
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnthracene–styrene-substituted m-carborane derivatives: insights into the electronic and structural effects of substituents on photoluminescence†
Mahdi
Chaari
 a,
Zsolt
Kelemen
a,
Duane
Choquesillo-Lazarte
b,
Francesc
Teixidor
a,
Clara
Viñas
a and
Rosario
Núñez
*a
a,
Zsolt
Kelemen
a,
Duane
Choquesillo-Lazarte
b,
Francesc
Teixidor
a,
Clara
Viñas
a and
Rosario
Núñez
*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus U.A.B., 08193, Bellaterra, Barcelona, Spain. E-mail: rosario@icmab.es; Fax: +3493 580 5729; Tel: +34 93 580 1853
bLaboratorio de Estudios Cristalográficos, IACT-CSIC, Avda. de las Palmeras 4, 18100 – Armilla, Granada, Spain
First published on 11th May 2020
Abstract
Two anthracenyl–styrenyl-m-carborane triads (one non-iodinated on B, 3, and one iodinated, 4) were synthesized and characterized to be further linked to octavinylsilsesquioxane (OVS) via cross-metathesis, giving rise to the corresponding hybrid materials 5 and 6. The crystal structure of the non-iodinated heterosubstituted-m-carborane 3 was analyzed by X-ray diffraction. Transmission electron microscopy images of pristine OVS and hybrids 5–6 show important differences in the morphology of the particles; whereas OVS forms cubic-like particles, 5–6 have a spherical shape with a broad particle-size distribution. All compounds showed similar vibronic emission spectra in solution, with maxima around 415 nm, assigned to the locally excited state (LE) emission of the anthracene moiety. The similarity with the spectra of the free anthracene (λem = 420 nm) suggested that only small electronic interactions between the anthracene units have taken place, and there is no influence of the iodo or styrene groups on the absorption properties. This is in agreement with the DFT calculations, where calculated oscillator strength corresponding to the transitions from iodo orbitals to the LUMO are weak and could not be observed experimentally. Noticeable, triads 3–4 exhibited exceptional fluorescence quantum yield values of around 100% in solution, that are comparable to those determined for their precursors 1–2, demonstrating that the influence of the styrene group is negligible. Linking these m-carborane derivatives to the OVS led to a significant decrease of quantum yields to 34–45% for 5–6 in solution. Moreover, the PL behavior in the aggregate state was investigated and the spectra of all compounds were very similar, showing emission red-shift with maxima around 455–459 nm. Remarkably, quite high fluorescence quantum yields were determined for 3–4 ((ϕF = 26–31%) and 5–6 (ϕF = 27–36%) in the aggregated state. These data confirm that the m-carborane platform enhances the quantum efficiency of the anthracene in solution, without losing the emission properties in the aggregate state. If this affirmation is associated to other scattered examples on other fluorophores also linked to m-carborane existing in the literature, the former conclusion is reinforced. m-Carborane enhances the fluorescence quantum yield of the free fluorophore, but does not alter the energy of the participating states in the photoluminescence in solution.
Introduction
Optoelectronic organic materials have been successfully applied in organic light-emitting diodes (OLEDs), thin-film-transistors, electrochromic devices, sensors, liquid crystal displays, among others, due to their low-cost, facile processability, variability in structural and property tuning, and the capability of producing large areas of a flexible thin film.1The incorporation of polyhedral oligomeric silsesquioxanes (POSSs) into optoelectronic materials to achieve improved performances has attracted particular attention due to the unique and interesting hybrid structures of POSS. In particular, octasilsesquioxanes are nanosized building blocks for organic/inorganic hybrid materials of general formulae [RSiO1.5]8 with a 3D scaffold that exhibit a huge versatility to be functionalized, high robustness and thermal stability.2 They can be modified to tailor chemical, mechanical, electrical, optical or electronic properties.3 These exceptional characteristics make them very useful for a wide variety of applications, including thermally and chemically resistant polymers and ceramics,4 as flame retardants,5 catalysis,6 nanomedicine,7 emitting layers in OLEDs,8 among others.
Carboranes are icosahedral clusters of formula C2B10H12,9 with three-dimensional (3D) σ-delocalization,10 and electron-acceptor ability through substitution at the Ccluster (Cc).11 The linking of different fluorophores to the carborane Cc through a –CH2– spacer results in carborane-containing dyes, which fluorescence emission depends on the cluster isomer (o- or m-) and the substituent at the second Cc atom.12,13 It was also noticed that those dyes with low or null emission in solution were able to exhibit moderate fluorescence efficiency in the solid-state.12d,14 In 2011, N. Hosmane et al. reported carborane-appended 1,3,5-triphenylbenzene (TB) and 1,3,5-tris(biphenyl-4-yl)benzene (TBB) containing three o-, m- and p-carborane clusters, showing that o-carborane produced a quenching of the fluorescence, whereas the m-isomer caused an increase of the fluorescence quantum yields (ϕF), from pristine TB (10%) and TBB (27%) to 25 and 46%, respectively, after bonding to m-carborane.15 Their interpretation was that the 3D structure prevents the π–π stacking interactions in the π-conjugated systems and enhances rigidity of the conjugated molecules. Later, H. Yan observed that anthracene derivatives functionalized with o-carborane exhibited a ϕF of 5%, whereas its homologous one with the m-isomer yielded a ϕF of 94%, even higher that the starting anthracene derivative (70%).16 These two examples were isolated cases and no further research was explored. Later, we also observed that m-carborane linked to stilbene groups achieves higher fluorescence efficiency than their homologous with o-carborane derivatives, being the ϕF values of m-carboranyl-stylbene 8–19% vs. o-carboranes (3–16%).12d So these isolated cases were moving into a general rule, but it had to be demonstrated more consistently. More recently, our group focused on m-carborane based anthracenes to demonstrate that triads of di-anthracenyl-m-carboranes gave quantum yield efficiencies of 63–66%, being in all cases significantly higher than the pristine anthracene (26%).17a Remarkably, when m-carborane is linked to only one anthracene to give C-monosubstituted compounds, the result is still more relevant and ϕF values close to 100% were obtained.17b Furthermore, for all of them, moderate to good quantum efficiencies in the aggregate state were also determined, demonstrating that m-carborane is a tool of choice to boost the photoluminescence (PL) properties of those organic π-conjugated systems bounded to it, as anthracenes, both in solution and solid-state.
Previously, our group has reported photoluminescent hybrid materials based on octasilsesquioxane cubic structures (POSS) decorated with terminal o-carborane clusters throughout organic π-conjugated systems.18 The final PL properties of these hybrids may be tailored by changing the substituents at the Cc atoms. In these hybrids the POSS cage acts as an organizing scaffold, causing restriction to the intramolecular movement of the arms and avoiding additional interactions between them that could cause the quenching of the fluorescence (ϕF = 0.02–0.44).18 Other sets of carboranyl-containing octasilsesquioxane hybrids were prepared linking o- and m-carborane to the POSS core by means of vinylstilbene units, exhibiting moderate to high fluorescent quantum yields in solution (ϕF = 0.21–0.59); being the most efficient, the one which contains non-substituted o-carborane units.19 However, a quenching of the fluorescence was observed in the solid-state attributed to intermolecular interactions.
