
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting cation aggregation in new magnesium amidohaloaluminate electrolytes for magnesium batteries†
Etienne V.
Brouillet
a,
Marco
Amores
 b,
Serena A.
Corr
*cd and
Stuart D.
Robertson
*a
b,
Serena A.
Corr
*cd and
Stuart D.
Robertson
*a
aWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow G1 1XL, UK. E-mail: stuart.d.robertson@strath.ac.uk
bDepartment of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1, Hongo, Bunkyo-Ku, Tokyo 113-0033, Japan
cDepartment of Chemical and Biological Engineering, University of Sheffield, Sheffield S1 3JD, UK. E-mail: s.corr@sheffield.ac.uk
dDepartment of Materials Science and Engineering, University of Sheffield, Sheffield S1 3JD, UK
First published on 1st May 2020
Abstract
Mg batteries present an attractive and sustainable alternative to Li-ion batteries, wherein magnesium metal as an anode displays a superior theoretical volumetric energy density of 3833 A h L−1versus 2062 A h L−1 for lithium. An outstanding crucial bottleneck in realising their more widespread uptake is the development of suitable electrolytes, where electrode passivation, a limited electrochemical window, conditioning requirements, low ion mobility and low coulombic efficiencies all contribute to current limitations in Mg batteries. In an area thus far dominated by the thermodynamically stable [Mg2Cl3]+ dinuclear cation, we present here a novel family of magnesium amidohaloaluminate electrolytes [(Dipp)(SiMe3)2NAlCl3]− [MgxCl2x−1]+ where the magnesium chloride cation aggregation has been tailored (x = 1, 2, 3) by substitution of the coordinating ligand to the Mg2+ centre, and show how directly altering this cation affects battery performance (Dipp = 2,6-diisopropylphenyl, Me = methyl). The electrochemical activity of these new electrolytes has been evaluated by cyclic voltammetry, galvanostatic cycling and impedance spectroscopy in Mg–metal symmetrical cells as well as in battery cells with the Mo6S8 Chevrel phase cathode material against magnesium metal. The mononuclear and dinuclear magnesium amidohaloaluminate electrolytes facilitate reversible Mg plating and stripping from the Mg–metal anode with excellent stability, withstanding over 70 hours of continuous cycling. We demonstrate the compatibility of these novel electrolytes with the Mo6S8 Chevrel intercalation cathode material, allowing cycling of a Mg–metal cell up to 100 cycles with coulombic efficiencies above 95%.
A. Introduction
The current rechargeable battery market is heavily reliant on lithium (Li)-ion chemistry, with Li-ion batteries ubiquitous in our portable devices and increasingly prevalent in powering (hybrid) electric vehicles.1,2 However, with increasing demands on our lithium supply driving up costs, it is vital alternatives to Li-ion chemistries are sought.3 Increasingly, efforts have been made in developing multivalent Group II battery chemistries that can achieve high volumetric capacities using lower cost, highly-abundant metals.4 Of these, magnesium is a particularly attractive candidate owing to a combination of its relatively light weight and massively higher natural abundance (compared to Li).5,6 The redox process of divalent magnesium ions (Mg2+) proceeds via a two-electron transfer, bestowing on magnesium a theoretical volumetric capacity almost twice that of lithium (3833 A h L−1cf. 2062 A h L−1). The negative reduction potential of magnesium at −2.37 V (vs. SHE) makes it a viable option for exploiting useful voltages. Despite these considerable advantages, impediments remain in the advancement of Mg-batteries and chief amongst these is the development of Mg-based electrolytes that enable reversible plating and stripping of Mg2+ on the Mg–metal anode. For example, the use of Mg inorganic salts, analogous to lithium electrolytes, in aprotic solvents leads to the formation of impermeable passivating layers at the electrode interface.7,8 Interestingly, the key to unlocking this early Mg electrolyte problem was the use of Grignard reagents, which enables passivating layer-free reversible electrodeposition in ethereal solvents.9 The strong reducing nature of Grignard reagents, which affects the electrochemical window dramatically, was simply overcome by addition of a group 13 Lewis acid, generating ionic bimetallic complexes such as magnesium aluminates.5 The active species of these compounds typically adopts the form of the solvent-separated ion pair [Mg2Cl3·6THF]+ [RxAlCl4−x]− in THF.10,11 Since the first prototype of a rechargeable magnesium battery and the characterisation of the purported active species of these magnesium aluminate electrolytes, a variety of analogous electrolytes have been proposed. The anion [RxAlCl4−x]− may be easily modified using different R groups, and by introducing halide, alkyl, aryl, amido or alkoxide ligands to the aluminium metal centre.10–16 These studies revealed evidence that the nature of the Al–R bond is key to a number of properties, such as the solubility, nucleophilicity and oxidative stability.17 In almost all of these case studies, the cation adopts the thermodynamically-favoured dinuclear magnesium cation [Mg2Cl3·6THF]+. We recently published a synthetic method to access magnesium aluminate compounds with a common aluminate anion and the ability to control the aggregation level of the magnesium cation through judicious choice of Lewis donor in both solid state and solution, observed by means of X-ray crystallography and electrospray-ionization mass spectrometry (ESI-MS) (Fig. 1).15 These cationic species include mononuclear [MgCl]+ and trinuclear [Mg3(μ3-Cl)2(μ2-Cl)3]+ aggregates, whose existence in the electrolyte solution were previously postulated as playing an important