Polyhydrido Complexes: Synthesis, Structure, and Catalytic Activity for the cis-1,4-Selective Polymerization of 1,3-Cyclohexadiene, Angew. Chem., Int. Ed., 2006, 45, 8184–8188 CrossRef CAS.
- H. B. Wang, X. Wu, Y. Yang, M. Nishiura and Z. M. Hou, Co-syndiospecific Alternating Copolymerization of Functionalized Propylenes and Styrene by Rare-Earth Catalysts, Angew. Chem., Int. Ed., 2020, 59, 7173–7177 CrossRef CAS.
- D. T. Liu, C. G. Yao, R. Wang, M. Y. Wang, Z. C. Wang, C. J. Wu, F. Lin, S. H. Li, X. H. Wan and D. M. Cui, Highly Isoselective Coordination Polymerization of ortho-Methoxystyrene with β-Diketiminato Rare-Earth-Metal Precursors, Angew. Chem., Int. Ed., 2015, 54, 5205–5209 CrossRef CAS.
- D. T. Liu, M. Y. Wang, Z. C. Wang, C. J. Wu, Y. P. Pan and D. M. Cui, Stereoselective Copolymerization of Unprotected Polar and Nonpolar Styrenes by an Yttrium Precursor: Control of Polar-Group Distribution and Mechanism, Angew. Chem., Int. Ed., 2017, 56, 2714–2719 CrossRef CAS.
- Z. C. Wang, M. Y. Wang, J. Y. Liu, D. T. Liu and D. M. Cui, Rapid Syndiospecific (Co)Polymerization of Fluorostyrene with High Monomer Conversion, Chem. – Eur. J., 2017, 23, 18151–18155 CrossRef CAS.
- B. Liu, K. N. Qiao, J. Fang, T. T. Wang, Z. C. Wang, D. T. Liu, Z. G. Xie, L. Maron and D. M. Cui, Mechanism and Effect of Polar Styrenes on Scandium-Catalyzed Copolymerization with Ethylene, Angew. Chem., Int. Ed., 2018, 57, 14896–14901 CrossRef CAS.
- S. H. Li, D. T. Liu, Z. C. Wang and D. M. Cui, Development of Group 3 Catalysts for Alternating Copolymerization of Ethylene and Styrene Derivatives, ACS Catal., 2018, 8, 6086–6093 CrossRef CAS.
- C. X. Wang, G. Luo, M. Nishiura, G. Y. Song, A. Yamamoto, Y. Luo and Z. M. Hou, Heteroatom-Assisted Olefin Polymerization by Rare-Earth Metal Catalysts, Sci. Adv., 2017, 3, e1701011 CrossRef.
- S. Y. Long, F. Lin, C. G. Yao and D. M. Cui, Highly cis-1,4 Selective Living Polymerization of Unmasked Polar 2-(2-Methylidenebut-3-enyl) Furan and Diels–Alder Addition, Macromol. Rapid Commun., 2017, 38, 1700227–1700232 CrossRef.
- Z. H. Shi, F. Guo, R. Meng, L. Jiang and Y. Li, Stereoselective Copolymerization of Amino-Functionalized Styrene with Butadiene Using a Half-Sandwich Scandium Complex, Polym. Chem., 2016, 7, 7365–7369 RSC.
- F. Guo, L. Jiang, K. Y. Diao and Z. M. Hou, Stereoselective Copolymerization of 4-(N, N-Diphenylamino) Styrene and Isoprene by a C5H5-Ligated Scandium Catalyst: Synthesis of Amino-Functionalized Crystalline Styrenic Thermoplastic Elastomers, Polym. Chem., 2020, 11, 1314–1320 RSC.
- J. S. Zigmond, K. A. Pollack, S. Smedley, J. E. Raymond, L. A. Link, A. Pavia-Sanders, M. A. Hickner and K. L. Wooley, Investigation of IntricateAmphiphilic Crosslinked Hyperbranched Fluoropolymers as Anti-icing Coatings for Extreme Environments, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 238–244 CrossRef CAS.
- A. Buonerba, C. Cuomo, V. Speranza and A. Grassi, Crystalline Syndiotactic Polystyrene as Reinforcing Agent of cis-1,4-Polybutadiene Rubber, Macromolecules, 2010, 43, 367–374 CrossRef CAS.
- Y. P. Pan, W. F. Rong, Z. B. Jian and D. M. Cui, Ligands Dominate Highly Syndioselective Polymerization of Styrene by Using Constrained-geometry-configuration Rare-earth Metal Precursors, Macromolecules, 2012, 45, 1248–1253 CrossRef CAS.
- Z. B. Jian, D. M. Cui and Z. M. Hou, Rare-Earth-Metal–Hydrocarbyl Complexes Bearing Linked Cyclopentadienyl or Fluorenyl Ligands: Synthesis, Catalyzed Styrene Polymerization, and Structure–Reactivity Relationship, Chem. – Eur. J., 2012, 18, 2674–2268 CrossRef CAS.
- Z. B. Jian, S. J. Tang and D. M. Cui, A Lutetium Allyl Complex That Bears a Pyridyl-Functionalized Cyclopentadienyl Ligand: Dual Catalysis on Highly Syndiospecific and cis-1,4-Selective (Co)Polymerizations of Styrene and Butadiene, Chem. – Eur. J., 2010, 16, 14007–14015 CrossRef CAS.
- J. M. Eagan, J. Xu, R. D. Girolamo, C. M. Thurber, C. W. Macosko, A. M. LaPointe, F. S. Bates and G. W. Coates, Combining Polyethylene and Polypropylene: Enhanced Performance with PE/iPP Multiblock Polymers, Science, 2017, 355, 814–816 CrossRef CAS.
- K. Nomura, X. Y. Peng, H. Kim, K. L. Jin, H. J. Kim, A. F. Bratton, C. R. Bond, A. E. Broman, K. M. Miller and C. J. Ellison, Multiblock Copolymers for Recycling Polyethylene−Poly (ethylene terephthalate) Mixed Waste, ACS Appl. Mater. Interfaces, 2020, 12, 9726–9735 CrossRef CAS.
- J. Yuk, S. Lee, A. F. Nugraha, H. Lee, S. H. Park, S. D. Yim and B. Bae, Synthesis and Characterization of Multi-Block Poly (arylene ether sulfone) Membranes with Highly Sulfonated Blocks for Use in Polymer Electrolyte Membrane Fuel Cells, J. Membr. Sci., 2016, 518, 50–59 CrossRef CAS.
- J. L. Tan, H. Z. Cheng, L. B. Wei, Y. W. Xin and X. H. Gui, Using Low-Rank Coal Slime as an Eco-Friendly Replacement for Carbon Black Filler in Styrene Butadiene Rubber, J. Cleaner Prod., 2019, 234, 949–960 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0qi00719f |
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a