Throughout our research, we consistently have observed that when fluorophores are linked to m-carborane, the quantum yields are dramatically enhanced, many of them approaching 100%, and usually much higher than the plain fluorophores. In the current work, the m-carborane has been heterosubstituted at both Cc atoms with two different substituents, one fluorophore (the anthracene) that provides the PL properties, and one styrene, which enables the linking of our system to another platform, i.e. OVS. To gain further insight into the substituent effects at the m-carborane cluster on the PL properties of the final fluorophores, herein we analyse the photophysical properties in solution and aggregate states of two heterosubstituted anthracenyl–styrenyl-m-carborane clusters and their respective POSS hybrids, demonstrating that even by attaching a second substituent to the Cc of the m-carborane, the fluorescence efficiency is kept very high.
Results and discussion
Synthesis and characterization of compounds 3–6
To prepare fluorescence emitting boron rich hybrids we have followed the next strategy: in a first step, the synthesis of heterosubstituted anthracenyl–styrenyl-m-carborane triads was performed, followed by their linking to octavinylsilsesquioxane via cross-metathesis.The starting anthracenyl-m-carborane derivatives 1–2 were prepared from 1,7-closo-C2B10H12 and 9-I-1,7-closo-C2B10H11,17b,20 respectively, following literature procedures. Then, a second functionalization at the adjoining Cc–H was done by nucleophilic substitution of 1–2 using 15% of the excess of 4-vinylbenzyl chloride at reflux overnight,12a giving rise to heterosubstituted compounds 3 and 4, in 61 and 67% yield, respectively (Scheme 1). This reaction was monitored by 11B{1H} NMR following the appreciable changes in the boron resonance distributions. In the second step, compounds 3 and 4 were reacted with the OVS via cross-metathesis, following the procedure previously reported by us,18 using CH2Cl2 and the first generation of Grubbs catalyst,21 to give hybrids 5 and 6, in a 52 and 50% yield, respectively. The reaction was monitored by 1H NMR and the complete conversion was confirmed upon a change in the aromatic proton resonance distributions, the disappearance of signals from the vinyl group, and the appearance of new resonances attributed to the two alkene protons.
 | ||
| Scheme 1 Synthetic procedures for (a) compounds 3–4 and (b) hybrids 5–6. | ||
The structures of 3–6 were established based on IR-ATR, 1H, 13C{1H} and 11B{1H} NMR spectroscopy and elemental analysis; triad 3 was confirmed by X-ray diffraction analysis. The IR-ATR spectra of all compounds show typical ν(B–H) strong bands of closo-clusters between 2557 and 2596 cm−1. The IR-ATR spectra of 5–6 show a band near 1600 cm−1 attributed to the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and the typical broad band around 1085 cm−1 due to the Si–O bond. 1H NMR spectra of 3–4 display one new singlet signal, at around δ 3.05 ppm, attributed to the Cc–CH2–styrenyl protons.12a Besides, other resonances attributed to the benzyl group protons are observed in the range 7.34–6.88 ppm, and the vinyl group in the range 6.82–5.31 ppm (see ESI†).
C) and the typical broad band around 1085 cm−1 due to the Si–O bond. 1H NMR spectra of 3–4 display one new singlet signal, at around δ 3.05 ppm, attributed to the Cc–CH2–styrenyl protons.12a Besides, other resonances attributed to the benzyl group protons are observed in the range 7.34–6.88 ppm, and the vinyl group in the range 6.82–5.31 ppm (see ESI†).
The 11B{1H} NMR spectra for 3–4 show the typical, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:2 and 2:5:1:1:1 pattern,17a respectively. As for previously reported iodinated carboranes,17,22 the B–I is identified as the highest field resonances at −23.96 ppm, which remains as a singlet in the 11B{1H} NMR. The 11B{1H} NMR spectra of 5–6 show overlapped boron resonances with patterns 16:64 or 16:40:16:8, respectively. The 13C{1H} NMR spectra of 3–4 show the aromatic resonances in the range 137–124 ppm and two signals attributed to Cc–CH2 carbons at around δ = 33 and 42 ppm. Two other additional resonances corresponding to the vinyl group appear in the range δ = 136 and 114 ppm. Moreover, the 13C{1H} NMR spectra of 5–6 show the aromatic and alkene resonances between 149 and 117 ppm, whereas peaks in the region 77–75 ppm are attributed to the Cc–C. The methylene groups resonances appear in the range δ 42–33 ppm.
:6:2 and 2:5:1:1:1 pattern,17a respectively. As for previously reported iodinated carboranes,17,22 the B–I is identified as the highest field resonances at −23.96 ppm, which remains as a singlet in the 11B{1H} NMR. The 11B{1H} NMR spectra of 5–6 show overlapped boron resonances with patterns 16:64 or 16:40:16:8, respectively. The 13C{1H} NMR spectra of 3–4 show the aromatic resonances in the range 137–124 ppm and two signals attributed to Cc–CH2 carbons at around δ = 33 and 42 ppm. Two other additional resonances corresponding to the vinyl group appear in the range δ = 136 and 114 ppm. Moreover, the 13C{1H} NMR spectra of 5–6 show the aromatic and alkene resonances between 149 and 117 ppm, whereas peaks in the region 77–75 ppm are attributed to the Cc–C. The methylene groups resonances appear in the range δ 42–33 ppm.
Finally, transmission electron microscopy (TEM) images of the octavinylsilsesquioxane (OVS) and 5–6 have been obtained (see Fig. 1) and some important differences in the morphology of the particles have been observed. The starting OVS forms cubic-like particles, whereas 5–6 have a spherical shape with a broad particle-size distribution. The shape of these spherical particles obtained for 5–6 is very similar to previous reported o-carboranyl–styrene decorated octasilsesquioxanes,18 but with a higher size, which suggests that the presence of the anthracenyl does not affect the shape of the hybrid.