role in Mg deposition when using a formally [Mg2Cl3]+ based electrolyte.18–20 With these three cation aggregation states to hand (accompanied by a common aluminate counteranion), the potential to investigate the effect of cation aggregation on electrolyte performance is now available. We now report the battery performances using these compounds as electrolytes in Mg–metal/Mo6S8 battery cells and interpret the different experimental results in order to identify which aggregation is more beneficial to Mg2+ ion transfer, plating/stripping and intercalation. We have selected the Mo6S8 Chevrel phase as a robust cathode material which has been widely studied in the literature, but it is interesting to note recent developments in high-performance cathodes such as MgTi2S4 which display improved properties.21,22 We show that all three electrolytes possess individual properties which affect the plating/stripping of Mg, electrochemical stability window, ionic conductivity, gravimetric specific capacity and overpotential of battery cells. The presence of different Lewis donors, particularly tetraamine Me6TREN (tris[2-(dimethylamino)ethyl]amine N(CH2CH2NMe2)3), is suspected to affect the electrochemical window by enhancing the oxidative stability of the electrolyte, with electrolyte conditioning required before achieving optimal performance.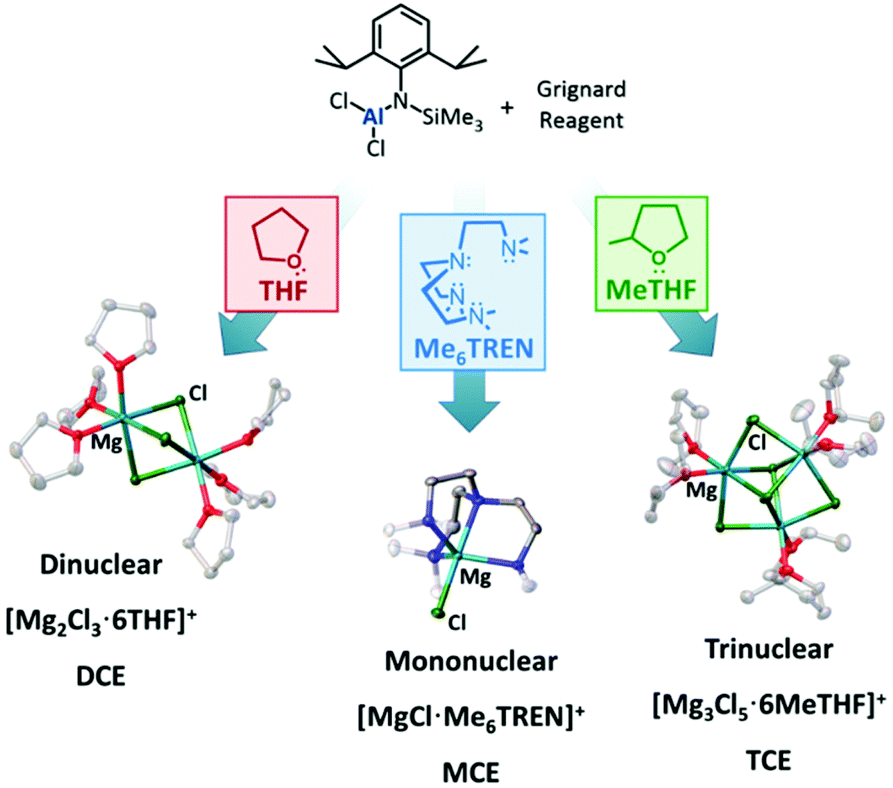 | ||
| Fig. 1 Schematic representation of the synthetic approach for the preparation of magnesium amidohaloaluminate complexes and the different aggregations of the [MgxCl2x−1]+ cations depending on the coordinating ligand. The [(Dipp)(SiMe3)NAlCl3]− counteranion is not shown for clarity. | ||
B. Results
Electrolyte complexes possessing the mononuclear (MCE), dinuclear (DCE) and trinuclear (TCE) magnesium chloride cation aggregations in conjunction with the common aluminate anion [(Dipp)(SiMe3)NAlCl3]−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 were synthesised according to the methodology previously developed in our group.15 The main prerequisite for a candidate electrolyte in a secondary metal-anode battery is the ability to reversibly plate and strip metal ions at the metal anode surface. To evaluate this plating and striping behaviour of the three magnesium amidohaloaluminate complexes presented here, polarisation tests of symmetrical cells with Mg–metal electrodes were performed (Fig. 2). During the first current pulse, the three electrolytes display a voltage peak which starts to decrease rapidly over the first 30 minutes. These observations may be attributed to electrolyte conditioning, where the electrolyte is believed to undergo structural adjustments during the cycling process, generating new species facilitating the reversible deposition of Mg. Further work is ongoing to elucidate these species. The conditioning of magnesium electrolytes has been reported in the literature, for example on the fully inorganic magnesium aluminium chloride-complex (MACC).19,24–27 In the subsequent pulses, the voltage of the cell using either MCE or DCE electrolytes stabilises at ca. 0.37 and 0.12 V respectively, while the TCE experiences a voltage at ca. 0.37 V which rises over cycling. The increase in cell voltage when TCE is used is indicative of a higher resistance toward plating and stripping of Mg which increases with the amount of Mg transferred from one electrode to the other. With MCE and DCE, the average cell polarisation voltage is constant during the experiment, indicating the excellent stability of the electrolyte upon Mg transference. The TCE electrolyte, however, suffers sudden voltage variations after 65 hours, followed by short-circuiting of the cell. Such soft short-circuiting, owing to dendrite growth, has also previously been observed for Mg(TFSI)2/glyme electrolytes via CV and EIS experiments.28 After disassembling the short-circuited battery cell, we observed unusual deposition occurring through the separator. We therefore link this short-circuit phenomenon to a higher predisposition of TCE to form localised deposits of Mg being entropically favoured by the higher aggregation number, as can be appreciated by metallic grey deposits on the separator after the polarisation test (Fig. S1†). Similar deposits were not observed in the case of the MCE or DCE electrolytes. We note here that Mg electrolytes are generally accepted as prohibiting dendrite formation although Banerjee et al. have recently reported dendrite formation when using Grignard reagents in symmetric Mg–Mg cells.29 All three cell measurements here have been performed using identical setup conditions in order to reliably and directly determine and compare the resistance properties of each electrolyte from the polarisation voltage. The results (in Fig. 2) clearly show the effect of the cation aggregation state on the impedance of the cell, where the cell resistance after cycling is significantly lower when the mononuclear and dinuclear cations are involved compared to the presence of the more highly aggregation-prone trinuclear cation species. The concentration of the magnesium aluminate salt used in the electrolyte solution is determined according to the concentration of Mg2+ ions, where a desired concentration of 0.35 M Mg2+ means a decreasing concentration of the salt with increasing cation aggregation (MCE 0.35 M; DCE 0.175 M; TCE 0.117 M). Considering these circumstances for the current study, we are aware that the higher dilution of the DCE and TCE salt could affect the transport and aggregation properties of the cationic species in solution. However, from a previous study using ESI-MS, the change of concentration only has a minor impact on the ratio of the higher aggregation states of the cation.15