 | ||
| Fig. 1 TEM photographs of the octavinylsilsesquioxane (OVS) and 5–6. | ||
X-ray structural analysis
Single-crystals of 3 suitable for X-ray structural determination were obtained by slow evaporation from a mixture of chloroform/n-heptane (9:1) at room temperature. The molecular structure for 3 was established by single-crystal X-ray diffraction (Fig. 2) and agree with the NMR data (vide supra). Experimental crystal data and structure refinement parameters for the structure are listed in Table 1. The triad 3 crystallizes in the monoclinic system P21/c space group. The molecular structure shows a typical icosahedral geometry with similar bond distances and angles to those m-carborane–anthracene dyads and triads previously reported.17
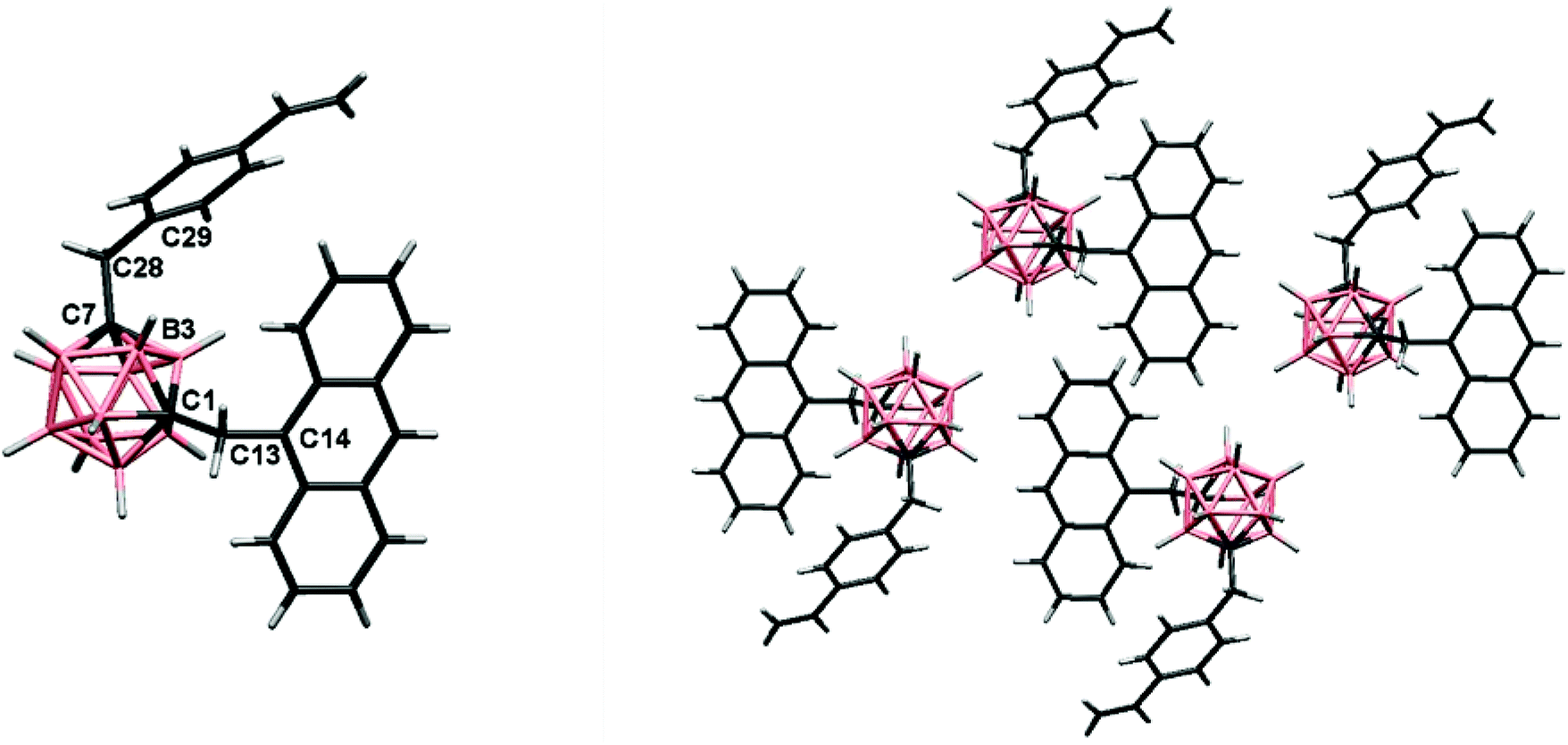 | ||
| Fig. 2 (Left) Molecular structure of 3. Torsion angles: C29C28C7B3, −40.4(2)° and C14C13C1B3, −38.3(2)°; (right) projections showing the organization of four molecules of 3 in the solid-state. Color code: B pink; C grey; H white. | ||
| 3 | |
|---|---|
| CCDC 1972767 (3) contain the supplementary crystallographic data for this paper.† | |
| Empirical formula | C26H30B10 |
| Formula weight | 450.60 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| Temperature/K | 293(2) |
| Wavelength/Å | 0.71073 |
| a/Å | 10.3008(18) |
| b/Å | 11.295(3) |
| c/Å | 22.279(4) |
| α/° | 90 |
| β/° | 97.941(7) |
| γ/° | 90 |
| Volume/Å3 | 2567.3(9) |
| Z | 4 |
| Density (calculated)/Mg m−3 | 1.166 |
| F(000) | 944 |
| Theta range for data collection/° | 2.581 to 27.484 |
| Absorption coefficient/mm−1 | 0.060 |
| Goodness-of-fit on F2 | 1.029 |
| R 1 [I > 2sigma(I)] | 0.0524 |
| wR2 [I > 2sigma(I)] | 0.1287 |
| R 1 (all data) | 0.0710 |
| wR 2 (all data) | 0.1405 |
In the structure of 3, the m-carborane moiety is linked from the Cc to one anthracene unit through a methylene spacer (–CH2–), and likewise from the adjoining Cc to one styrene unit. As shown in Fig. 2, the rotation of the anthracene and benzene rings through the (H)(H)C–C(anthracene–benzene) bond allows more or less similar conformations in the solid-state. Conformations found in the solid-state and torsion angles can be seen in Fig. 2 caption.
The solid-state structure in 3 is dominated by intermolecular C–H⋯π interactions (Fig. S1† and Table 2), mainly between (H)C–H⋯Canthracene (Fig. S1† and Table 2). Moreover, interactions between –CH2– hydrogens and vinyl carbons are found. Other weak B–H⋯H–C contact is listed in Table 2. From the crystal packing of 3 (Fig. 2, Fig. S1†), we may determine that there are not π–π stacking between two anthracenes or between anthracene and benzene units, due to the presence of the –CH2– spacer, from which one hydrogen atom interacts with an aromatic Canthracene (∼2.9 Å) avoiding the intermolecular π–π interactions.
| Compound | D–H⋯A | d(H⋯A) | <(DHA) | <(HHB) |
|---|---|---|---|---|
| Symmetry codes (i) −x, 1/2 + y, 3/2 − z (ii) x, 3/2 − y, 1/2 + z (iii) 1 − x, 1/2 + y, 3/2 − z (iv) 1 − x, 1/2 + y, 3/2 − z. | ||||
| 3 | C(13)–H(13B)⋯C(23)i | 2.892 | 168.27 | |
| C(13)–H(13A)⋯C(34)ii | 2.977 | 150.4 | ||
| C(28)–H(28B)⋯C(21)iii | 2.937 | 171 | ||
| C(28)–H(28A)⋯H(4)–B(4)iv | 2.437 | 123.2 | 109.31 | |
From the fingerprint plots analysis, it immediately emerges that H⋯H contacts comprise nearly 72% of the total Hirshfeld surface area for compound 3. The H⋯C contacts contribute around 28% to the total Hirshfeld surface area, whereas the contribution of C⋯C contacts (e.g., π⋯π interactions) is less than 1% (see more details in ESI†).
Photophysical properties and TD-DFT calculations
The photophysical properties of 3–6 were determined by UV-Vis absorption and fluorescence spectroscopies in THF, as well as in a mixture of THF/water (v/v = 1/99) to form aggregates (Table 3). Electronic properties of 3–6 in the ground state were assessed by UV-Vis absorption measurements (∼10−5 M for 3–4 and ∼10−6 M for 5–6, Fig. 3). All compounds exhibit similar absorption spectra with vibrational structures and the four characteristic peaks at around 334, 351, 369 and 389 nm assigned to the π–π* transition band of the anthracene moiety, as was previously observed for precursors 1–2.17 This suggests that the incorporation of the methyl–styrenyl group through the neighboring Cc does not alter the absorption patterns of the anthracene. The molar extinction coefficients (ε) are significantly different between monomers 3–4 and hybrids 5–6. For 3–4 the ε values were in the range from 94 × 102 to 99 × 102 M−1 cm−1, slightly lower than their precursors 1–2 (102 × 102 and 106 × 102 M−1 cm−1, respectively);17b whereas for 5–6 higher values in the range from 553 × 102 to 630 × 102 M−1 cm−1 were obtained. The molar extinction coefficients for the carboranyl-containing POSS hybrids are almost six times larger than the values found for the individual precursors (3–4), so that the ratio of absorption per carboranyl triad is higher when this unit is attached to the POSS cage, which assembles eight fluorophores per molecule (Table 3).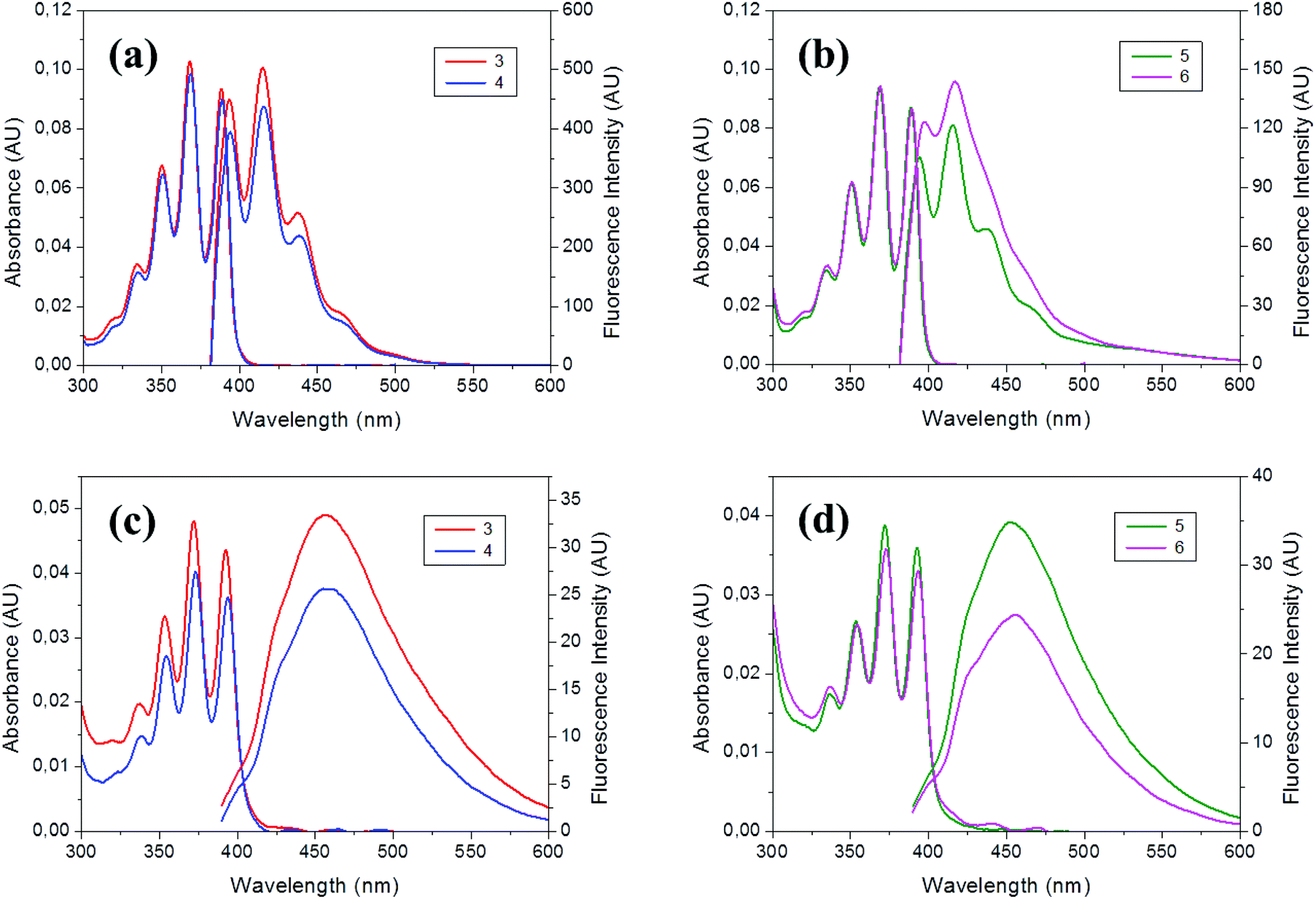 | ||
| Fig. 3 Absorption and emission spectra of 3–6 in THF solutions (a–b) and aggregates (THF/H2O, v/v = 1/99) (c–d). AU: arbitrary units. | ||
| Compounds | THF | THF/water (1/99) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| λ abs (nm) | ε/105 (M−1 cm−1) | λ em (nm) |
ϕ
Fa |
Stokes shift (nm) | λ abs (nm) | λ em (nm) |
ϕ
Fa |
Stokes shift (nm) | |
| a Reference compound quinine sulfate (0.5 M H2SO4, ϕF = 0.54). | |||||||||
| 3 | 369 | 0.099 | 414 | 1 | 45 | 371 | 457 | 0.31 | 86 |
| 4 | 369 | 0.094 | 415 | 0.98 | 46 | 373 | 455 | 0.26 | 82 |
| 5 | 369 | 0.553 | 415 | 0.34 | 46 | 373 | 457 | 0.36 | 84 |
| 6 | 369 | 0.630 | 416 | 0.45 | 47 | 373 | 456 | 0.27 | 83 |
Photoluminescence emission spectra of 3–6 were also measured in THF. All of them show similar vibronic emission spectra in solution, with maxima around λem = 415 nm (Fig. 3a), that can be assigned to the locally excited estate (LE) emission of the anthracene moiety. The similarity between spectra of 3–4 in solution to the one from anthracene itself (λem = 420 nm) suggests that only small electronic interactions between anthracene units take place, and there is no electronic or steric influence of the iodo or styrene groups, which is in full agreement with the DFT calculations (vide infra). This is proven because 3–4 exhibit fluorescence quantum yield values (ϕF) near 100% in solution, that is comparable to the ϕF of precursors 1–2,17b demonstrating that the fluorescence efficiency is mostly given by the anthracene/carboranyl group, whereas the influence of the styrene is negligible. Moreover, these results are consistent with previous reports,15–17 where m-carborane bound to other fluorophores produced an important increase of the quantum efficiency.
Moreover, when triads 3–4 were grafted to the OVS cube structure, the quantum yields drop to 34–45% for 5–6 in solution. If we compare these values with others reported for similar structures bearing o-carborane, Me-o-carborane and Ph-o-carborane, with ϕF 0.02, 0.09 and 0.44, respectively,18 we may conclude that the synergistic combination of the anthracene luminophore with the m-carborare, which is a less electron-acceptor group that the o-carborane, results in higher luminescent POSS hybrids. Table 3 summarizes the photophysical data for all the compounds.
Finally, we investigated the PL behavior in the aggregate state (THF/H2O = 1/99 (v/v), 1 × 10−5 M for 3–4 and 1 × 10−6 M for 5–6). The PL spectra for these compounds are very similar, showing non-vibronic structures and a maximum emission around 455–459 nm (Table 3 and Fig. 3c and d), red-shifted about 40 nm, with regard to the THF solutions. Consequently, larger Stokes shifts are also observed. Noticeably, similar moderate fluorescence quantum yields were determined for hybrids 5–6 (ϕF = 27–36%) and monomers 3–4 (26–31%), suggesting that neither the free monomers nor the hybrids obtained after their coupling to OVS have π–π interactions in solid-state, as it was discussed above for the crystal packing of 3. Nevertheless, a significant drop of the fluorescence efficiency occurs in aggregates 3–4 (26–31%) with regard to the solution (98–100%), probably due to the restriction of the rotation of the anthracene units.