23 were synthesised according to the methodology previously developed in our group.15 The main prerequisite for a candidate electrolyte in a secondary metal-anode battery is the ability to reversibly plate and strip metal ions at the metal anode surface. To evaluate this plating and striping behaviour of the three magnesium amidohaloaluminate complexes presented here, polarisation tests of symmetrical cells with Mg–metal electrodes were performed (Fig. 2). During the first current pulse, the three electrolytes display a voltage peak which starts to decrease rapidly over the first 30 minutes. These observations may be attributed to electrolyte conditioning, where the electrolyte is believed to undergo structural adjustments during the cycling process, generating new species facilitating the reversible deposition of Mg. Further work is ongoing to elucidate these species. The conditioning of magnesium electrolytes has been reported in the literature, for example on the fully inorganic magnesium aluminium chloride-complex (MACC).19,24–27 In the subsequent pulses, the voltage of the cell using either MCE or DCE electrolytes stabilises at ca. 0.37 and 0.12 V respectively, while the TCE experiences a voltage at ca. 0.37 V which rises over cycling. The increase in cell voltage when TCE is used is indicative of a higher resistance toward plating and stripping of Mg which increases with the amount of Mg transferred from one electrode to the other. With MCE and DCE, the average cell polarisation voltage is constant during the experiment, indicating the excellent stability of the electrolyte upon Mg transference. The TCE electrolyte, however, suffers sudden voltage variations after 65 hours, followed by short-circuiting of the cell. Such soft short-circuiting, owing to dendrite growth, has also previously been observed for Mg(TFSI)2/glyme electrolytes via CV and EIS experiments.28 After disassembling the short-circuited battery cell, we observed unusual deposition occurring through the separator. We therefore link this short-circuit phenomenon to a higher predisposition of TCE to form localised deposits of Mg being entropically favoured by the higher aggregation number, as can be appreciated by metallic grey deposits on the separator after the polarisation test (Fig. S1†). Similar deposits were not observed in the case of the MCE or DCE electrolytes. We note here that Mg electrolytes are generally accepted as prohibiting dendrite formation although Banerjee et al. have recently reported dendrite formation when using Grignard reagents in symmetric Mg–Mg cells.29 All three cell measurements here have been performed using identical setup conditions in order to reliably and directly determine and compare the resistance properties of each electrolyte from the polarisation voltage. The results (in Fig. 2) clearly show the effect of the cation aggregation state on the impedance of the cell, where the cell resistance after cycling is significantly lower when the mononuclear and dinuclear cations are involved compared to the presence of the more highly aggregation-prone trinuclear cation species. The concentration of the magnesium aluminate salt used in the electrolyte solution is determined according to the concentration of Mg2+ ions, where a desired concentration of 0.35 M Mg2+ means a decreasing concentration of the salt with increasing cation aggregation (MCE 0.35 M; DCE 0.175 M; TCE 0.117 M). Considering these circumstances for the current study, we are aware that the higher dilution of the DCE and TCE salt could affect the transport and aggregation properties of the cationic species in solution. However, from a previous study using ESI-MS, the change of concentration only has a minor impact on the ratio of the higher aggregation states of the cation.15
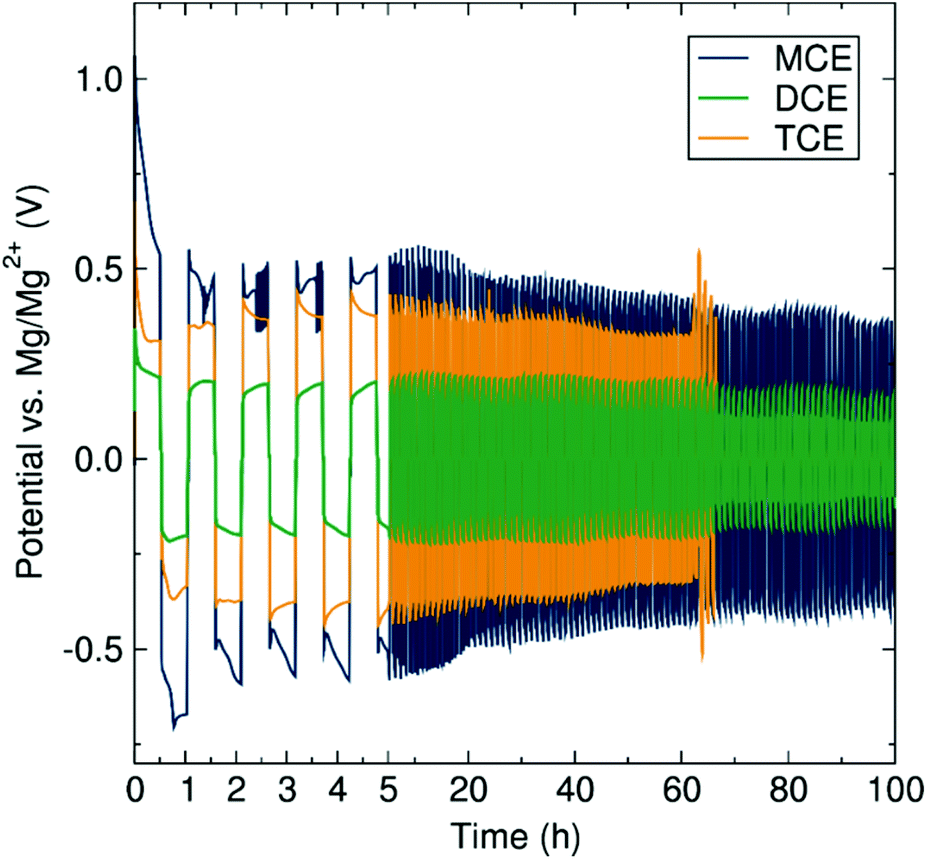 | ||
| Fig. 2 Polarisation measurements of symmetrical cells with Mg–metal electrodes and the three magnesium aluminate electrolytes (MCE, dark blue; DCE green; TCE orange) with different Mg2+ aggregation states at current densities of 200 μA cm−2. | ||
Electrochemical Impedance Spectroscopy (EIS) analyses were carried out before and after cycling of the symmetrical cells to fully analyse and differentiate the transport properties of the electrolytes. From the Nyquist plots displayed in Fig. 3, the charge transfer process occurring at the Mg–metal/electrolyte interface is represented by a semicircle. The intercept of the semicircle at high frequencies corresponds to the pure Ohmic resistance of the separators soaked in the electrolyte solutions. At lower frequencies, the starting point of the semicircle represents the ionic transport properties of the electrolyte. While the ionic transport properties of the electrolyte remain on the same order of magnitude for the three electrolytes before and after the polarisation test (40–80 Ohms), the charge transfer resistance experiences a clear decrease after cell cycling, indicating that the electrolyte conditioning is directly related to charge transfer at the Mg–metal surface. This large decrease can be correlated to a variety of possible electrolyte rearrangements at the Mg–metal/electrolyte interface in order to facilitate the plating/stripping mechanism. Such rearrangements would include previously reported processes responsible for the promotion of Mg deposition, mainly Cl− adsorption onto the Mg anode but also the different aggregation states of the magnesium chloride cations found in solution where a mononuclear cation is favourable.19,25–27,30,31 In agreement with polarisation tests, DCE displays the lowest impedance both before and after the polarisation measurements (note the impedance of TCE in Fig. 3b is that of the short-circuited cell).