To get a better insight into the nature of the photophysical properties of the investigated compounds, DFT calculations were performed (more details in ESI†). Several rotamers were considered and between the rotamers, the energy differences are small (Fig. S2 in the ESI†). In the case of 3, the structure obtained from the single-crystal X-ray diffraction was optimized at ω-B97XD/6-31+G* level not showing any significant difference from the structure from X-ray diffraction. As can be expected this structure exhibits the highest stability among the investigated rotamers (ΔE = 4.0–5.1 kcal mol−1 at ω-B97XD/6-31G* level of theory, Fig. S2†). Investigating the Kohn–Sahm frontier orbitals of the different rotamers it can be established that, the energy (Δε < 0.2 eV) and the shape of the orbitals does not depend strongly on the orientation of the substituents in agreement with the small energy differences between the rotamers. In the case of 3 both the HOMO and LUMO are localized at the anthracene unit (Fig. 4 and Fig. S3†), but the orbitals of the styrenyl moiety also appear in the frontier orbital region (HOMO−1, LUMO+1, see Fig. 4 and Fig. S3†). The π-systems of anthracene and styrene units do not interact with each other and the energy of the above-mentioned orbitals are very close to the energy of the corresponding orbitals of the parent anthracenyl-m-carborane (1) and stirenyl-m-carborane (Fig. 4); this further supports the lack of interaction between them. In the case of 4 the shape and energy level of HOMO and LUMO are similar to 3 and the lone pairs of the iodine atoms appear as HOMO−2 and HOMO−3 (Fig. S3†).
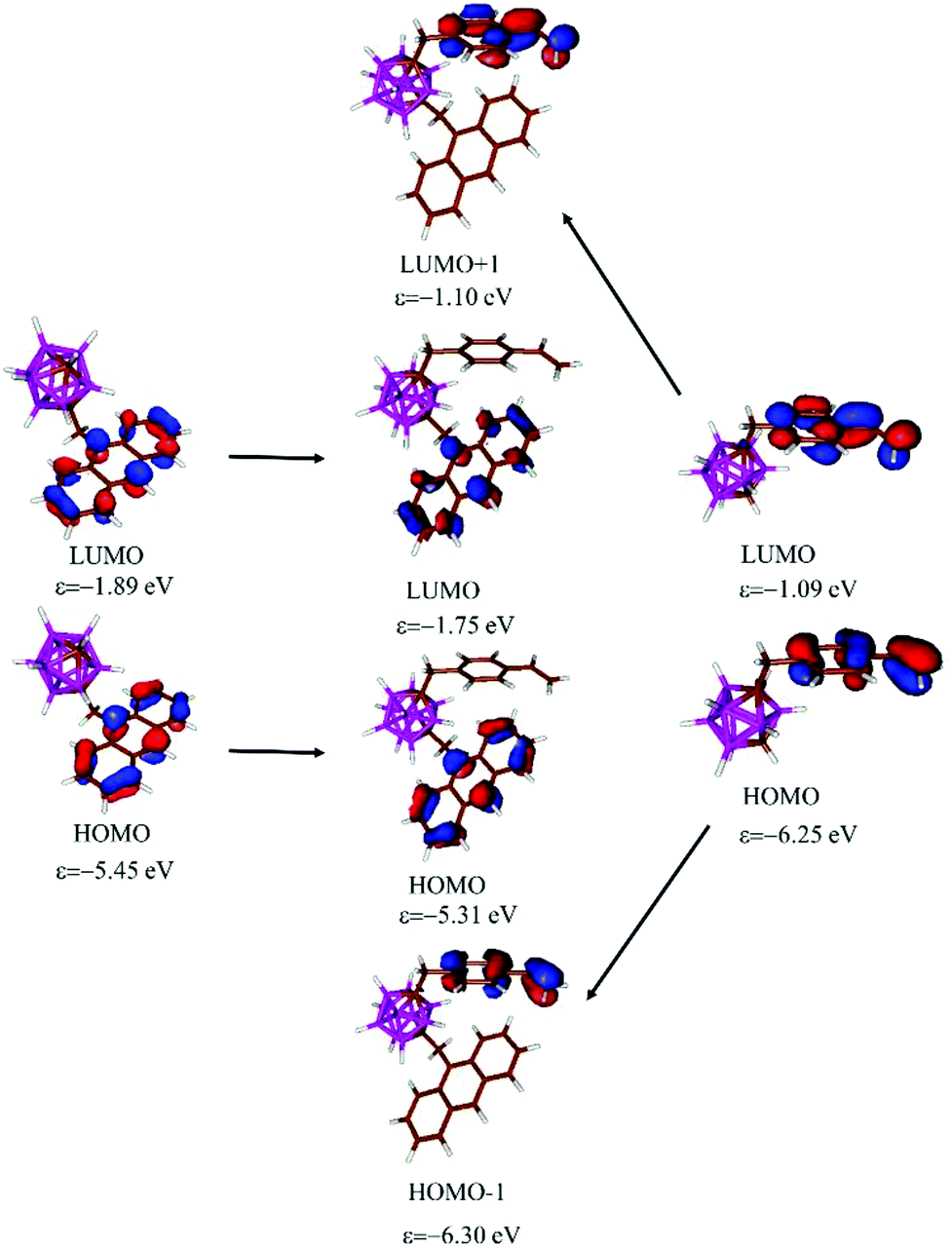 | ||
| Fig. 4 Kohn–Sahm molecular orbitals of anthacenyl-m-carborane 1 (left), 3 (middle) and styrenyl-m-carborane (right) at B3LYP/6-31G*//ω-B97XD/6-31+G* level of theory. | ||
TD-DFT calculations were performed at different level of theory. The geometry was obtained at ω-B97XD/6-31+G* level of theory and this geometry was used for the further TD-DFT calculations. Several methods and basis sets were tested (Table S1 in ESI†) and, in agreement with our previous results,17,23 the B3LYP method with the rather small 6-31G basis set gives the best numerical results, only 5 nm error for both 3 and 4 (calculated: 384 nm vs. experimental: 389 nm, note that simple TD-DFT calculations could not interpret the vibrational fine structure, which is unique for rigid aromatic system such as anthracene). These results confirmed that the transition corresponds to the local π–π* (HOMO–LUMO) transition of the anthracene moiety.
For 4, it should be noted that the lone pairs of the iodo atoms are in the border orbitals’ region (HOMO−2, HOMO−3 in the ESI†). Moreover, TD-DFT calculations show transitions from these orbitals to the LUMO (fifth and sixth excited states, see in Table S3†), and the calculated oscillator strength (which corresponds to the intensity of the peaks in the experimental spectra) of these transitions are low (<0.002), thus they could not be observed experimentally. Therefore, the presence of iodo groups does not affect the photophysical properties of the parent compound.
The geometry optimization of 5 and the TD-DFT calculations were performed as well (more details in Table S4†); they gave questionable geometry and overestimated the absorption peak by 30 nm (it should be highlighted that these systems are quite huge for simple DFT calculations (more than 500 atoms)). To obtain a better description of the effect of the octasilsesquioxane substitutions, one of its arms (trimethoxysilyl substituted derivatives see Fig. S4 in the ESI†) was calculated. As it can be expected the substitution at the styrenyl unit does not affect the shape and the energy of the frontier orbitals (localised at the anthracene unit, Fig. S4†), which is in agreement with the experimentally observed spectra.