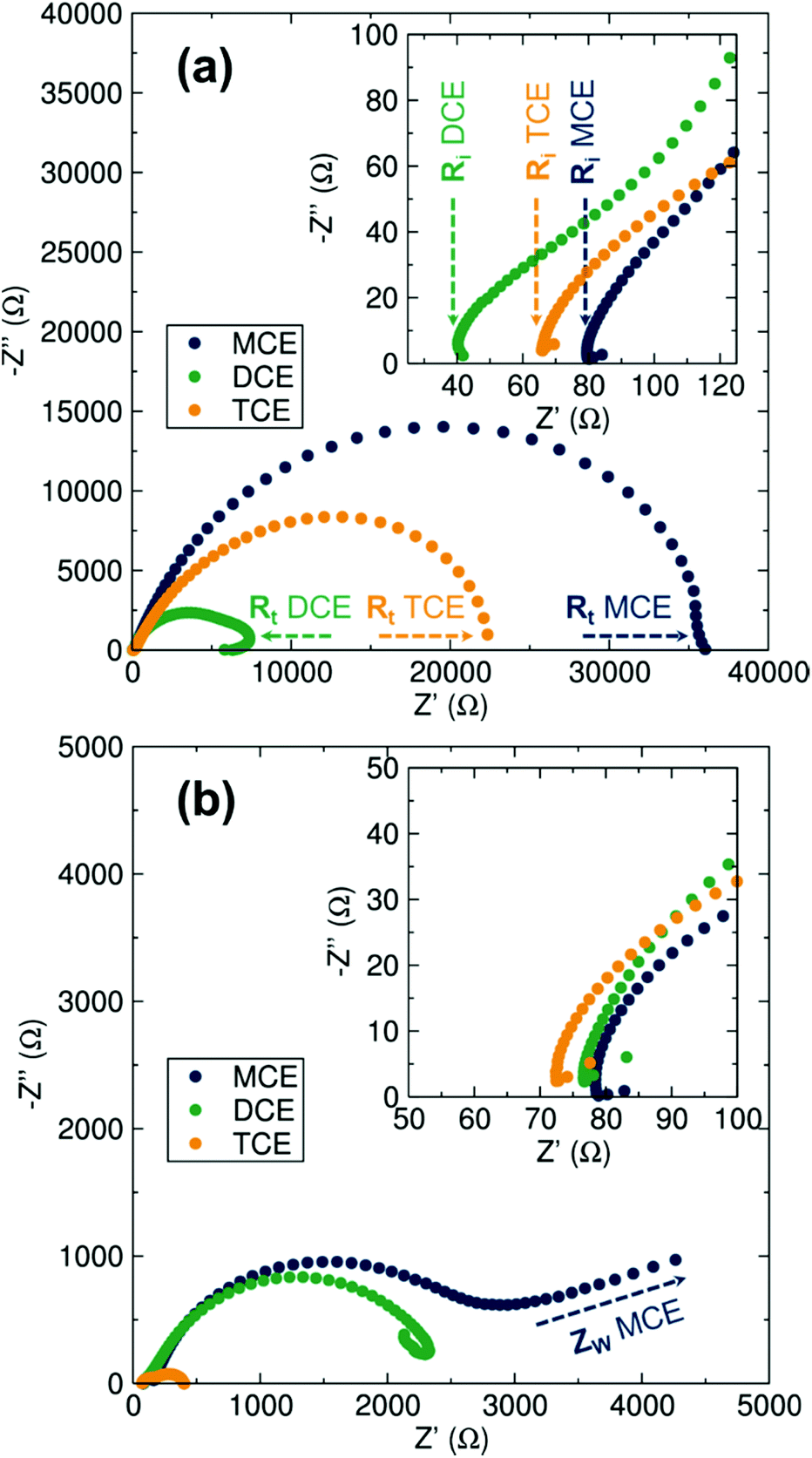 | ||
| Fig. 3 Nyquist plot of the EIS data of three different electrolytes (MCE, dark blue; DCE green; TCE orange) in Mg–metal symmetrical cells before (a) and after (b) the polarisation tests. The electrolytes resistance in the separators is indicated by the Ri label, while the total resistance of the cell including the charge transfer and any contribution from possible passivation layer is labelled as Rt. The appearance of a Warburg impedance contribution is labelled as Zw. | ||
To establish the performance of these electrolytes for future Mg battery applications, battery cells comprising each candidate electrolyte, together with a Mg–metal anode and the Mo6S8 Chevrel phase cathode material, were analysed by cyclic voltammetry (CV) and galvanostatic cycling measurements. The previously studied [Mg2Cl3·6THF]+ [(HMDS)AlCl3]− electrolyte (HMDS = hexamethyldisilazide, N(SiMe3)2) was also studied in identical full cells as a benchmark for the electrochemical properties of the Chevrel phase (Fig. S2†).11,14
The stability and compatibility of each electrolyte with the Mo6S8 Chevrel phase electrode material was first examined by means of CV experiments. MCE exhibits a robust stability up to 2.5 V with a relatively slow conditioning requirement, showing the defined redox peak of the Chevrel phase at cycle 40 (Fig. 4a). As previously noted, the electrolyte conditioning of the fully inorganic dinuclear cation-based electrolyte MACC has been reported and was attributed to the adjustment of the Mg:Al stoichiometry which allows for a degree of control over magnesium chloride cation aggregate formation.19,25
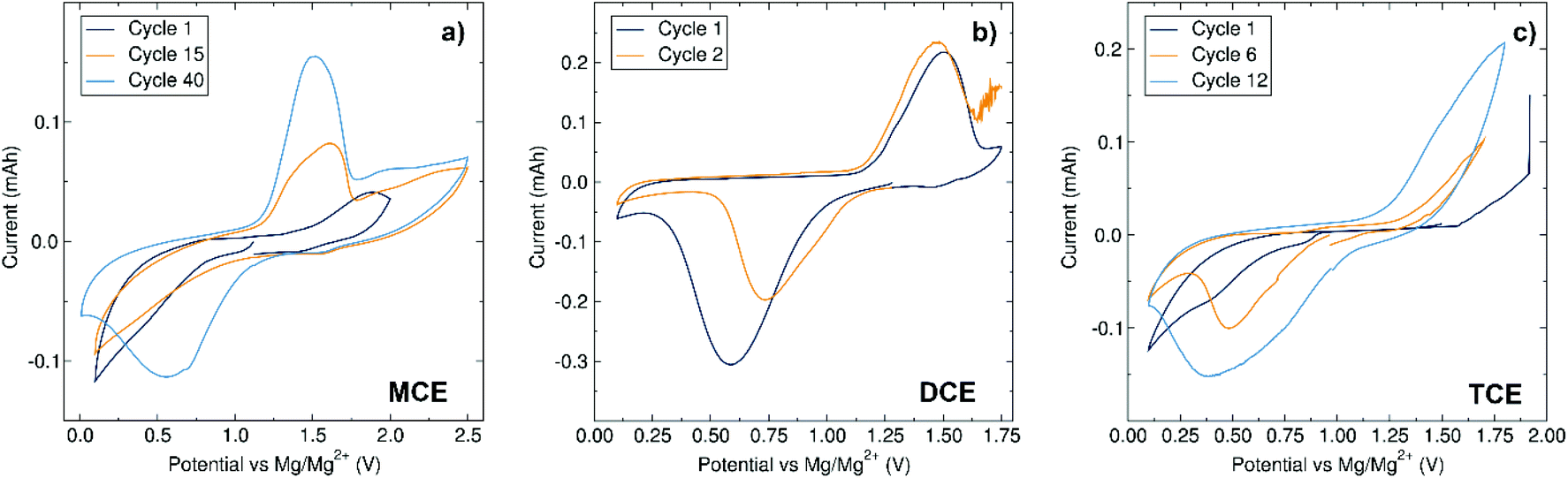 | ||
| Fig. 4 Cyclic voltammograms of Mg/Mo6S8 cells at scan rates of 0.1 mV s−1 using (a) MCE 0.35 M in THF, (b) DCE 0.175 M in THF and (c) TCE 0.117 M in MeTHF. | ||
While the chloride ligands are almost exclusively involved in driving towards equilibrium in the MACC system, we postulate that the conditioning of MCE here proceeds via a different route involving rearrangements associated with both the (Dipp)N(SiMe3) amide and the Me6TREN species. Our previous work identified the species present in a THF solution of MCE by ESI-MS where, apart from the X-ray characterised mononuclear cation [MgCl·Me6TREN]+, the Me6TREN coordinated dinuclear cation [Mg2Cl3·2Me6TREN]+ was also observed. The formation of a trinuclear magnesium cation aggregate in the presence of Me6TREN seems unlikely, but the accessibility of the dinuclear cation, presumably due to the lability of the Me6TREN arms, could potentially play a role in this conditioning mechanism.32–34 In the case of DCE, a sudden increase in the current was observed above 1.8 V, and is likely due to electrolyte decomposition (Fig. 4b). The electrochemical stability window of DCE is large enough to cycle the Chevrel phase, which displays an average charging voltage of 1.5 V vs. Mg/Mg2+. When operating the cell within this window, the redox peaks of the Chevrel phase with DCE appear well defined from the second cycle, indicative of a conditioning-free electrolyte. In regard to TCE, the CV reveals a decomposition voltage comparable to DCE at ca. 2 V (Fig. 4c), but after a short conditioning phase well-defined redox peaks are observed. The conditioning requirements are in line with observations from impedance measurements in symmetrical cells prior to cycling, following the trend of highest resistance requiring greater conditioning need: MCE displays the highest resistance of 36 kΩ and requires the strongest need for conditioning, followed by TCE which possesses a more modest need for conditioning and a resistance of 22 kΩ and finally DCE which has no apparent need for conditioning and a resistance of 7.4 kΩ. The oxidative stability of DCE and TCE is lower than its closest kin in the literature, [Mg2Cl3·6THF]+ [(HMDS)AlCl3]− (with a CV of this electrolyte presented in Fig. S2 in the ESI†). We suspect that the increased steric profile of our aryl/silyl amide with respect to HMDS weakens both the Al–N and N–Si bonds and decreases the stability of the complex, while the different electronic properties contribute further to this since it lacks the stabilisation of the two N–Si bonds emerging from the symmetry of the disilazide moiety present in HMDS. MCE possesses an oxidative stability approximately 1.0 V higher than both DCE and TCE and we associate this with the presence of Me6TREN, which can potentially interact with the aluminium anion during electrolyte conditioning. The interactive aptitude of the tetraamine ligands can be illustrated by previous studies which showed that in the presence of acid traces, new species such as [MgCl·Me6TREN]·HCl+ were observed.15 Knowing the strong Lewis acidic nature of AlCl3 and related compounds, it would not be unexpected to consider an interaction between a Me6TREN unit and [(Dipp)(SiMe3)NAlCl3]−. Future work includes investigating the 29Si NMR of the electrolytes after cycling to investigate these structural arrangements.
To evaluate the aggregation effect on the resulting capacities, galvanostatic cycling at a rate of C/10 was performed at room temperature (Fig. 5). All three cells were cycled between 0.2 V and 1.5 V to inhibit decomposition of the electrolytes and directly compare their capacities. The first discharge cycles for all three electrolytes give similar capacity values, ranging from ca. 125 to 145 mA h g−1 which are close to the theoretical capacity value for Mo6S8 (128 mA h g−1). The small additional capacity could arise from electrolyte decomposition or capacitive behaviour at the electrode surfaces during the first cycle. The capacity of the subsequent cycles is reduced, a phenomenon commonly observed with the Chevrel phase which is known to trap some Mg2+ ions within the host structure after the first cycle.35–37 In agreement with the results observed from CV experiments, MCE displays slow conditioning behaviour, with capacities below 20 mA h g−1 in the first cycles before increasing to ∼70 mA h g−1 after 50 cycles and maintaining a robust profile over 100 cycles. DCE shows a slow increase in discharge capacity from ∼45 mA h g−1 up to ∼60 mA h g−1 after 100 cycles. Comparatively, the charge/discharge performance in terms of capacity is lowest for TCE in MeTHF, with capacities not exceeding 50 mA h g−1 after 100 cycles. The TCE system shows the least pronounced increase in specific capacity with conditioning, from 27 to 50 mA h g−1. This electrolyte displays a slower conditioning process and less consistent capacity retention compared to MCE and DCE. The larger size of the trinuclear cation [Mg3Cl5·6MeTHF]+ is a potential hindrance to ionic diffusion and mobility of the active species in MeTHF. The volume of the crystallographically characterised cations increases as follows: [MgCl·Me6TREN]+ (256.9 Å3), [Mg2Cl3·6THF]+ (484.8 Å3) and [Mg3Cl5·6MeTHF]+ (621.6 Å3). The size of the ionic species in an electrolytic solution is often responsible for lowering of the ionic diffusion of the ions, reducing their mobility, hence affecting charge transfer during charging and discharging of the battery. This low ionic diffusion is compensated at higher voltage, thus the reason for the higher oxidation peak and more sluggish kinetics observed in the CV of TCE. In addition to ion mobility challenges, greater cation aggregation at the electrode/electrolyte interface can also negatively affect the battery performance, as greater energy input will be required to deposit/intercalate Mg2+ cations at this more aggregated interface.