Conclusions
A set of two new C-heterosubstituted m-carborane triads bearing one anthracene and one styrene group (3–4) along with octasilsesquioxane-based hybrids (5–6) decorated with eight of those triads have been successfully synthesized, isolated and fully characterized. TEM images of OVS and 5–6 show important differences in the morphology of the particles; whereas OVS forms cubic-like particles, 5–6 have spherical shapes with a broad particle-size distribution. The crystal structure of 3 was analyzed by X-ray diffraction. According to their emission spectra, all compounds show similar vibronic emissions in solution assigned to the locally excited estate (LE) emission of the anthracene moiety. The similarity between these spectra to the anthracene itself suggested that only small electronic interactions between the anthracene units occurred, as it was confirmed by TD-DFT calculations. The calculated oscillator strengths corresponding to the transitions from iodo orbitals to the LUMO were low and could not be observed experimentally. Thus, the presence of iodo units did not affect the photophysical properties of our systems. On the other hand, due to its large dimension for simple DFT calculations, only one arm of 5 and 6 has been calculated as a model system, suggesting that the substitution at the styrenyl unit did not affect the shape and the energy of the frontier orbitals. Extraordinary fluorescence efficiencies, around the unity, were exhibited for triads 3–4 in solution, being lower for hybrids 5–6. Noticeably, all of them exhibited quite high fluorescence quantum yield values in the aggregate state. In agreement with earlier scattered data on m-carborane related to other fluorofores, herein we provide extra results that evidence that m-carborane is a suitable scaffold for coupling to anthracene to produce excellent emitters in solution, while maintaining the emission properties in the aggregate state. The ϕF on anthracene has increased to near 100% upon the linkage to the m-carborane. An explanation that is consistent with the experimental and computational details is that m-carborane does not alter the energy levels participating in the fluorescence, mainly due to anthracene, however it prevents alternative non-radiative transitions. DFT and TDDFT calculations have given support to the non-influence of m-carborane in the orbitals participating in the PL, but have not provided any hint for the high quantum yield. Taking into account the potential of m-carborane based fluorophores, in the future we plan to explore other related compounds and carry out calculations at higher level of theory (e.g. CASPT2) that require much more computational time and do molecular dynamic calculations (MD) that may give more precise information. In addition, we will focus in exploiting and studying the potential applications of m-carborane-containing fluorophores.Experimental
Instrumentation
Elemental analyses were performed using a Carlo Erba EA1108 microanalyzer. ATR-IR spectra were recorded on JASCO FT/IR-4700 spectrometer on a high-resolution. The 1H NMR (300.13 MHz), 11B{1H} (96.29 MHz) and 13C{1H} NMR (75.47 MHz) spectra were recorded on a Bruker ARX 300 spectrometer. All NMR spectra were recorded in CDCl3 solutions at 25 °C. Chemical shift values for 11B {1H} NMR spectra were referenced to external BF3·OEt2, and those for 1H and 13C {1H} NMR were referenced to SiMe4 (TMS). Chemical shifts are reported in units of parts per million downfield from the reference, and all coupling constants are reported in Hertz. UV-Vis spectra were recorded on VARIANT Cary 5 UV-Vis-NIR spectrophotometer, using spectroscopic grade THF (Sigma-Aldrich), in normal quartz cuvette having 1 cm path length, for different solutions for each compound in the range 5 × 10−5 to 1 × 10−5 M to calculate the molar extinction coefficients (ε). The fluorescence emission spectra and excitation spectra for all samples were recorded in a VARIANT Cary Eclipse Fluorescence spectrometer. No fluorescent contaminants were detected on excitation in the wavelength region of experimental interest. The fluorescence quantum yields were determined by the “single point method” and repeated three times with similar optical density for reproducibility,24 against quinine sulfate in 0.5 M aqueous sulfuric acid with ϕF = 0.54 as a standard.25 For the suspensions in THF/water (1/99, v/v) the refractive index was assumed to be that of pure water (1.33).Materials
All reactions were performed under an atmosphere of dinitrogen employing standard Schlenk techniques. Tetrahydrofuran and dichloromethane were purchased from Merck and distilled from sodium benzophenone before to use. Commercial grade diethyl ether, hexane, petroleum ether, n-heptane, and chloroform were used without further purification. Compounds 1–2 were synthesized according to the literature.17bn-BuLi solution (1.6 M in hexane), Grubbs 1st generation catalyst [RuCl2(CHPh)-(PCy3)2], Octavinylsilsesquioxane (OVS) were purchased from Aldrich. 4-Vinylbenzyl chloride was purchased from ACROS Organics.X-ray single-crystal structure determination
The crystal structure of compound 3 was determined by single-crystal X-ray crystallography. A suitable crystal of this material was mounted on a MITEGEN Micro mount™ and used for the data collection on a Bruker D8 Venture diffractometer using MoKα radiation (λ = 0.71073 Å). Absorption correction was applied using SADABS.26 The structure was solved by direct methods which revealed the position of all non-hydrogen atoms and refined with full-matrix least-squares calculations on F2,27 using Olex2 as the graphical interface.28 Anisotropic displacement parameters were assigned to all atoms except for hydrogen atoms, which are riding their parent atoms with an isotropic temperature factor chosen as 1.2 times those of their parent atoms. Details of the structure determination and refinement of the studied compounds are summarized in Table 1. Crystallographic data for the structure reported in this paper have been deposited in the Cambridge Crystallographic Data Centre, CCDC 1972767.†CH2), 5.83 (d, 3J(H,H) = 18 Hz, 1H, CHCH2), 5.35 (d, 3J(H,H) = 12 Hz, 1H, CHCH2), 4.22 (s, 2H, CH2–C14H9), 3.06 (s, 2H, CH2–C6H4); 1H{11B} NMR, δ (ppm): 8.44 (s, 1H, C14H9), 8.12 (d, 3J(H,H) = 9 Hz, 2H, C14H9), 8.03 (d, 3J(H,H) = 9 Hz, 2H, C14H9), 7.58–7.54 (m, 2H, C14H9), 7.53–7.49 (m, 2H, C14H9), 7.33 (d, 3J(H,H) = 6 Hz, 2H, C6H4), 6.94 (d, 3J(H,H) = 9 Hz, 2H; C6H4), 6.77 (dd, 3J(H,H) = 18 Hz, 3J(H,H) = 12 Hz, 1H, CHCH2), 5.83 (d, 3J(H,H) = 18 Hz, 1H, CHCH2), 5.35 (d, 3J(H,H) = 12 Hz, 1H, CHCH2), 4.22 (s, 2H, CH2–C14H9), 3.06 (s, 2H, CH2–C6H4), 2.61 (s, 1H, B–H), 2.51 (s, 2H, B–H), 2.28 (s, 3H, B–H), 2.18 (s, 2H, B–H), 2.14 (s, 2H, B–H); 11B{1H} NMR, δ (ppm): −6.61 (s, 2B), −11.09 (s, 6B), −13.61 (s, 2B); 13C{1H} NMR, δ (ppm): 136.80 (s, C6H4), 136.51 (s, CHCH2), 131.40 (s, C14H9), 130.82 (s, C14H9), 130.03 (s, C6H4), 129.49 (s, C14H9), 129.25 (s, C14H9), 128.19 (s, C14H9), 126.23 (s, C14H9 and C6H14), 124.99 (s, C14H9), 124.78 (s, C14H9), 114.06 (s, CHCH2), 76.30 (s, Cc–CH2), 75.84 (s, Cc–CH2), 42.70 (s, CH2–C6H4), 33.77 (s, CH2–C14H9); ATR-IR (cm−1): ν = 3052, 2927, 2853 (Car–H), 2596, 2587, 2572, 2557 (B–H), 1624 (CC); elemental analysis calcd (%) for C26H30B10: C, 69.30; H, 6.71. Found: C, 68.80; H, 6.74.