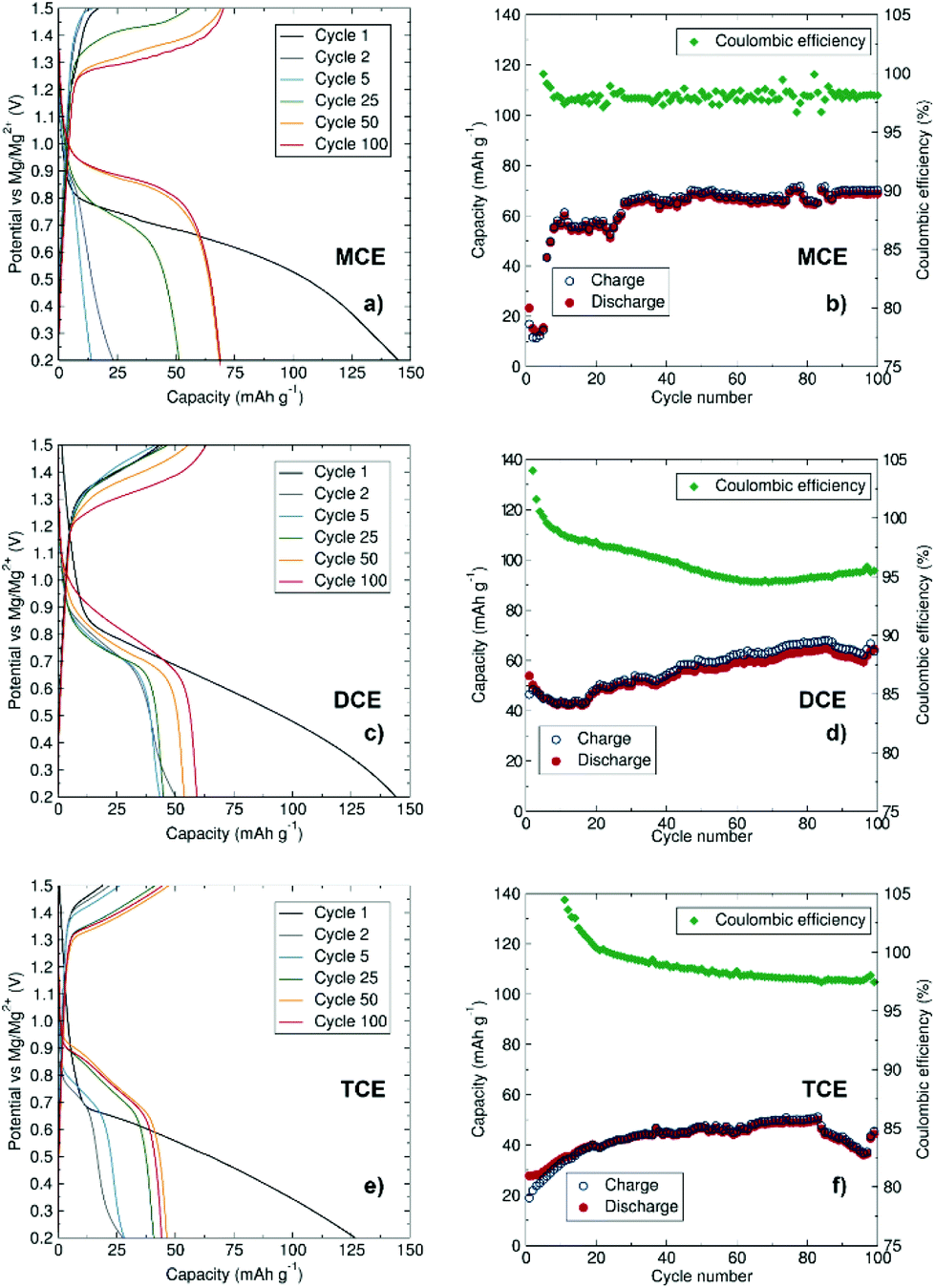 | ||
| Fig. 5 Mg/Mo6S8 cells, at a C/10 rate (12.8 mA g−1) (a) charge/discharge profile and (b) cyclic efficiency of 0.35 M MCE in THF; (c) charge/discharge profile and (d) cyclic efficiency of 0.175 M DCE in THF; (e) charge/discharge profile and (f) cyclic efficiency of 0.117 M TCE in MeTHF. | ||
We observe lower conductivity values for MCE, likely due to the Me6TREN ligand coordinating more strongly compared to the THF and MeTHF molecules in DCE and TCE, respectively, thereby restricting Mg2+ diffusion in the MCE electrolyte. Similar impeded diffusion has been reported for Mg2+–BH4− clusters.38 Setting a consistent Mg2+ concentration for DCE and TCE naturally implies a higher concentration of coordinating Cl− ions, which may result in a larger cluster size and hence a lower diffusion coefficient. In the case of TCE, the MeTHF solvent has a lower dielectric constant than THF, implying a poorer ability to solvate Mg2+ cations which may result in a larger Mg–Cl type cluster that may impinge on the diffusion properties.26 A complete analysis of the complex dynamics in these systems which affect Mg2+ diffusion properties in different electrolytic systems is still required, but for the purposes of our study, consistent Mg2+ concentrations were selected as a basis for examining cation aggregation properties across these three systems.
To further analyse the performance of MCE and DCE, the upper voltage limit was increased to 2 V (Fig. S3 and S4†). In this case, the capacity of the cell with MCE shows a quicker conditioning at the expense of a lower overall capacity which can be attributed to premature decomposition of the electrolyte. From the early charge/discharge cycles of DCE, it is clear that the higher maximum voltage improves the overpotential and enhances the specific capacity of the cell, reaching 87 mA h g−1 on the second discharge which is higher than the specific capacity observed with the benchmark [(HMDS)AlCl3]–[Mg2Cl3·6THF]+ material (77 mA h g−1 in Fig. S4†). This higher performance is likely due to the increased stability of the electrolyte at higher voltages after conditioning which enables further deintercalation of Mg ions from the Chevrel cathode material above 1.6 V, without the need to modify the material surface as recently reported.39Ex situ PXRD of the cycled cathode in DCE (Fig. S5†) confirms Mg2+ intercalation into the Chevrel crystal structure with a displacement of the Bragg reflections towards higher d-spacing and the presence of additional reflections corresponding to the MgMo6S8 phase.
A major advantage of Mg-ion batteries over their lithium counterparts is the apparent absence of dendrite formation. Recent studies have explored the influence of Mg electrolyte nature and composition on the plating topology at the surface of the Mg–metal anode.28,40–42 For example, the morphology of Mg deposition using bis(trifluoromethane)sulfonylimide (TFSI) based electrolytes is driven by the decomposition of TFSI at the electrode surface, which inhibits Mg growth in some areas due to the formation of a passivating layer and affects the regularity of the plating.42 The effect on Mg deposition with our three electrolyte systems was evaluated by SEM and EDX analysis. While SEM of the cycled magnesium anode shows no evidence for surface dendritic Mg growth for any of our electrolytes, there are electrolyte-dependant differences observable after 100 cycles (Fig. 6). The surface morphology of Mg reversible deposition is relatively smooth across all three samples, with the presence of small porous cavities which do not compromise the safety of the battery, as well as trapped glass fibres from the glass fibre separator post-cycling. The difference between electrolytes lies in the elemental composition of the deposit identified by EDX mapping. Al deposition is exclusively observed when MCE and DCE are used as electrolytes, but this is not the case with TCE (Fig. S8–S10†). The participation of Al in the reversible deposition of Mg in Mg-ion batteries is a phenomenon previously reported in the literature and is not unexpected when considering the more positive reduction potential of Al (−1.67 V vs. NHE) compared to Mg (−2.35 V vs. NHE). Aluminium deposition in rechargeable Mg-ion batteries is associated with a redistribution of the original Al:Mg ratio during conditioning. In this case, the Al:Mg ratio of 1:3 for TCE is likely too deficient in aluminium to allow the Al3+ cations to participate meaningfully in the deposition process.19
 | ||
| Fig. 6 SEM images of the Mg–metal electrode surface after galvanostatic cycling of the Mg/Mo6S8 cells containing the MCE (a, b), DCE (c, d) and TCE (e, f) electrolytes. | ||
C. Conclusions
The nature of cation aggregation of the electrolyte species has a pronounced effect on the resulting Mg-ion performance and it is clear that developing our understanding of these aggregate species can provide insights into improving these performances. Compared to the more widely observed dinuclear cation [Mg2Cl3·6THF]+, the mononuclear cation [MgCl·Me6TREN]+ affords higher specific capacity after electrolyte conditioning. The conditioning of MCE is suspected to originate from the presence of the Me6TREN Lewis donor, whose tetradenticity results in a tighter hold on the Mg metal centre compared to the monodentate, labile THF ligands. The active redox species in the MCE system is thus likely to be different to the crystallographically characterised species and further in situ investigations are underway to determine the nature of the active species. We attribute the observed greater oxidative stability of the MCE electrolyte over the DCE and TCE species to the formation of weaker Al–N and N–Si bonds. Since the anion species is common across these three systems, this suggests that the anion itself likely undergoes some degree of transformation during cycling. The versatility of Me6TREN affords the ability to adapt to structural needs owing to its coordinative flexibility arising from the lability of its arms. The Me6TREN basicity can promote interactions with acidic species, making it our prime eligible suspect for the enhanced MCE oxidative stability. This discovery could be a cornerstone in studying the effect of donor ligands such as Me6TREN as additives to rechargeable magnesium battery electrolytes to improve their oxidative stability, and enable their application with high voltage cathode materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Carnegie Trust for the Universities of Scotland for a Collaborative Research Grant (No. 50425) for generously supporting this research. SC gratefully acknowledges EPSRC funding (EP/N001982/2).Notes and references
- M. Armand and J.-M. Tarascon, Building better batteries, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- D. Larcher and J.-M. Tarascon, Towards greener and more sustainable batteries for electrical energy storage, Nat. Chem., 2015, 7, 19 CrossRef CAS PubMed.