CH2), 5.80 (d, 3J(H,H) = 18 Hz, 1H, CHCH2), 5.33 (d, 3J(H,H) = 12 Hz, 1H, CHCH2), 4.24 (s, 2H, CH2–C14H9), 3.05 (s, 2H, CH2–C6H4); 11B{1H} NMR, δ (ppm): −5.60 (s, 2B), −10.04 (s, 5B), −13.56 (s, 1B), −15.48 (s, 1B), −23.96 (s, 1B); 13C{1H} NMR, δ (ppm): 137.01 (s, C6H4), 136.33 (s, CHCH2), 135.79 (s, C6H4), 131.34 (s, C14H9), 130.67 (s, C14H9), 129.88 (s, C6H4), 129.30 (s, C14H9), 128.73 (s, C14H9), 128.42 (s, C14H9), 126.41 (s, C14H9), 126.32 (s, C6H4), 125.02 (s, C14H9), 124.37 (s, C14H9), 114.24 (s, CHCH2), 42.49 (s, CH2–C6H4), 33.60 (s, CH2–C14H9); ATR-IR (cm−1): ν = 3083, 3035, 2927, 2852 (Car–H), 2593 (B–H), 1623 (CC); elemental analysis calcd (%) for C26H29B10I: C, 54.17; H, 5.07. Found: C, 54.10; H, 5.07.
:10) to obtain a grey solid. Further purification was performed by preparative layer chromatography (CH2Cl2/hexane, 7:3) to obtain compound 5 as yellowish solid. Yield: 49 mg, 52%. 1H NMR, δ (ppm): 8.28 (s, 8H, C14H9), 7.97 (d, 3J(H,H) = 10 Hz, 16H, C14H9), 7.89 (d, 3J(H,H) = 8 Hz, 16H, C14H9), 7.54 (d, 3J(H,H) = 18 Hz, 8H, CHCH–Si), 7.46–7.40 (m, 48H, C6H4 and C14H9), 6.91 (d, 3J(H,H) = 8 Hz, 16H, C6H4), 6.51 (d, 3J(H,H) = 20 Hz, 8H, CHCH–Si), 4.04 (s, 16H, CH2–C14H9), 3.00 (s, 16H, CH2–C6H4); 11B{1H} NMR, δ (ppm): −6.52 (br, 16B), −10.81 (br, 48B), −13.33 (br, 16B); 13C{1H} NMR, δ (ppm): 148.68 (s, CHCH–Si), 137.54 (s, C6H4), 136.10 (s, C6H4), 130.94 (s, C14H9), 130.36 (s, C14H9), 129.86 (s, C6H4), 129.03 (s, C6H4), 128.88 (s, C14H9), 127.79 (s, C6H4), 126.70 (s, C14H9), 125.85 (s, C14H9), 124.61 (s, C14H9), 124.33 (s, C14H9), 117.23 (s, CHCH–Si), 75.94 (s, Cc–CH2), 75.17 (s, Cc–CH2), 42.32 (s, CH2–C6H4), 33.29 (s, CH2–C14H9); 29Si NMR (CDCl3): δ = −78.09; ATR-IR (cm−1): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3034, 2957, 2929, 2867 (Car–H), 2589 (B–H), 1605 (CC), 1087 (Si–O); elemental analysis calcd (%) for C208H232B80O12Si8 + 2 C6H14: C, 63.12; H, 6.26. Found: C, 63.18; H, 6.07.
CH–Si), 7.46–7.38 (m, 48H, C6H4 and C14H9), 6.87 (d, 3J(H,H) = 8 Hz, 16H, C6H4), 6.50 (d, 3J(H,H) = 20 Hz, 8H, CHCH–Si), 4.04 (s, 16H, CH2–C14H9), 3.00 (s, 16H, CH2–C6H4); 11B{1H} NMR, δ (ppm): −5.35 (br, 16B), −9.36 (br, 40B), −14.97 (br, 16B), −23.64 (br, 8B); 13C{1H} NMR, δ (ppm): 148.57 (s, CHCH–Si), 136.96 (s, C6H4), 136.28 (s, C6H4), 130.93 (s, C14H9), 130.27 (s, C14H9), 129.77 (s, C6H4), 129.00 (s, C6H4), 128.32 (s, C14H9), 128.09 (s, C6H4), 126.81 (s, C14H9), 126.12 (s, C14H9), 124.72 (s, C14H9), 123.98 (s, C14H9), 117.43 (s, CHCH–Si), 77.17 (s, Cc–CH2), 76.52 (s, Cc–CH2), 42.10 (s, CH2–C6H4), 33.14 (s, CH2–C14H9); 29Si NMR (CDCl3): δ = −78.15; ATR-IR (cm−1): = 2955, 2922, 2851 (Car–H), 2593 (B–H), 1604 (CC), 1085 (Si–O); elemental analysis calcd (%) for C208H224B80I8O12Si8 + 2 C6H14 + CH2Cl2: C, 50.29; H, 4.85. Found: C, 50.30; H, 5.09.
= 3034, 2957, 2929, 2867 (Car–H), 2589 (B–H), 1605 (CC), 1087 (Si–O); elemental analysis calcd (%) for C208H232B80O12Si8 + 2 C6H14: C, 63.12; H, 6.26. Found: C, 63.18; H, 6.07.