- C. P. Grey and J.-M. Tarascon, Sustainability and in situ monitoring in battery development, Nat. Mater., 2017, 16, 45 CrossRef PubMed.
- J. Muldoon, C. B. Bucur and T. Gregory, Quest for nonaqueous multivalent secondary batteries: magnesium and beyond, Chem. Rev., 2014, 114, 11683 CrossRef CAS PubMed.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Prototype systems for rechargeable magnesium batteries, Nature, 2000, 407, 724 CrossRef CAS PubMed.
- J. Muldoon, C. B. Bucur and T. Gregory, Fervent Hype behind Magnesium Batteries: An Open Call to Synthetic Chemists-Electrolytes and Cathodes Needed, Angew. Chem., Int. Ed., 2017, 56, 12064 CrossRef CAS PubMed.
- Z. Lu, A. Schechter, M. Moshkovich and D. Aurbach, On the electrochemical behavior of magnesium electrodes in polar aprotic electrolyte solutions, J. Electroanal. Chem., 1999, 466, 203 CrossRef CAS.
- H. Kuwata, M. Matsui and N. Imanishi, Passivation Layer Formation of Magnesium Metal Negative Electrodes for Rechargeable Magnesium Batteries, J. Electrochem. Soc., 2017, 164, A3229 CrossRef CAS.
- C. Liebenow, Reversibility of electrochemical magnesium deposition from Grignard solutions, J. Appl. Electrochem., 1997, 27, 221 CrossRef CAS.
- N. Pour, Y. Gofer, D. T. Major and D. Aurbach, Structural Analysis of Electrolyte Solutions for Rechargeable Mg Batteries by Stereoscopic Means and DFT Calculations, J. Am. Chem. Soc., 2011, 133, 6270 CrossRef CAS PubMed.
- Z. Zhao-Karger, X. Zhao, O. Fuhr and M. Fichtner, Bisamide based non-nucleophilic electrolytes for rechargeable magnesium batteries, RSC Adv., 2013, 3, 16330 RSC.
- T. Liu, Y. Shao, G. Li, M. Gu, J. Hu, S. Xu, Z. Nie, X. Chen, C. Wang and J. Liu, A facile approach using MgCl2 to formulate high performance Mg2+ electrolytes for rechargeable Mg batteries, J. Mater. Chem. A, 2014, 2, 3430 RSC.
- Z. Zhao-Karger, J. E. Mueller, X. Zhao, O. Fuhr, T. Jacob and M. Fichtner, Novel transmetalation reaction for electrolyte synthesis for rechargeable magnesium batteries, RSC Adv., 2014, 4, 26924 RSC.
- H. S. Kim, T. S. Arthur, G. D. Allred, J. Zajicek, J. G. Newman, A. E. Rodnyansky, A. G. Oliver, W. C. Boggess and J. Muldoon, Structure and compatibility of a magnesium electrolyte with a sulphur cathode, Nat. Commun., 2011, 2, 427 CrossRef PubMed.
- E. V. Brouillet, A. R. Kennedy, K. Koszinowski, R. McLellan, R. E. Mulvey and S. D. Robertson, Exposing elusive cationic magnesium–chloro aggregates in aluminate complexes through donor control, Dalton Trans., 2016, 45, 5590 RSC.
- E. G. Nelson, J. W. Kampf and B. M. Bartlett, Enhanced oxidative stability of non-Grignard magnesium electrolytes through ligand modification, Chem. Commun., 2014, 50, 5193 RSC.
- J. Zhu, Y. Guo, J. Yang, Y. Nuli, F. Zhang, J. Wang and S. I. Hirano, Halogen-free boron based electrolyte solution for rechargeable magnesium batteries, J. Power Sources, 2014, 248, 690 CrossRef CAS.
- T. Liu, J. T. Cox, D. Hu, X. Deng, J. Hu, M. Y. Hu, J. Xiao, Y. Shao, K. Tang and J. Liu, A fundamental study on the [(μ-Cl)3Mg2(THF)6]+ dimer electrolytes for rechargeable Mg batteries, Chem. Commun., 2015, 51, 2312 RSC.
- C. J. Barile, E. C. Barile, K. R. Zavadil, R. G. Nuzzo and A. A. Gewirth, Electrolytic Conditioning of a Magnesium Aluminum Chloride Complex for Reversible Magnesium Deposition, J. Phys. Chem. C, 2014, 118, 27623 CrossRef CAS.
- S. He, K. V. Nielson, J. Luo and T. L. Liu, Recent advances on MgCl2 based electrolytes for rechargeable Mg batteries, Energy Storage Mater., 2017, 8, 184 CrossRef.
- P. Canepa, G. Sai Gautam, D. C. Hannah, R. Malik, M. Liu, K. G. Gallagher, K. A. Persson and G. Ceder, Odyssey of Multivalent Cathode Materials: Open Questions and Future Challenges, Chem. Rev., 2017, 117, 4287 CrossRef CAS PubMed.
- X. Sun, P. Bonnick, V. Duffort, M. Liu, Z. Rong, K. A. Persson, G. Ceder and L. F. Nazar, A high capacity thiospinel cathode for Mg batteries, Energy Environ. Sci., 2016, 9, 2273 RSC.
- M. Á. Fuentes, A. Zabala, A. R. Kennedy and R. E. Mulvey, Structural Diversity in Alkali Metal and Alkali Metal Magnesiate Chemistry of the Bulky 2,6-Diisopropyl-N-(trimethylsilyl)anilino Ligand, Chem. – Eur. J., 2016, 22, 14968 CrossRef CAS PubMed.
- I. Shterenberg, M. Salama, H. D. Yoo, Y. Gofer, J.-B. Park, Y.-K. Sun and D. Aurbach, Evaluation of (CF3SO2)2N− (TFSI) Based Electrolyte Solutions for Mg Batteries, J. Electrochem. Soc., 2015, 162, A7118 CrossRef CAS.
- K. A. See, K. W. Chapman, L. Zhu, K. M. Wiaderek, O. J. Borkiewicz, C. J. Barile, P. J. Chupas and A. A. Gewirth, The Interplay of Al and Mg Speciation in Advanced Mg Battery Electrolyte Solutions, J. Am. Chem. Soc., 2016, 138, 328 CrossRef CAS PubMed.
- P. Canepa, S. Jayaraman, L. Cheng, N. N. Rajput, W. D. Richards, G. S. Gautam, L. A. Curtiss, K. A. Persson and G. Ceder, Elucidating the structure of the magnesium aluminum chloride complex electrolyte for magnesium-ion batteries, Energy Environ. Sci., 2015, 8, 3718 RSC.
- K. A. See, Y.-M. Liu, Y. Ha, C. J. Barile and A. A. Gewirth, Effect of Concentration on the Electrochemistry and Speciation of the Magnesium Aluminum Chloride Complex Electrolyte Solution, ACS Appl. Mater. Interfaces, 2017, 9, 35729 CrossRef CAS PubMed.
- M. S. Ding, T. Diemant, R. Jürgen Behm, S. Passerini and G. A. Giffin, Dendrite Growth in Mg Metal Cells Containing Mg(TFSI)2/Glyme Electrolytes, J. Electrochem. Soc., 2018, 165, A1983 CrossRef CAS.
- R. Davidson, A. Verma, D. Santos, F. Hao, C. Fincher, S. Xiang, J. Van Buskirk, K. Xie, M. Pharr, P. P. Mukherjee and S. Banerjee, Formation of Magnesium Dendrites during Electrodeposition, ACS Energy Lett., 2019, 4, 375 CrossRef CAS.
- P. Canepa, G. S. Gautam, R. Malik, S. Jayaraman, Z. Rong, K. R. Zavadil, K. Persson and G. Ceder, Understanding the Initial Stages of Reversible Mg Deposition and Stripping in Inorganic Nonaqueous Electrolytes, Chem. Mater., 2015, 27, 3317 CrossRef CAS.
- J. H. Ha, B. Adams, J.-H. Cho, V. Duffort, J. H. Kim, K. Y. Chung, B. W. Cho, L. F. Nazar and S. H. Oh, A conditioning-free magnesium chloride complex electrolyte for rechargeable magnesium batteries, J. Mater. Chem. A, 2016, 4, 7160 RSC.
- T. Cadenbach, E. Hevia, A. R. Kennedy, R. E. Mulvey, J.-A. Pickrell and S. D. Robertson, Accessing low denticity coordination modes of a high denticity tripodal ligand to complete its coordinative repertoire, Dalton Trans., 2012, 41, 10141 RSC.
- D. R. Armstrong, M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Monomerizing alkali-metal 3,5-dimethylbenzyl salts with tris(N, N-dimethyl-2-aminoethyl)amine (Me6TREN): structural and bonding implications, Inorg. Chem., 2013, 52, 12023 CrossRef CAS PubMed.
- D. R. Armstrong, C. M. M. Harris, A. R. Kennedy, J. J. Liggat, R. McLellan, R. E. Mulvey, M. D. T. Urquhart and S. D. Robertson, Developing lithium chemistry of 1,2-dihydropyridines: from kinetic intermediates to isolable characterized compounds, Chem. – Eur. J., 2015, 21, 14410 CrossRef CAS.
- M. D. Levi, E. Lancry, H. Gizbar, Z. Lu, E. Levi, Y. Gofer and D. Aurbach, Kinetic and Thermodynamic Studies of Mg2+ and Li+ Ion Insertion into the Mo6S8 Chevrel Phase, J. Electrochem. Soc., 2004, 151, A1044 CrossRef CAS.
- M. D. Levi, H. Gizbar, E. Lancry, Y. Gofer, E. Levi and D. Aurbach, A comparative study of Mg2+ and Li+ ion insertions into the Mo6S8 Chevrel phase using electrochemical impedance spectroscopy, J. Electroanal. Chem., 2004, 569, 211 CrossRef CAS.
- J. Richard, A. Benayad, J. F. Colin and S. Martinet, Charge Transfer Mechanism into the Chevrel Phase Mo6S8 during Mg Intercalation, J. Phys. Chem. C, 2017, 121, 17096 CrossRef CAS.
- D. Samuel, C. Steinhauser, J. G. Smith, A. Kaufman, M. D. Radin, J. Naruse, H. Hiramatsu and D. J. Siegel, Ion Pairing and Diffusion in Magnesium Electrolytes Based on Magnesium Borohydride, ACS Appl. Mater. Interfaces, 2017, 9, 43755 CrossRef CAS PubMed.
- D. Muthuraj and S. Mitra, Reversible Mg insertion into chevrel phase Mo6S8 cathode: Preparation, electrochemistry and X-ray photoelectron spectroscopy study, Mater. Res. Bull., 2018, 101, 167 CrossRef CAS.
- S. Y. Ha, Y. W. Lee, S. W. Woo, B. Koo, J. S. Kim, J. Cho, K. T. Lee and N. S. Choi, Magnesium(II) bis(trifluoromethane sulfonyl) imide-based electrolytes with wide electrochemical windows for rechargeable magnesium batteries, ACS Appl. Mater. Interfaces, 2014, 6, 4063 CrossRef CAS PubMed.
- R. E. Doe, R. Han, J. Hwang, A. J. Gmitter, I. Shterenberg, H. D. Yoo, N. Pour and D. Aurbach, Novel, electrolyte solutions comprising fully inorganic salts with high anodic stability for rechargeable magnesium batteries, Chem. Commun., 2014, 50, 243 RSC.
- H. D. Yoo, S.-D. Han, I. L. Bolotin, G. M. Nolis, R. D. Bayliss, A. K. Burrell, J. T. Vaughey and J. Cabana, Degradation Mechanisms of Magnesium Metal Anodes in Electrolytes Based on (CF3SO2)2N− at High Current Densities, Langmuir, 2017, 33, 9398 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, further electrochemical characterisation, X-ray diffraction, electron microscopy and EDX mapping. See DOI: 10.1039/c9qi01606f |
| This journal is © the Partner Organisations 2020 |