CH–Si), 7.46–7.38 (m, 48H, C6H4 and C14H9), 6.87 (d, 3J(H,H) = 8 Hz, 16H, C6H4), 6.50 (d, 3J(H,H) = 20 Hz, 8H, CHCH–Si), 4.04 (s, 16H, CH2–C14H9), 3.00 (s, 16H, CH2–C6H4); 11B{1H} NMR, δ (ppm): −5.35 (br, 16B), −9.36 (br, 40B), −14.97 (br, 16B), −23.64 (br, 8B); 13C{1H} NMR, δ (ppm): 148.57 (s, CHCH–Si), 136.96 (s, C6H4), 136.28 (s, C6H4), 130.93 (s, C14H9), 130.27 (s, C14H9), 129.77 (s, C6H4), 129.00 (s, C6H4), 128.32 (s, C14H9), 128.09 (s, C6H4), 126.81 (s, C14H9), 126.12 (s, C14H9), 124.72 (s, C14H9), 123.98 (s, C14H9), 117.43 (s, CHCH–Si), 77.17 (s, Cc–CH2), 76.52 (s, Cc–CH2), 42.10 (s, CH2–C6H4), 33.14 (s, CH2–C14H9); 29Si NMR (CDCl3): δ = −78.15; ATR-IR (cm−1): = 2955, 2922, 2851 (Car–H), 2593 (B–H), 1604 (CC), 1085 (Si–O); elemental analysis calcd (%) for C208H224B80I8O12Si8 + 2 C6H14 + CH2Cl2: C, 50.29; H, 4.85. Found: C, 50.30; H, 5.09.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The work was supported by Spanish Ministerio de Economía y Competitividad, MINEICO (CTQ2016-75150-R and “Severo Ochoa” Program for Centers of Excellence in R&D SEV-2015-0496) and Generalitat de Catalunya (2017/SGR/1720). Z. K. is grateful for the general support of the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement MSCA-IF-2016-751587. D. Ch.-L. acknowledges funding by project No. PGC2018-102047-B-I00 (MCIU/AEI/FEDER, UE). Theoretical calculations have been achieved using computers from the Supercomputing Centre of Catalonia (CESCA).References
-
(a) H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda and C. Adachi, High-efficiency organic light-emitting diodes with fluorescent emitters, Nat. Commun., 2014, 5, 4016 CrossRef CAS PubMed
; (b) H. Xu, R. F. Chen, Q. Sun, W. Y. Lai, Q. Q. Su, W. Huang and X. G. Liu, Recent progress in metal–organic complexes for optoelectronic applications, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC
-
(a) Q. Ye, H. Zhou and J. Xu, Cubic Polyhedral Oligomeric Silsesquioxane Based Functional Materials: Synthesis, Assembly, and Applications, Chem. – Asian J., 2016, 11, 1322–1337 CrossRef CAS PubMed
-
C. Hartmann-Thompson, Applications of Polyhedral Oligomeric Silsesquioxanes, Springer, Netherlands, Midland, 2011, vol. 3 Search PubMed
-
(a) T. Maegawa, O. Miyashita, Y. Irie, H. Imoto and K. Naka, Synthesis and properties of polyimides containinghexaisobutyl-substituted T8cages in their mainchains, RSC Adv., 2016, 6, 31751–31757 RSC
-
(a) Y. R. Liu, Y. D. Huang and L. Liu, Effects of TriSilanolIsobutyl-POSS on thermal stability of methylsilicone resin, Polym. Degrad. Stab., 2006, 91, 2731–2738 CrossRef CAS
- Y. Kim, K. Koh, M. F. Roll, R. M. Laine and A. J. Matzger, Porous Networks Assembled from Octaphenylsilsesquioxane Building Blocks, Macromolecules, 2010, 43, 6995–7000 CrossRef CAS
- H. Ghanbari, B. G. Cousins and A. M. Seifalian, A nanocage for nanomedicine: polyhedral oligomeric silsesquioxane (POSS), Macromol. Rapid. Commun., 2011, 32, 1032–1046 CrossRef CAS PubMed
-
(a) M. Bahrami, J. C. Furgal, H. Hashemi, M. Ehsani, Y. Jahani, T. Goodson, J. Kieffer and R. M. Laine, Synthesis and Characterization of Nanobuilding Blocks [o-RStyrPhSiO1.5]10,12 (R = Me, MeO, NBoc, and CN). Unexpected Photophysical Properties Arising from Apparent Asymmetric Cage Functionalization as Supported by Modeling Studies, J. Phys. Chem. C, 2015, 119(28), 15846–15858 CrossRef CAS
-
(a) V. I. Bregadze, Dicarba-closo-dodecaboranes C2B10H12 and their derivatives, Chem. Rev., 1992, 92, 209–223 CrossRef CAS
-
(a) J. Poater, M. Solà, C. Viñas and F. Teixidor, π-Aromaticity and Three-Dimensional Aromaticity: Two sides of the Same Coin?, Angew. Chem., Int. Ed., 2014, 53, 12191–12195 CrossRef CAS PubMed
-
(a) R. Núñez, P. Farràs, F. Teixidor, C. Viñas, R. Sillanpää and R. Kivekäs, A Discrete P⋯I-I⋯P Assembly: The Large Influence of Weak Interactions on the 31P NMR Spectra of Phosphane–Diiodine Complexes, Angew. Chem., Int. Ed., 2006, 45, 1270–1272 CrossRef PubMed
-
(a) A. Ferrer-Ugalde, E. J. Juárez-Pérez, F. Teixidor, C. Viñas, R. Sillanpää, E. Pérez-Inestrosa and R. Núñez, Synthesis and Characterization of New Fluorescent Styrene-Containing Carborane Derivatives: The Singular Quenching Role of a Phenyl Substituent, Chem. – Eur. J., 2012, 18, 544–553 CrossRef CAS PubMed
-
(a) A. González-Campo, A. Ferrer-Ugalde, C. Viñas, F. Teixidor, R. Sillanpää, J. Rodríguez-Romero, R. Santillan, N. Farfán and R. Núñez, A versatile methodology for the controlled synthesis of photoluminescent high-boron-content dendrimers, Chem. – Eur. J., 2013, 19, 6299–6312 CrossRef PubMed
- J. Cabrera-González, C. Viñas, M. Haukka, S. Bhattacharyya, J. Gierschner and R. Núñez, Photoluminescence in carborane–stilbene triads: A structural, spectroscopic, and computational study, Chem. – Eur. J., 2016, 22, 13588–13598 CrossRef PubMed
- B. P. Dash, R. Satapathy, E. R. Gaillard, K. M. Norton, J. A. Maguire, N. Chug and N. S. Hosmane, Enhanced π-Conjugation and Emission via Icosahedral Carboranes: Synthetic and Spectroscopic Investigation, Inorg. Chem., 2011, 50, 5485–5493 CrossRef CAS PubMed
- D. Tu, P. Leong, Z. Li, R. Hu, C. Shi, K. Y. Zhang, H. Yan and Q. Zhao, Carborane-triggered metastable charge transfer state leading to spontaneous recovery of mechanochromic luminescence, Chem. Commun., 2016, 52, 12494 RSC
-
(a) M. Chaari, Z. Kelemen, J. Giner-Planas, F. Teixidor, D. Choquesillo-Lazarte, A. Ben Salah, C. Viñas and R. Núñez, Photoluminescence in m-carborane–anthracene triads: a combined experimental and computational study, J. Mater.
Chem. C, 2018, 6, 11336–11347 RSC
- A. Ferrer-Ugalde, E. J. Juárez-Pérez, F. Teixidor, C. Viñas and R. Núñez, Synthesis, characterization, and thermal behavior of carboranyl–styrene decorated octasilsesquioxanes: Influence of the carborane clusters on photoluminescence, Chem. – Eur. J., 2013, 19, 17021–17030 CrossRef CAS PubMed
- J. Cabrera-González, A. Ferrer-Ugalde, S. Bhattacharyya, M. Chaari, F. Teixidor, J. Gierschner and R. Núñez, Fluorescent carborane–vinylstilbene functionalised octasilsesquioxanes: synthesis, structural, thermal and photophysical properties, J. Mater. Chem. C, 2017, 5, 10211–10219 RSC
-
(a)
M. A. Fox, Icosahedral carborane derivatives, Durham thesis, Durham University, 1991 Search PubMed
-
R. H. Grubbs, in Handbook of Metathesis, Wiley-VCH, Weinheim, 2003 Search PubMed
- A. V. Puga, F. Teixidor, R. Sillanpää, R. Kivekääs, M. Arca, G. Barberà and C. Viñas, From Mono- to Poly-Substituted Frameworks: A Way of Tuning the Acidic Character of Cc-H in o-Carborane Derivatives, Chem. – Eur. J., 2009, 15, 9755–9763 CrossRef CAS PubMed
- M. Chaari, J. Cabrera-Gonzáñez, Z. Kelemen, C. Viñas, A. Ferrere-Ugalde, D. Choquesillo-Lazarte, A. Ben Salah, F. Teixidor and R. Núñez, Luminescence properties of carborane-containing distyrylaromatic systems, J. Organomet. Chem., 2018, 865, 206–213 CrossRef CAS
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer US, Baltimore, Maryland, USA, 2007 Search PubMed
- A. M. Brouwer, Standards for photoluminescence quantum yield measurements in solution (IUPAC Technical Report), Pure Appl. Chem., 2011, 83, 2213–2228 CAS
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 11B{1H}, 13C{1H} NMR and IR-ATR spectra, computational details, crystallographic data and Hirshfeld surface analyses for all the compounds. CCDC 1972767. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qi00127a |
| This journal is © the Partner Organisations 2020 |